


Abstract
In the context of the renewed interest of peptides as therapeutics, it is important to have an on-line resource for 3D structure prediction of peptides with well-defined structures in aqueous solution. We present an updated version of PEP-FOLD allowing the treatment of both linear and disulphide bonded cyclic peptides with 9–36 amino acids. The server makes possible to define disulphide bonds and any residue–residue proximity under the guidance of the biologists. Using a benchmark of 34 cyclic peptides with one, two and three disulphide bonds, the best PEP-FOLD models deviate by an average RMS of 2.75 Å from the full NMR structures. Using a benchmark of 37 linear peptides, PEP-FOLD locates lowest-energy conformations deviating by 3 Å RMS from the NMR rigid cores. The evolution of PEP-FOLD comes as a new on-line service to supersede the previous server. The server is available at: http://bioserv.rpbs.univ-paris-diderot.fr/PEP-FOLD.
INTRODUCTION
The recent years have seen a renewal of peptides as candidate therapeutics for several reasons. First, recent advances in peptide chemistry and delivery have overcome the traditional limitations of peptides as drug candidates (1). Second, the shift of therapeutic strategies towards the network of protein interactions, particularly the search for protein–protein interaction inhibitors, has pushed forward the limits of small chemical molecules, whereas advances in protein recombinant technologies provide evidence that larger therapeutics such as peptides or peptide derivatives could offer plausible alternatives (2). Another motivation also comes from the large reservoir of natural peptides that have diverse and specific biological activities, and among these, bacterial small proteins (3) and venom peptides (4) raise more interest. Finally, peptides are also described as promising candidates for the treatment of central nervous system disorders (5).
To assist peptide lead identification and optimization, robust computational methods are clearly expected to bring significant contributions (6,7). Recent efforts from the community of computer scientists have tackled various aspects including the design of generic databases devoted to peptide–protein interactions such as PepX (8), the problem of protein–peptide docking (9), the search for peptidomimetics (10) and the development of fast peptide structure prediction methods (e.g. PEP-FOLD, Bhageerath, PEPstr, Peplook, I-Tasser, Rosetta) (11–16).
In 2009, we introduced the PEP-FOLD service (11) for de novo peptide structure prediction. Though this first rapid on-line version has been used by external users for structural characterization of peptides or protein fragments (17,18) and peptide or vaccine design (19,20), the maximal length of 25 amino acids limits the number of applications. In addition, like the Bhageerath (12) and PepStr (13) servers, PEP-FOLD was only available for linear peptides, whereas there are many natural cyclic peptides with disulfide bonds such as conotoxins or cyclotides (21) and disulfide bonds increase peptide stability (22). Recently, the Peplook procedure (not available on-line) brought some improvements in this direction (14). Here, we introduce an improved version of the service open to the community that (i) extends the length of linear peptides to 36 amino acids and (ii) accepts cyclic peptides using disulfide bonds defined by the user.
MATERIALS AND METHODS
The 3D prediction scheme is very similar to that reported in (11) and (23). A general overview of the service is presented in Figure 1. It is based on a Hidden Markov Model derived Structural Alphabet (SA) (24), i.e. a kind of generalized secondary structure extending the number of states from 3 (helix, coil, strand) to 27 in our case. The core of PEP-FOLD consists in three steps. The first step predicts SA letters from the amino acid sequence. From the amino acid sequence, a psi-blast profile is generated and is used as input of a SVM that returns a probability profile of each SA letter at each position of the sequence. This SA profile is then analysed to select some letters at each position. The second step performs the 3D assembly of the prototype fragments associated with the letters selected. It relies on the sOPEP coarse grained force field (25), which uses a six bead representation (full backbone except the α-hydrogen and one bead for each side chain). The 3D generation is achieved by an enhanced greedy procedure (26) that builds the peptide residue by residue. It is followed by a Monte-Carlo procedure for final refinement. This build-up procedure works using a rigid assembly scheme and thus does not explore the full conformational space but only a discrete subset. This stochastic procedure is repeated 100 times starting from various positions in the sequence. The third step generates all-atom conformations from the coarse grained models returned by the 100 simulations and performs a clustering procedure.
Figure 1.

PEP-FOLD 2012 flowchart.
