


Abstract
We recently proposed to classify proteins by their functional surfaces. Using the structural attributes of functional surfaces, we inferred the pairwise relationships of proteins and constructed an expandable database of protein surface classification (PSC). As the functional surface(s) of a protein is the local region where the protein performs its function, our classification may reflect the functional relationships among proteins. Currently, PSC contains a library of 1974 surface types that include 25 857 functional surfaces identified from 24 170 bound structures. The search tool in PSC empowers users to explore related surfaces that share similar local structures and core functions. Each functional surface is characterized by structural attributes, which are geometric, physicochemical or evolutionary features. The attributes have been normalized as descriptors and integrated to produce a profile for each functional surface in PSC. In addition, binding ligands are recorded for comparisons among homologs. PSC allows users to exploit related binding surfaces to reveal the changes in functionally important residues on homologs that have led to functional divergence during evolution. The substitutions at the key residues of a spatial pattern may determine the functional evolution of a protein. In PSC (http://pocket.uchicago.edu/psc/), a pool of changes in residues on similar functional surfaces is provided.
INTRODUCTION
Characterizing protein function and classifying proteins into proper families are two major goals in the study of proteins. The commonly accepted definition of a protein family is a group of proteins that share similar sequences, structures and functions that are derived from a common ancestor. Well-known classifications, such as Pfam (1), COG (2), structural classification of proteins (SCOP) (3) and class, architecture, topology, homologous superfamily (CATH) (4) have provided biological insights into protein structure, function and evolution. However, two proteins may have diverged so much, such that their homology is no longer evident at the sequence or global structural level, making it challenging to decide if the two proteins are functionally related. This underscores the importance of identifying local structural regions that are well conserved in evolution (5,6).
Protein classification has important missions, such as the identification of binding sites involved in biochemical reactions, characterization of related proteins that share common core functions and identification of the evolutionary forces that affect functional divergence during protein evolution. Using protein functional surfaces as the basis for classification may achieve these purposes (7). Functional surfaces are local structures which may give immediate clues to functionally important protein regions. Most importantly, they are central units in proteins and provide site-specific information as to how a protein interacts with small molecules and other proteins. Evolutionarily, they tend to be better conserved than primary sequences. Therefore, they can be used to classify more distantly related proteins (8). Indeed, functional surfaces can even reveal relationships among proteins that belong to different folds (8–10). On the other hand, functional surfaces can also be used to detect subtle functional differences among proteins with the same fold. For example, oxophytodienoate reductase and NADPH dehydrogenase have the same fold identification of CATH 3.20.20.70 (Aldolase class I). However, their Enzyme Commission (EC) annotations are EC 1.3.1.42 and EC 1.6.99.1, so they actually have different enzymatic functions.
Our approach relies on pairwise surface structural similarities (7,8,11,12). As the computational cost is extremely heavy for an exhaustive pairwise comparison of all local putative surfaces, we focused on the functional surfaces of bound forms (i.e. proteins with ligands), because they provide not only abundant biological information but also fixed binding shapes. We first carried out a coarse classification by pairwise local RMSD measures and grouped approximately 24 000 bound structures into approximately 2000 surface types. Each surface type was then refined into surface subtypes by structural attributes. A major strength of our approach is that we consider the characteristics of spatial patterns, physiochemical texture and evolutionary conservation. We called it protein surface classification (PSC). PSC includes the largest database of protein functional surface classification and it has been expandable. Each surface in PSC includes geometric measurements and structural attributes, which form a profile (i.e. a surface signature). We calculated the local structural relationships of functional homologs in protein families using a functional inference technique. These features can be used to exploit similar functional surfaces for revealing interchangeability between functionally important residues (see an example below). In addition, the binding ligands of homologs can provide structural information as to how a protein potentially interacts with a variety of ligands, which may give a clue for developing therapeutic drugs. Finally, PSC provides a framework for classifying unbound structures.
PSC LIBARAY AND DATA ACCESS
The PSC database was constructed as follows. First, we collected the bound structures from 24 170 entries of Protein Data Bank (PDB) (13), which included a total of 25 857 chains. Then, using an automated pipeline, we identified the binding surfaces of each bound form (9,14) and calculated their geometric measurements, including the composition of a spatial pattern, solvent accessible area and molecular volume. In addition, we provided biological annotations via cross-links to UniProt (15). Enzyme annotations from EC (16) and fold terms from CATH are provided. We also allow users to access all putative binding surfaces along with their corresponding evolutionary conservation and geometric measurements. Most importantly, structurally similar or functionally related binding surfaces across species are associated with each other and characterized by structural attributes.
PSC is freely accessible at http://pocket.uchicago.edu/psc/ and the detailed file format is also provided.
