


Abstract
The recent discoveries of regulatory non-coding RNAs changed our view of RNA as a simple information transfer molecule. Understanding the architecture and function of active RNA molecules requires methods for comparing and analyzing their 3D structures. While structural alignment of short RNAs is achievable in a reasonable amount of time, large structures represent much bigger challenge. Here, we present the SETTER web server for the RNA structure pairwise comparison utilizing the SETTER (SEcondary sTructure-based TERtiary Structure Similarity Algorithm) algorithm. The SETTER method divides an RNA structure into the set of non-overlapping structural elements called generalized secondary structure units (GSSUs). The SETTER algorithm scales as O(n2) with the size of a GSSUs and as O(n) with the number of GSSUs in the structure. This scaling gives SETTER its high speed as the average size of the GSSU remains constant irrespective of the size of the structure. However, the favorable speed of the algorithm does not compromise its accuracy. The SETTER web server together with the stand-alone implementation of the SETTER algorithm are freely accessible at http://siret.cz/setter.
INTRODUCTION
The transfer of genetic information was for a long time considered as the main biological function of RNA. However, recent evidence shows that all organisms contain a wealth of small untranslated RNAs (so-called non-protein coding RNAs—ncRNAs) that function in a variety of cellular processes (1,2). These findings have directly challenged our understanding of biological regulation, and extended our view of RNA as an important player in the development of complex organisms. Similar to proteins, understanding the function of these active RNA molecules requires methods for analyzing their primary (i.e. sequence), secondary (i.e. RNA motifs) and tertiary (i.e. 3D) structures. While alignment of homologous sequences can bring a great insight into RNA similarity and evolution, such methods are not satisfactory for significantly diverged sequences. As homologous molecules share conserved secondary and tertiary structures, the demand for fast and accurate methods for structural analysis at the secondary and tertiary structural levels increased substantially in the past years.
Many methods have been developed to predict, characterize, identify and analyze secondary motifs in RNA (3–6). Main limitation of these methods is their inability to predict and annotate tertiary interactions that are established between secondary structural elements, and that are important for forming the global fold of RNA (7,8). However, as both the number and size of solved 3D RNA structures dramatically increased in recent years, various computational tools for 3D RNA structural analysis started to pop up (9,10). A good starting point to assign the function of known 3D RNA structure is a similarity searching in functionally annotated databases of 3D RNA structures (11–13). The key component of any structural database is the 3D structural alignment. A number of research groups developed programs for RNA structural alignment including DIAL (14), SARA (15), SARSA (16), iPARTS (17), ARTS (18), LaJolla (19) or R3D Align (20). The common limitations of these approaches include the restriction on the size of aligned structures or the low speed when aligning large RNA molecules. In this article, we introduce a SETTER web server utilizing our novel SETTER (SEcondary sTructure-based TERtiary Structure Similarity Algorithm) algorithm (21) for RNA structure pairwise comparison. The most relevant feature of the method is that it performs at significant speeds even for largest RNA structures (no size limit is imposed on aligned structures) without compromising accuracy of the alignment.
MATERIALS AND METHODS
A principal idea of the SETTER method is to divide each RNA structure into smaller non-overlapping structural elements called generalized secondary structure units (GSSUs). A GSSU generally consists of a loop, a neck and a stem (see Figure 1). However, this is not a strict rule and some of these elements may be missing. A formal description of the GSSU is given by the following definition.
Definition 1. —
Let
be an RNA structure with a nucleotide sequence
and let
denote its subset participating in a Watson–Crick base pair. By a generalized secondary structure unit (GSSU)
we understand a pair of substrings of
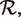
and
(i1 ≤ i2 < j1 ≤ j2, i2 = j1 − 1) of maximum lengths such that each nucleotide ntx ∈
:
i1 ≤ x ≤ i2 :
or ntx is paired with ny where j1 ≤ y ≤ j2
j1 ≤ x ≤ j2 :
or ntx is paired with ny where i1 ≤ y ≤ i2
In case of ambiguity, a maximum length is assigned to the substring occurring earlier in the sequence. Let imax and jmin be the highest/lowest indexes of the Watson–Crick paired bases in
. We define a loop as
, a stem as
∖
and a neck as the pair {ntimax,ntjmin}.
Figure 1.