Two major improvements have been brought to this scheme in the new version of the service. First, the selection of the SA letters from the profile has been revisited so as to remove the letters with too low probabilities. As a result, whereas the initial PEP-FOLD version used eight letters at each position, the new version uses 5.5 letters on average. We also removed the secondary structure constraints predicted by PSI-PRED because the SA profile contains this information. A second major modification involves the use of TM scores (27) to cluster the full generated structures and then the sOPEP energies or the predicted TMscores of Apollo (28)—an extension of (29) for the prediction GDT_TS from structural features—to rank the clusters and the conformations within the clusters. The first version only used the RMSD between the models for clustering. Another variation involves the formulation of sOPEP for S–S bonds. Here, the disulfide bonds are not simply constrained to a typical bond distance because we grow the peptide from any position of the structure and thus all oxidized cysteines are not known in the early steps of the assembly. Rather, the interaction between two oxidized cysteines i and j is described by:
| (1) |
where
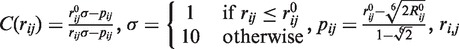 denotes the distance between the side-chain centroids,
denotes the distance between the side-chain centroids, 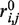 = 3.36 Å is the distance where the energy is the lowest (−15 kcal/mol),
= 3.36 Å is the distance where the energy is the lowest (−15 kcal/mol), 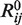 = 2.39 Å is the distance where
= 2.39 Å is the distance where 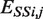 is 0, and E0 is the energy value for
is 0, and E0 is the energy value for 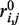 . The left side term is identical to sOPEP former term and the right side results in a sharper behaviour preventing energy at longer distances. The same formulation has been generalized to residue contacts when specified by the user. Finally, other minor modifications have been brought to the side chain positioning in the all-atom generation step—it is now achieved using OSCAR-star (30)—and the disulfide bond specifications are passed to the quick all-atom minimization based on Gromacs (31).
. The left side term is identical to sOPEP former term and the right side results in a sharper behaviour preventing energy at longer distances. The same formulation has been generalized to residue contacts when specified by the user. Finally, other minor modifications have been brought to the side chain positioning in the all-atom generation step—it is now achieved using OSCAR-star (30)—and the disulfide bond specifications are passed to the quick all-atom minimization based on Gromacs (31).
SERVICE OVERVIEW
As described in Figure 1, the service is now fully embedded in the Mobyle framework (32), providing automated form generation, command execution and result display. The input fields include the primary sequence (only standard amino acids) and the specification of constraints (disulfide bonds, residue proximities). In addition to building models up to 36 amino acids, it is possible to treat sequences of 50 amino acids and specify the maximal 36-residue region subjected to 3D modelling. This facility is useful for peptides where the N- and C-terminal regions are known to be disordered and the user wishes to focus on the modelling of the structured core. To this end, our tests show that the prediction of the SA profile is best using the complete sequence rather than a truncated sequence. The output fields have also been modified as a result of the new clustering procedure. Although our results suggest that sorting the clusters using sOPEP remains best, they can also be sorted according to the predicted TM scores (27) obtained by Apollo (28). Finally, the use of the Mobyle framework comes with facilities to visualize the best models using the openastex (33) or Jmol (34) applets and perform other analyses such as the secondary structure, side-chain conformations, etc. This number of analyses is expected to increase progressively.
RESULTS
Linear peptides with 9–36 amino acids
The modifications brought to PEP-FOLD have a limited impact on the linear peptides with 9–25 amino acids. A table of the results on 24 peptides is presented on the on-line documentation of the service. We see that the sharper SA letter selection does not prevent PEP-FOLD to generate near-native conformations, with the best rigid core (RC) conformations deviating on average by only 1.5 Å from the NMR structures, versus 1.7 Å using the earlier PEP-FOLD service. Note that the PDB entry 1wz4 is excluded from analysis since its NMR structure displays a clash between the backbone oxygens of Glu7 and Asp11. The updated PEP-FOLD procedure also generates lowest-energy conformations deviating on average by 2.5 Å from the NMR RCs. The results on a set of linear peptides with 25–36 amino acids are presented in Table 1. Averaged over the 13 systems, PEP-FOLD returns lowest-energy conformations deviating from the first NMR model by 4.8 and 3.4 Å using the full structure (FS) and the RC. Looking at the five clusters of lowest energy, the average RMSD is 3.6 Å (FS) and 2.8 Å (RC). Overall, PEP-FOLD generates near-native conformations (RC-d < 4 Å) for 11 among 13 structurally diverse proteins with secondary structure compositions varying from α, α2 (2l0g Figure 2C), β2, αβ2, βα, βαβ to β3. Of particular interest is the high quality prediction of the βαβ topology for the 36-residue 2ki0 protein with a RC-d of 2.0 Å based on the sOPEP energy (Figure 2E) which is stabilized by long-range interactions in the amino acid sequence. Similarly, the updated PEP-FOLD version returns the native β3 conformation of the 36-residue 1e0n protein (Figure 2A), while the earlier version, using the 27 amino acids which are not random coil in the NMR structure, predicted a β-strand packed against two helices (11). For the 28-residue 1psv and 31-residue 2gdl, the lowest-energy conformations differ by 7.4 and 4.8 Å RMS from NMR using the RC. In Figure 2B, the high RMSD for 1psv comes from the β-hairpin. Interestingly, experimental and computational studies on homologous peptides have shown that the β-hairpin is only marginally stable (35). In contrast, for the 2gdl target (Figure 2G) which displays an α-coil-α topology, the predicted β-strand at the C-terminal is not compatible with the α-helix structure observed by NMR.