CLUSTERING METHOD BASED ON AN AGGLOMERATIVE APPROACH
To establish PSC, we applied a clustering analysis on these 25 857 identified binding surfaces by an agglomerative approach (7). We first conducted exhaustive pairwise local surface comparisons. We then grouped similar surfaces into a surface type at a threshold of structural similarity based on the local RMSD P ≤ 10−4. Each surface type is uniquely represented by a center which is the member with the highest degree of connections and with the smallest mean RMSD that possesses the most generic spatial pattern for the surface type. As a result, we classified these 25 857 binding surfaces into 1974 surface types by clustering local structures.
DISCOVERING STRUCTURAL HOMOLOGS IN A SURFACE TYPE
A user can submit a PDB code as a query to PSC. A functional surface hit will be displayed on the pre-computed result page and can be visualized interactively through the JMOL plugin (17). For example, it is fully customized for selecting site-specific residues on a spatial pattern. Each surface in PSC contains the detailed geometric measurements and structural attributes, including the residue composition, polar solvent accessible area, apolar solvent accessible area, sphericity, anisotropic, surface density, skewness and kurtosis. These selected structural attributes are extracted and integrated to produce a profile for the query.
PSC can also compute the local structural relationships among the homologs within a surface type. These pre-computed structural homologs contain both their EC annotations and CATH identifications. Users may launch a new browser window to reconstruct a structural phylogeny and compute pairwise distances based on RMSD measures with P-values for statistical evaluation. Importantly, these local pairwise relationships allow building a structural phylogeny to understand protein functional divergence.
We use a familiar protein, human alcohol dehydrogenase (ADH, PDB1htb with chain A), as an example to show what information PSC provides. PSC first gives an overview of geometric measurements and produces a structural visualization as shown in Figure 1. ADH interacts with a cofactor nicotinamide adenine dinucleotide (NAD). The identified functional surface contains a spatial pattern of 38 residues, a solvent accessible area of 676.95 Å2 and a molecular volume of 827.54 Å3. R47, T48, H51 and L57 are the catalytic residues involved in the reactivity of ADH, which is annotated with EC 1.1.1.1 and a fold identification of CATH 3.90.180.10 and 3.40.50.720. The detailed biological annotation from UniProt can be accessed through the accession number of P00325. The R47H mutant of ADH destabilizes the interaction with the cofactor NAD and affects the ability of catalyzing alcohol. This phenotypic mutant explains a low risk of alcoholism (18). Moreover, the mass center of the functional surface is located at (−5.76, 11.59, −28.48), while the global mass center of the protein is located at (1.21, 11.13, −26.71). The distance between the two centers, called the anisotropic distance, is 7.2 Å (19). Previous studies (7,19) have shown that protein binding sites tend to be close to the mass center of a protein. In a large-scale computation, we found that a functional surface has an average anisotropic distance of 10.28 Å with a standard deviation of 5.15 Å. This well-characterized distance, therefore, is useful for predicting the binding site of a protein.
Figure 1.

Identification and characterization of the functional surface of human alcohol dehydrogenase (ADH). The geometric features and functional, fold and biological annotations are highlighted. The binding pocket with the cofactor NAD (red) was predicted using the SplitPocket algorithm (10). The pocket contains a cluster of 38 binding residues (green) with catalytic residues (pink) such as R47, T48, H51 and L57. Among the pocket residues, the halo-gold color means a selected residue and halo-blue indicates the currently selected residue, for example, R47. The UniProt and EC annotations, and the CATH terms are also provided.
PSC also provides a surface signature for a query. In Figure 2, it contains a profile from the structural attributes in terms of geometric features, physicochemical textures and evolutionary conservation. These structural attributes have been normalized to be between −1 and 1 to serve as descriptors. This computed profile captures the surface characteristics of a protein. For example, the shape of the whole ADH structure with a sphericity of 0.51 is almost the same as that of its binding pocket despite of different orientations. However, the distribution of the atoms of ADH has a perfect symmetric shape with a skewness of 0, while that of its binding pocket has a skewness of 0.3. We also found that the apolar solvent accessible area of 1172.54 Å2 in the binding pocket is much wider than the polar area of 601.49 Å2, which may give favorable hydrophobic interaction with the cofactor NAD. By comparing the two computed profiles, one may make a functional inference in shape analysis through the assessment of similarity between two binding surfaces and determine whether their surface types have come from a common ancestor.
Figure 2.