Three GSSUs extracted from an RNA structure. The GSSU generation process starts at the 5′ end of the sequence. The numbers denote the order at which the GSSUs were generated.
The structure is processed in a sequence order, and each nucleotide is assigned unambiguously to only one GSSU. In the first step, nucleotides are stored on the stack until a nucleotide nti Watson–Crick (WC) bonded with a nucleotide ntj is encountered. These two residue form the neck and all nucleotides lying between them represent the loop. Non-WC pairs are not considered because they often mediate RNA tertiary contacts which would break an unambiguity in the GSSU assignment. In the next step the stem is formed from all residues lying before the residue WC bonded to the residue not stored on the stack. A detailed description of the process including all its subtleties is given in the original publication (21) and the pseudocode of the algorithm can be found in Section S1 of Supplementary Data.
Once each RNA structure in the structure pair is decomposed into GSSUs, the alignment algorithm is invoked. For the purposes of the alignment, each residue is represented by its P atom. Each pair of GSSUs is aligned independently utilizing the Kabsch RMSD algorithm (22) and each such superposition is scored by an S-distance (21) defined by Equation 1.
 |
(1) |
where  and
and 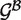 represent two GSSUs to be compared,
represent two GSSUs to be compared, 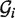 stands for the i-th nucleotide in the sequence of
stands for the i-th nucleotide in the sequence of 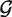 , and
, and 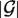 stands for the length of
stands for the length of 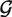 . S-distance is a sum of Euclidean distances
. S-distance is a sum of Euclidean distances 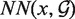 of all nearest-neighbor nucleotides modified by the factor ζ taking into account the nucleotide type (i.e. U, G, C, A), and normalized by the number of nearest neighbors γ in distance at most ϵ. The lower the S-distance, the better the superposition.
of all nearest-neighbor nucleotides modified by the factor ζ taking into account the nucleotide type (i.e. U, G, C, A), and normalized by the number of nearest neighbors γ in distance at most ϵ. The lower the S-distance, the better the superposition.
The SETTER alignment algorithm involves several steps described in Section S2 of Supplementary Data. The aligned structures are scored by the overall similarity measure denoted as 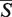 -distance. The statistical significance of the
-distance. The statistical significance of the 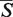 -distance is estimated by computing its P-value. The smaller the P-value, the more statistically significant the
-distance is estimated by computing its P-value. The smaller the P-value, the more statistically significant the 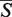 -distance, i.e. the more confident one can be that the alignment does not arise by a chance. The S-distance follows the log-normal distribution parameters of which (location parameter m and scale parameter s) depend on the length of the aligned structures. To determine the μ and σ parameters, a set of 50 000 structure pairs was prepared for each length N = 5, 10, 15, … , 300 by randomly cutting regions of the given length from 1972 RNA structures available in the PDB database by the March 2012. Details about this procedure are given in Section S3 of Supplementary Data.
-distance, i.e. the more confident one can be that the alignment does not arise by a chance. The S-distance follows the log-normal distribution parameters of which (location parameter m and scale parameter s) depend on the length of the aligned structures. To determine the μ and σ parameters, a set of 50 000 structure pairs was prepared for each length N = 5, 10, 15, … , 300 by randomly cutting regions of the given length from 1972 RNA structures available in the PDB database by the March 2012. Details about this procedure are given in Section S3 of Supplementary Data.
The 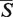 -distances were determined for all possible alignments in the data set containing the structures of the given length, and the μ and σ parameters were found by a maximum likelihood fitting of the
-distances were determined for all possible alignments in the data set containing the structures of the given length, and the μ and σ parameters were found by a maximum likelihood fitting of the 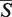 -distances distributions. The dependence of the μ and σ parameters on the length of the structure N is given by the following equations (21):
-distances distributions. The dependence of the μ and σ parameters on the length of the structure N is given by the following equations (21):
| (2) |
where a = 0.1729 and b = 14.3753.
| (3) |
where a = 7.3325 and b = 0.0136. The relations between Equations (2) and (3) provide a simple way to calculate μ and σ parameters for any sequence length N.
The performance data regarding the SETTER's speed and accuracy are available in the original paper (21). Specifically, the runtimes of the SETTER were compared with that of SARA and iPARTS (using their web server versions) with SETTER clearly outperforming these approaches. As SETTER can be used both for the pairwise structural alignment, as well as for the functional annotation of a new RNA structure, both these functionalities were also benchmarked.