Table 1.
Results obtained for 13 linear peptides with 25–36 residues
| PDB id | Top | L | RC | sOPEP |
best5 |
|||
|---|---|---|---|---|---|---|---|---|
| FS-d | RC-d | FS-d | RC-d | Rnk | ||||
| 1by0 | a | 27 | 1:23 | 4.36 | 1.75 | 2.94 | 1.74 | 1 |
| 1yyb | a | 27 | 1:20 | 6.49 | 1.47 | 3.4 | 1.42 | 1 |
| 2kbl | b2 | 29 | 3:4|6:27 | 4.68 | 3.91 | 4.68 | 3.91 | 1 |
| 1fsd | ab2 | 28 | 1:26 | 4.14 | 3.88 | 3.91 | 3.66 | 1 |
| 1psva | ab2 | 28 | 2:25 | 7.19 | 7.44 | 4.61 | 4.6 | 4 |
| 2k76 | ba | 30 | 4:29 | 3.15 | 2.75 | 2.1 | 1.88 | 1 |
| 2gdla | aca | 31 | 8:8|10:11|14:15|21:29 | 7.57 | 4.84 | 6.53 | 4.57 | 3 |
| 2l0ga | a2 | 32 | 5:32 | 4.53 | 3.32 | 2.1 | 1.64 | 1 |
| 2bn6 | a2 | 33 | 4:29 | 4.49 | 3.18 | 3.09 | 2.14 | 1 |
| 1e0na | b3 | 35 | 1:25 | 2.16 | 2.16 | 1.68 | 1.68 | 2 |
| 1wr3 | b3 | 36 | 5:15 | 5.72 | 3.74 | 4.26 | 3.5 | 1 |
| 1wr4 | b3 | 36 | 5:34 | 4.68 | 3.26 | 4.68 | 3.26 | 1 |
| 2ki0a | bab | 36 | 5:11|13:36 | 3.56 | 1.99 | 2.66 | 2.43 | 1 |
| Mean | 4.8 | 3.4 | 3.6 | 2.8 | ||||
PDB id, PDB identifier; Top, secondary structure topology (a for helix, b for strand, c for coil); L, peptide length; RC, the definition of the rigid core (PDB positions start at 1); FS-d (RC-d), full structure (rigid core) RMS deviation (Å) for the model of lowest energy (sOPEP) and the best model among the five clusters of lowest energy (best5); Rnk, the rank of the cluster containing the best model.
aThe lowest-energy models for 1e0n, 1psv, 2l0g, 2ki0 and 2gdl are shown on Figure 2.
Figure 2.

PEP-FOLD models. The experimental conformation is in green. PEP-FOLD models are in cyan. From left to right, top to bottom: 1e0n (A), 1psv (B), 2l0g (C), 1n0a (D), 2ki0 (E), 1kwd (F) and 2gdl (G) lowest-energy models and 1wm8 (H) best model.
Disulfide-bonded cyclic peptides
Table 2 presents the results of 34 peptides containing one, two or three disulfide bonds using the Peplook test set (14) except the peptides 2p7r, 1qvl and 1foz <8 amino acids, and the peptide 1ixu free of any disulfide bond in the NMR structure. For each peptide, we describe the models of lowest energy and of lowest RMSD (best) with respect to the NMR structure. We also give the number of SS bonds formed after the coarse-grained and the all-atom procedures. On average, the updated PEP-FOLD generates best models deviating by only 2.7 and 2.5 Å RMS and lowest-energy models deviating by 4.2 and 3.7 Å RMS using the FSs and RCs, respectively. This indicates that sOPEP is not optimal yet for recognizing near-native from higher RMSd conformations. Compared to the results of the earlier PEP-FOLD version, which did not consider the disulfide bonds explicitly (11), we reach an improvement of 1.3 Å RMS. Compared to Peplook performances, the PEP-FOLD best models are also closer by 1 Å RMS to the NMR conformations. Whereas the server leads to good results for peptides containing 1 and 2 disulfide bonds (Figure 2F and D for 1kwd and 1n0a) with 61.2% of the disulfide bonds formed at the coarse-grained level (Table 1), the performances are degraded for the nine peptides with three disulfide bonds (Figure 2H for 1wm8). For these peptides, the lowest-energy conformations deviate on average by 5.4 Å RMS from the NMR structures and a similar behaviour is observed with Peplook (5.6 Å RMS). This discrepancy between prediction and experiment comes in part from the non-optimization of the sOPEP force field for cyclic peptides, but mainly we observe that the structures with three disulfide bonds are very constrained, leading to large deviations at the local level between the conformations observed in the NMR structures and the structural alphabet conformations predicted from the sequence free of any disulfide bond constraints. Similar results are obtained if we vary the number of SA letters from 5.5 to 8 during assembly.