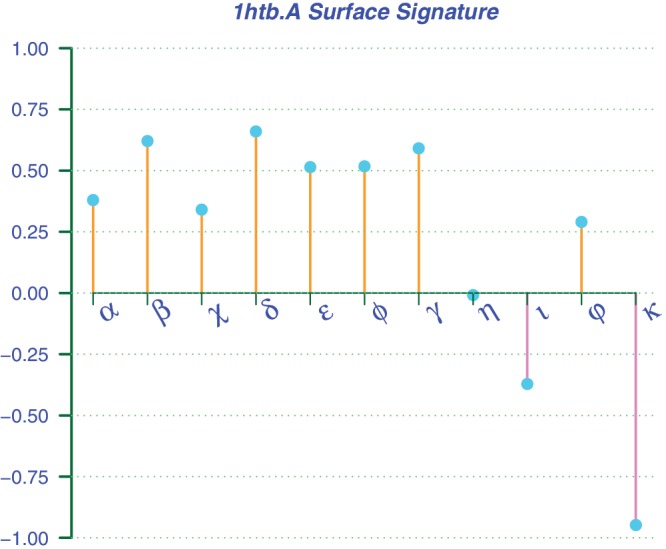
The surface signature of human alcohol dehydrogenase (PDB1htb.A). The structural attributes as descriptors have been normalized, so that their values are between 1 and −1. The selected descriptors include global polar solvent accessible area (α), global apolar solvent accessible area (β), local polar solvent accessible area (χ), local apolar solvent accessible area (δ), global sphericity (ε), local sphericity (φ), local surface density (γ), global skewness (η), global kurtosis (ι), local skewness (ρ) and local kurtosis (κ). From this profile, one can see that the symmetric shape (η = 0) of the whole structure is similar to that of its functional surface (ε = φ ≈ 0.51), which also contains a much wider apolar solvent accessible area (δ = 0.66) than the polar area (χ = 0.34).
We have set our goal to achieve a better understanding of protein molecular function and structural evolution. Therefore, PSC provides a pre-computed list of related members from the same surface type. For example, the surface type of ADH includes 75 structural homologs across many species including human, horse, mouse, Gadus callarias (fish), Rana perezi (frog), Scaptodrosophila lebanonensis (fly), Arabidopsis thaliana (plant), Sulfolobus solfataricus (archaea) and Pseudomonas aeruginosa (bacteria). These homologs share a core function which was already present in their common ancestor. The core function contains EC annotation(s) and CATH identification if it is identified. One may follow the link to access the profile of a member. We recorded their spatial patterns and functionally important residues as shown Figure 3, so that users can enumerate possible combinatory compositions of a binding shape similar to the query. That is, geometric considerations are taken for mapping spatial patterns to the diverse shapes of binding sites produced by evolution. Such geometric and physicochemical features are invaluable for users who are interested in drug design and directed enzyme evolution. This is because these related surfaces provide cheminformatic clues of actual binding sites from structural homologs under physicochemical constraints that have been acting on functionally important residues. The immediate benefit is to exploit similar binding surfaces to reveal the interchangeability between important residues and the patterns of how a protein surface type with essential biological functions has evolved. From these related patterns, for example, one can find the residue variants of R47 in ADH: H, P, and G (Figure 3), which have been identified in fish (H), mouse (P) and frog (G), respectively. Residue preference is also observed across species. Through screening evolutionary variants, one can effectively engineer a protein to gain a desired function, and design drugs or inhibitors in a rational manner. Moreover, this site-specific analysis gives a potential mean to study human disease associated non-synonymous single nucleotide polymorphisms (nsSNPs) through the Online Mendelian Inheritance in Man (OMIM, http://www.ncbi.nlm.nih.gov/omim/), if their geometric locations could be structurally identified. The spatial patterns provide a set of residue variants to study functional diversification and disease-associated nsSNPs (20,21). Finally, a comparison of surface members with EC annotations and CATH identifications allows users to gain structural insights into the relationship between shape and function.
Figure 3.

Multiple surface alignment of eight ADH orthologs by the first 14 binding residues of spatial pattern. The structural orthologs: (a) human (PDB1htb.A), (b) horse (PDB1a71.A), (c) mouse (PDB1e3e.A), (d) fish (PDB1cdo.A), (e) frog (PDB1p0f.A), (f) plant (PDB2cf6.A), (g) archaea (PDB1r37.A) and (h) bacteria (PDB1llu.A) with their binding cofactors (red) are shown immediately below. Their identified binding residues are colored in green, while their catalytic residues are colored in pink. In PSC, a surface type gives a collection of binding surfaces similar to a query to reveal the change in the functionally important residue variants. For example, R47 in human can potentially mutate to H, P and G as shown in (a) R47, (b) R47, (c) P47, (d) H47, (e) G1047, (f) H48, (g) H45 and (h) H39. Among them, their aligned residues are indicated in halo-blue color.