The quality of the structural alignment was assessed by a comparison with the R3D Align, ARTS, SARA and DIAL methods. Various measures [an RMSD, a percentage of structural identity (PSI), a percentage of sequence identity (PID) (15,23) and a number of nucleotides aligned and a number of exact base matches (20)] reflecting the quality of the alignment of two 16S rRNA structures and of the alignment of the sarcin/ricin domain from 28S rRNA with the central part of the 5S rRNA were calculated. The results (Supplementary Data, Section S4) demonstrate that SETTER produces the structural alignments of quality comparable to these approaches. A detailed description of all benchmarks is available in (21). In addition, speed and accuracy of SETTER were compared with ARTS (18) using a set consisting of the following RNA structures: tRNA (≈70 residues), 5S rRNA (≈120 residues), 16S rRNA (≈1500 residues) and 23S rRNA (≈2700 residues). The results available in Section S5 of Supplementary Data, show that SETTER and ARTS are of comparable quality in terms of both speed and alignment accuracy.
The functional assignment was benchmarked against iPARTS and SARA utilizing the classification accuracy (ACC) and the area under the ROC curve (AUC) measures for three data sets from the SCOR database. SETTER performs better than iPARTS and SARA in terms of AUC and is comparable with SARA in terms of ACC (21).
WEB SERVER
SETTER web server is designed as a user-friendly and straightforward interface to the SETTER algorithm. RNA structures can be compared in two modes: in a single mode and in a batch mode. In the single mode, two RNA structures are aligned, and in the batch mode the query structure is aligned with every structure in a target data set. Temporary files related to finished queries are stored at the server for 10 days, and every user receives a unique URL address to check the results within this period of time.
The SETTER algorithm is written in the C++ programming language and is available as a standalone binary. The SETTER web server is implemented in the Python web framework Django. All communication between the server and the binary is realized through XML files. Server's front-end is implemented in the standard HTML markup language using Javascript programming language and AJAX technologies utilizing the jQuery library. The functionality of the web server is fully described in an integrated help system, and the server can be easily tested using the provided example data (Figure 2F). The server runs under the Linux operating system on a virtual machine with a dual core 3.00 GHz Intel(R) Xeon(R) processor. The SETTER web server as well as the accompanied binary package are freely available at http://siret.cz/setter.
Figure 2.

User interface of the SETTER web server. (A) Structure input. (B) The batch mode input. (C) Checkbox to force the use of up-to-date PDB files. (D) SETTER parameters. (E) Information about the progress of the SETTER job. (F) Example data and help system controls.
Input
In the single mode, structures can be submitted either as PDB IDs or they can be uploaded as user-defined PDB files (Figure 2A). In the batch mode, the target set can be defined using the PDB IDs only (Figure 2B). If structures are identified by the PDB IDs, a local cache is checked for the presence of the input PDB files. If they are not found they are downloaded from the PDB database (24). The download of the up-to-date files from the PDB database may also be forced by a user (Figure 2C).
Once the structures are loaded to the system, the WC hydrogen-bonded base pairs are located using the 3DNA program (25) run with its implicit settings. The 3DNA output files are parsed and the SETTER input XML files are generated. The SETTER application performs the required calculation, and the output XML files are created. If the alignment was not successful, no XML files are generated, the job stops and a user is notified. The progress of the running job can be tracked using the unique URL provided for every job (Figure 2E). Based on the information stored in the SETTER output XML files the Results screen (Figure 3) is generated.
Figure 3.

The results screen. (A) The web server tab system. (B) Left panel contains many details about the alignment. (C) Aligned structures are visualized using the Jmol applet. (D) The controls of the visualization. (E) Visualization of the decomposition of the structures into individual GSSUs. (F) Visualization of aligned residues. (G) Highlighting of the nearest neighbor nucleotides defined by the adjustable distance range in Å. (H) Results of the batch mode are displayed as vertically stacked boxes.
SETTER parameters
The SETTER algorithm is controlled by several parameters which values can influence the quality of the structural alignment, the speed of the method or both. Their default values (Figure 2D) were determined empirically during the extensive testing of the method. These parameters work reasonably well for a majority of tested structures, however a better alignment can sometimes be achieved by fine tuning their values (Figure 2D). The μ and σ parameters of the log-normal distribution were fitted only for the default SETTER's parameters in which case both the 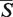 -distance and its corresponding P-value can be calculated. For non-default parameters only the
-distance and its corresponding P-value can be calculated. For non-default parameters only the 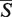 -distance is reported. A list of parameters, their meaning, their reasonable ranges and their default values are now given in Section S6 of Supplementary Data.