Table 2.
Results obtained for 34 cyclic peptides with disulfide bonds
| pdb Id | L | #SS | RC | sOPEP |
best |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FS-d | RC-d | CGa | AA | FS-d | RC-d | CG | AA | ||||
| 1im7 | 21 | 1 | 3:9::15:21 | 3.9 | 3.9 | 100 | 0 | 2.5 | 2.5 | 100 | 1 |
| 1jbl | 16 | 1 | 2:14 | 3.3 | 3.3 | 100 | 1 | 1.8 | 1.8 | 0 | 0 |
| 1n0ab | 17 | 1 | 7:15 | 0.7 | 0.7 | 0 | 1 | 0.4 | 0.4 | 0 | 0 |
| 1n0c | 24 | 1 | 2:11::14:24 | 2.3 | 2.3 | 100 | 1 | 0.4 | 0.4 | 0 | 1 |
| 1nim | 24 | 1 | 1:23 | 3.8 | 1.8 | 0 | 0 | 3.4 | 2.8 | 0 | 1 |
| 1gnb | 14 | 2 | 2:9::12:14 | 4.8 | 4.1 | 50 | 1 | 4 | 3.8 | 50 | 0 |
| 1b45 | 13 | 2 | – | 3.6 | 3.3 | 100 | 1 | 2.3 | 2.2 | 0 | 0 |
| 1etl | 22 | 2 | – | 3.1 | 3.1 | 100 | 1 | 1.6 | 1.6 | 0 | 0 |
| 1hje | 19 | 2 | – | 3.6 | 3.6 | 50 | 1 | 2.2 | 2.2 | 50 | 2 |
| 1hp9 | 12 | 2 | 2:11 | 3.5 | 3.5 | 50 | 1 | 2.2 | 2.2 | 50 | 1 |
| 1ien | 13 | 2 | – | 4.8 | 4.8 | 50 | 0 | 2.5 | 2.5 | 0 | 0 |
| 1im1 | 14 | 2 | – | 1.3 | 1.2 | 0 | 1 | 1.2 | 1.2 | 50 | 0 |
| 1kcn | 16 | 2 | – | 5.7 | 5.6 | 100 | 0 | 3.5 | 2.8 | 50 | 1 |
| 1kwdb | 30 | 2 | 3:29 | 2.9 | 2.1 | 100 | 2 | 2 | 1.3 | 0 | 1 |
| 1mii | 11 | 2 | – | 1.8 | 1.8 | 50 | 0 | 1.6 | 1.6 | 0 | 0 |
| 1oig | 15 | 2 | 2:7::11:14 | 6.5 | 6.1 | 50 | 1 | 4.8 | 4.7 | 50 | 1 |
| 1r8t | 24 | 2 | 1:22 | 4.5 | 3.4 | 100 | 1 | 2.6 | 2.2 | 50 | 1 |
| 1rpc | 21 | 2 | 3:21 | 6.9 | 6.3 | 50 | 1 | 4.4 | 4 | 0 | 0 |
| 1ter | 21 | 2 | – | 7 | 5.9 | 50 | 1 | 3 | 2.8 | 50 | 0 |
| 1v6r | 26 | 2 | 1:23 | 5.1 | 5.1 | 100 | 2 | 3.9 | 3.9 | 0 | 0 |
| 1wqc | 13 | 2 | 2:13 | 1.7 | 1.3 | 100 | 2 | 1.2 | 1.1 | 100 | 2 |
| 1x7k | 22 | 2 | 1:17 | 4.2 | 2.5 | 0 | 0 | 4.2 | 2.5 | 0 | 0 |
| 1xgb | 16 | 2 | – | 3.5 | 3 | 0 | 0 | 3 | 3 | 50 | 2 |
| 2ajw | 13 | 2 | – | 2.4 | 0.9 | 50 | 2 | 1.3 | 1 | 0 | 0 |
| 2i28 | 28 | 2 | – | 2.1 | 2.1 | 100 | 1 | 1.5 | 1.5 | 0 | 0 |
| 2oq9 | 24 | 2 | 8:24 | 9.2 | 4 | 100 | 1 | 3.5 | 3.3 | 50 | 1 |
| 2efz | 12 | 3 | – | 4.2 | 4.2 | 0 | 1 | 2.1 | 2.1 | 66.7 | 1 |
| 2nx7 | 13 | 3 | 2:13 | 6.9 | 6.9 | 33.3 | 1 | 4.1 | 4 | 33.3 | 1 |
| 1mmc | 10 | 3 | – | 7.1 | 6.1 | 66.7 | 2 | 4.6 | 4.6 | 33.3 | 0 |
| 1orx | 22 | 3 | 1:19 | 4.8 | 4.8 | 33.3 | 1 | 2.9 | 2.7 | 0 | 0 |
| 1sp7 | 28 | 3 | 1:10::12:28 | 4.5 | 4.3 | 66.7 | 2 | 2.6 | 2 | 33.3 | 0 |
| 1v5a | 28 | 3 | – | 5 | 5.1 | 66.7 | 1 | 3.7 | 3.8 | 0 | 0 |
| 1wm8b | 19 | 3 | 6:16 | 7 | 7 | 66.7 | 2 | 3.6 | 3.6 | 66.7 | 2 |
| 2it7 | 28 | 3 | 2:28 | 4 | 4 | 66.7 | 1 | 3.7 | 3.7 | 66.7 | 1 |
| Mean | 4.2 | 3.7 | 61.2 | 2.7 | 2.5 | 29.5 | |||||
PDB id, PDB identifier; L, peptide length; SS, number of disulfide bond; RC, the definition of the rigid core (PDB positions start at 1); FS-d (RC-d), full structure (rigid core) RMS deviation (Å) for the model of lowest energy (sOPEP) and the model of lowest RMSD (best); CG, SS bonds formed in the Coarse Grained representation; AA, SS bonds formed in the final all-atom representation.