FUTURE DEVELOPMENTS
In the near future, we intend to apply the framework to unbound structures in order to establish a comprehensive surface classification. For this purpose, we have been developing a surface matching algorithm (7,8,22) to do the task of surface alignment between a bound and an unbound form. The new development will allow users to use a surface alignment method with a P-value statistical evaluation. This development should invite further exploration of structural insights into protein function, classification and evolution.
FUNDING
University of Chicago and Academia Sinica, Taiwan. Funding for open access charge: Academia Sinica, Taiwan.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Dr. Jie Liang, the University of Illinois at Chicago.
REFERENCES
- 1.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, et al. The Pfam protein families database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- 4.Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH–a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 5.Tseng YY, Liang J. Estimation of amino acid residue substitution rates at local spatial regions and application in protein function inference: a Bayesian Monte Carlo approach. Mol. Biol. Evol. 2006;23:421–436. doi: 10.1093/molbev/msj048. [DOI] [PubMed] [Google Scholar]
- 6.Tourasse NJ, Li WH. Selective constraints, amino acid composition, and the rate of protein evolution. Mol. Biol. Evol. 2000;17:656–664. doi: 10.1093/oxfordjournals.molbev.a026344. [DOI] [PubMed] [Google Scholar]
- 7.Tseng YY, Li WH. Classification of protein functional surfaces using structural characteristics. Proc. Natl Acad. Sci. USA. 2012;109:1170–1175. doi: 10.1073/pnas.1119684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng YY, Chen ZJ, Li WH. fPOP: footprinting functional pockets of proteins by comparative spatial patterns. Nucleic Acids Res. 2010;38:D288–D295. doi: 10.1093/nar/gkp900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tseng YY, Dundas J, Liang J. Predicting protein function and binding profile via matching of local evolutionary and geometric surface patterns. J. Mol. Biol. 2009;387:451–464. doi: 10.1016/j.jmb.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tseng YY, Li WH. Identification of protein functional surfaces by the concept of a split pocket. Proteins. 2009;76:959–976. doi: 10.1002/prot.22402. [DOI] [PubMed] [Google Scholar]
- 11.Binkowski TA, Adamian L, Liang J. Inferring functional relationships of proteins from local sequence and spatial surface patterns. J. Mol. Biol. 2003;332:505–526. doi: 10.1016/s0022-2836(03)00882-9. [DOI] [PubMed] [Google Scholar]
- 12.Dundas J, Adamian L, Liang J. Structural signatures of enzyme binding pockets from order-independent surface alignment: a study of metalloendopeptidase and NAD binding proteins. J. Mol. Biol. 2011;406:713–729. doi: 10.1016/j.jmb.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tseng YY, Dupree C, Chen ZJ, Li WH. SplitPocket: identification of protein functional surfaces and characterization of their spatial patterns. Nucleic Acids Res. 2009;37:W384–W389. doi: 10.1093/nar/gkp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Webb EC. Enzyme Nomenclature 1992: Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes / Prepared for NC-IUBMB by Edwin C. Webb. San Diego: Academic Press; 1992. International Union of Biochemistry and Molecular Biology. [Google Scholar]
- 17.Jmol. an open-source Java viewer for chemical structures in 3D. Available at: http://www.jmol.org/ [Google Scholar]
- 18.Borras E, Coutelle C, Rosell A, Fernandez-Muixi F, Broch M, Crosas B, Hjelmqvist L, Lorenzo A, Gutierrez C, Santos M, et al. Genetic polymorphism of alcohol dehydrogenase in Europeans: the ADH2*2 allele decreases the risk for alcoholism and is associated with ADH3*1. Hepatology. 2000;31:984–989. doi: 10.1053/he.2000.5978. [DOI] [PubMed] [Google Scholar]
- 19.Nicola G, Vakser IA. A simple shape characteristic of protein-protein recognition. Bioinformatics. 2007;23:789–792. doi: 10.1093/bioinformatics/btm018. [DOI] [PubMed] [Google Scholar]
- 20.Stitziel NO, Tseng YY, Pervouchine D, Goddeau D, Kasif S, Liang J. Structural location of disease-associated single-nucleotide polymorphisms. J. Mol. Biol. 2003;327:1021–1030. doi: 10.1016/s0022-2836(03)00240-7. [DOI] [PubMed] [Google Scholar]
- 21.Stitziel NO, Binkowski TA, Tseng YY, Kasif S, Liang J. topoSNP: a topographic database of non-synonymous single nucleotide polymorphisms with and without known disease association. Nucleic Acids Res. 2004;32:D520–D522. doi: 10.1093/nar/gkh104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tseng YY, Li WH. Evolutionary approach to predicting the binding site residues of a protein from its primary sequence. Proc. Natl Acad. Sci. USA. 2011;108:5313–5318. doi: 10.1073/pnas.1102210108. [DOI] [PMC free article] [PubMed] [Google Scholar]