-distance is reported. A list of parameters, their meaning, their reasonable ranges and their default values are now given in Section S6 of Supplementary Data.
Output
To simplify the navigation and to allow an easy modification of a previously submitted job, a system of tabs is implemented (Figure 3A).
The details about the computed alignment are displayed in the Results tab. The left panel of the Results tab (Figure 3B) contains an 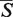 -distance, a statistical significance of the alignment given as its P-value, and a running time of the algorithm. For each structure in the alignment, its PDB header is parsed and details about the publication of the structure can be inspected by hovering over the ‘PDB header’ link (Figure 3B). By clicking on the ‘details’ hyperlink, the structure entry in the PDB database (24) is opened in a new window. The structure can be downloaded either in PDB or in mmCIF formats (26) under the ‘format’ link. In addition to the
-distance, a statistical significance of the alignment given as its P-value, and a running time of the algorithm. For each structure in the alignment, its PDB header is parsed and details about the publication of the structure can be inspected by hovering over the ‘PDB header’ link (Figure 3B). By clicking on the ‘details’ hyperlink, the structure entry in the PDB database (24) is opened in a new window. The structure can be downloaded either in PDB or in mmCIF formats (26) under the ‘format’ link. In addition to the 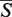 -distance, several commonly used measures of the alignment quality are reported under the ‘alignment quality measures’ link. These include RMSD between the phosphate atoms, a PSI (a percentage of superimposed residues within 4.0 Å with respect to the length of the shorter of the two structures), a PID (a percentage of aligned nucleotides of the same type with respect to the length of the shorter of the two structures) (15,23), number of aligned nucleotides and number of exact base matches (20). Next the table showing the number of GSSU units and the number of nucleotides in each structure is displayed. Links below this table allow to download the alignment report either in the plain text or in the PDF format. In addition, the superimposed structures can be downloaded in the PDB format or the visualization of the aligned structures can be obtained as the JPEG image.
-distance, several commonly used measures of the alignment quality are reported under the ‘alignment quality measures’ link. These include RMSD between the phosphate atoms, a PSI (a percentage of superimposed residues within 4.0 Å with respect to the length of the shorter of the two structures), a PID (a percentage of aligned nucleotides of the same type with respect to the length of the shorter of the two structures) (15,23), number of aligned nucleotides and number of exact base matches (20). Next the table showing the number of GSSU units and the number of nucleotides in each structure is displayed. Links below this table allow to download the alignment report either in the plain text or in the PDF format. In addition, the superimposed structures can be downloaded in the PDB format or the visualization of the aligned structures can be obtained as the JPEG image.
Two superimposed structures are visualized on the right side (Figure 3C) utilizing the Jmol (27) Java applet. The visualization can be controlled using the top panel (Figure 3D) which allows to easily adjust color, molecular display scheme and to turn each of the models on or off independently. In addition, the standard Jmol's menu allowing a more detailed handling of various aspects of the visualization is available upon the right click in the Jmol window. Individual GSSUs in both structures can be inspected by checking the box in the ‘Show GSSU pairs’ panel (Figure 3E). Two aligned GSSUs are shown in different colors (red and blue). The displayed GSSUs can either be cycled through by clicking on the left/right arrows or they can be selected from the drop-down box (Figure 3E).
SETTER is based on the superposition of individual GSSUs and does not rely on a sequence alignment algorithm. Thus, the information about corresponding (i.e. aligned) residues is missing. However, such information is necessary to calculate commonly used alignment quality measures such as e.g. RMSD. Therefore, a list of aligned residues was generated utilizing a simple geometric approach. For each nucleotide a in a structure A, its nearest neighbor in a structure B was identified. Similarly, for each nucleotide b in a structure B, its nearest neighbor in a structure A was found. A nucleotide pair is considered to be aligned if a is the nearest neighbor of b and, at the same time, b is the nearest neighbor of a.
The aligned residues are displayed by checking the ‘Show aligned residues’ panel (Figure 3F). The alignment precision can further be studied by highlighting the nearest neighbor nucleotides defined by the adjustable distance range in Angstrom units (Figure 3G).
In the batch mode, the resulting alignments are displayed as vertically stacked boxes (Figure 3H). The boxes are ordered in a descending order by their 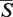 -distances. Alignment results are expanded in the active window after clicking the corresponding box.
-distances. Alignment results are expanded in the active window after clicking the corresponding box.
SUMMARY
A web server for the RNA 3D pairwise structural alignment was developed and is available at http://siret.cz/setter. The web server utilizes an efficient and accurate SETTER algorithm (21). The SETTER method divides the RNA structure into the set of non-overlapping structural elements called generalized secondary structure units (GSSUs). The structural alignment is then based on the pairwise comparison utilizing 3D similarity of the GSSUs. The quality of the alignment is given by the measure called 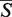 -distance; the smaller the
-distance; the smaller the 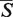 -distance the better the alignment. SETTER can be used both for the pairwise structural alignment, as well as for the functional annotation of a new RNA structure.
-distance the better the alignment. SETTER can be used both for the pairwise structural alignment, as well as for the functional annotation of a new RNA structure.
The web server receives as an input a pair of 3D RNA structures or a query structure and a list of target structures. The server outputs the 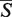 -distance and its statistical significance given as a p-value, running time, and various common measures of the alignment quality. Picture of two superimposed structures can be downloaded in the JPEG format. The aligned structures can also be downloaded in the PDB format. In addition, the alignment of is visualized directly by the server utilizing the Jmol Java applet. Using the visualization, user can easily inspect the resulting alignment switching between the following views: aligned structures, decomposition of the structures into the GSSUs, a visualization of the aligned residues and a visualization of nearest neighbors in the given distance range. SETTER method is capable of aligning even the largest RNA structures deposited in the PDB database in a reasonable amount of time (e.g. two structures of the 25S rRNA each having 3396 nt and represented by 89 GSSUs aligned in 1 min and 20 s) and represents thus an important addition to the portfolio of automatic RNA structural analysis tools.
-distance and its statistical significance given as a p-value, running time, and various common measures of the alignment quality. Picture of two superimposed structures can be downloaded in the JPEG format. The aligned structures can also be downloaded in the PDB format. In addition, the alignment of is visualized directly by the server utilizing the Jmol Java applet. Using the visualization, user can easily inspect the resulting alignment switching between the following views: aligned structures, decomposition of the structures into the GSSUs, a visualization of the aligned residues and a visualization of nearest neighbors in the given distance range. SETTER method is capable of aligning even the largest RNA structures deposited in the PDB database in a reasonable amount of time (e.g. two structures of the 25S rRNA each having 3396 nt and represented by 89 GSSUs aligned in 1 min and 20 s) and represents thus an important addition to the portfolio of automatic RNA structural analysis tools.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Sections 1–6.
FUNDING
Funding for open access charge: Ministry of Education of the Czech Republic [MSM6046137302, MSM 6046137306]; Czech Science Foundation (GAČR) [P202/11/0968]; Charles University project P46 Informatics.
Conflict of interest statement. None declared.
REFERENCES
- 1.Mattick JS, Makunin IV. Non-coding RNA. Hum. Mol. Genet. 2006;15:17–29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 2.Taft RJ, Pang KC, Mercer TR, Dinger M, Mattick JS. Non-coding RNAs: regulators of disease. J. Pathol. 2010;220:126–139. doi: 10.1002/path.2638. [DOI] [PubMed] [Google Scholar]
- 3.Apostolico A, Ciriello G, Guerra C, Heitsch CE, Hsiao C, Williams LDD. Finding 3D motifs in ribosomal RNA structures. Nucleic Acids Res. 2009;37:e29. doi: 10.1093/nar/gkn1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wadley LM, Pyle AM. The identification of novel RNA structural motifs using COMPADRES: an automated approach to structural discovery. Nucleic Acids Res. 2004;32:6650–6659. doi: 10.1093/nar/gkh1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duarte CM, Wadley LM, Pyle AM. RNA structure comparison, motif search and discovery using a reduced representation of RNA conformational space. Nucleic Acids Res. 2003;31:4755–4761. doi: 10.1093/nar/gkg682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong C, Tang H, Zhang S. RNAMotifScan: automatic identification of RNA structural motifs using secondary structural alignment. Nucleic Acids Res. 2010;38:e176. doi: 10.1093/nar/gkq672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Holbrook SR. Structural principles from large RNAs. Ann. Rev. Biophys. 2008;37:445–464. doi: 10.1146/annurev.biophys.36.040306.132755. [DOI] [PubMed] [Google Scholar]
- 8.Batey RT, Rambo RP, Doudna JA. Tertiary motifs in RNA structure and folding. Angew. Chem. Ed Intil Engl. 1999;38:2326–2343. doi: 10.1002/(sici)1521-3773(19990816)38:16<2326::aid-anie2326>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Laing C, Schlick T. Computational approaches to 3D modeling of RNA. J. Phys. Condens. Mat. 2010;22:283101. doi: 10.1088/0953-8984/22/28/283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laing C, Schlick T. Computational approaches to RNA structure prediction, analysis, and design. Curr. Opin. Struct. Biol. 2011;21:306–18. doi: 10.1016/j.sbi.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tamura M, Hendrix DK, Klosterman PS, Schimmelman NR, Brenner SE, Holbrook SR. SCOR: Structural Classification of RNA, version 2.0. Nucleic Acids Res. 2004;32:D182–D184. doi: 10.1093/nar/gkh080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murthy VL, Rose GD. RNABase: an annotated database of RNA structures. Nucleic Acids Res. 2003;31:502–504. doi: 10.1093/nar/gkg012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abraham M, Dror O, Nussinov R, Wolfson HJ. Analysis and classification of RNA tertiary structures. RNA. 2008;14:2274–2289. doi: 10.1261/rna.853208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferrè F, Ponty Y, Lorenz WA, Clote P. DIAL: a web server for the pairwise alignment of two RNA three-dimensional structures using nucleotide, dihedral angle and base-pairing similarities. Nucleic Acids Res. 2007;35:W659–W668. doi: 10.1093/nar/gkm334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capriotti E, Marti-Renom MA. SARA: a server for function annotation of RNA structures. Nucleic Acids Res. 2009;37:W260–W265. doi: 10.1093/nar/gkp433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang Y-F, Huang Y-L, Lu CL. SARSA: a web tool for structural alignment of RNA using a structural alphabet. Nucleic Acids Res. 2008;36:W19–W24. doi: 10.1093/nar/gkn327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C-W, Chen K-T, Lu CL. iPARTS: an improved tool of pairwise alignment of RNA tertiary structures. Nucleic Acids Res. 2010;38:W340–W347. doi: 10.1093/nar/gkq483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dror O, Nussinov R, Wolfson H. ARTS: alignment of RNA tertiary structures. Bioinformatics. 2005;21(Suppl. 2):ii47–ii53. doi: 10.1093/bioinformatics/bti1108. [DOI] [PubMed] [Google Scholar]
- 19.Bauer RA, Rother K, Moor P, Reinert K, Steinke T, Bujnicki JM, Preissner R. Fast structural alignment of biomolecules using a hash table, N-grams and string descriptors. Algorithms. 2009;2:692–709. [Google Scholar]
- 20.Rahrig RR, Leontis NB, Zirbel CL. R3D Align: global pairwise alignment of RNA 3D structures using local superpositions. Bioinformatics. 2010;26:2689–2697. doi: 10.1093/bioinformatics/btq506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoksza D, Svozil D. Efficient RNA pairwise structure comparison by SETTER method. Bioinformatics. 2012;28:1858–1864. doi: 10.1093/bioinformatics/bts301. [DOI] [PubMed] [Google Scholar]
- 22.Kabsch W. A solution for the best rotation to relate two sets of vectors. Acta Crystallog. A. 1976;32:922–923. [Google Scholar]
- 23.Capriotti E, Marti-Renom MA. RNA structure alignment by a unit-vector approach. Bioinformatics. 2008;24:i112–i118. doi: 10.1093/bioinformatics/btn288. [DOI] [PubMed] [Google Scholar]
- 24.Berman HM, Westbrook JD, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu X-J, Olson WK. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 2008;3:1213–1227. doi: 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westbrook JD, Fitzgerald PM. The PDB format, mmCIF, and other data formats. Method. Biochem. Anal. 2003;44:161–179. [PubMed] [Google Scholar]
- 27. Jmol: An Open-source Java Viewer for Chemical Structures in 3D. http://www.jmol.org/ (4 June 2012, date last accessed) [Google Scholar]