aOur criterion for evaluating a disulfide bond formed in the coarse grained structure is that rij in equation (1) lies within 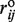 ± 0.7 Å. The discrepancy between the number of disulfide bonds in the atomistic and coarse grained structures comes from the fact that a very stringent condition on the S–S distance must be satisfied (2.0 ± 0.2 Å) in all-atom structures.
± 0.7 Å. The discrepancy between the number of disulfide bonds in the atomistic and coarse grained structures comes from the fact that a very stringent condition on the S–S distance must be satisfied (2.0 ± 0.2 Å) in all-atom structures.
bThe lowest-energy models for 1n0a and 1kwd, and the best RMSD model for 1wm8 are shown on Figure 2.
DISCUSSION
The update of our PEP-FOLD server had two goals. The first goal was to extend the length of linear peptides so as to develop a server able to explore systems with higher thermodynamic stabilities, and with new topologies such as the βαβ and β3 folds. Using a benchmark of 37 peptides with 9–36 amino acids, and a total of 100 simulations for each system, the updated PEP-FOLD version fails to generate the native conformation for only one system, where a β-strand is preferred over an α-helix at the C-terminus. Note that our results are reproducible and do not change if we use 200 simulations. Preliminary simulations also indicate that PEP-FOLD performs well for sequences upwards of 50 amino acids where the FS is modelled (data not shown), but opening the service for this length is under consideration given the computer resources needed. This increase in peptide size from 25 to 50 amino acids comes from the reduction of the SA letters considered at each position. The structure simulations of even larger systems within the framework of a greedy procedure remain to be evaluated.
The second goal was to provide structural predictions for cylic peptides with disulfide bonds. For systems with one and two disulfide bonds, PEP-FOLD performs better than Peplook. For systems with more disulfide bonds, such as natural toxins, improvements in the SA letter prediction and atomistic construction are needed. One solution for construction, under investigation, is to enforce disulfide bond formation in the Gromacs procedure and to relax the models by short molecular dynamics simulations.
In summary, the updated version of PEP-FOLD provides a fast and convenient on-line approach for the de novo design of peptides up to 36 residues, and it also supports the possibility to tackle peptide engineering by the insertion of disulfide bonds, a classical way to increase stability. The typical computational time for a 36-residue peptide is 40 min using 40 cores. The server also supports, using the same formalism as disulfide bonds, the possibility to specify additional constraints such as residue proximities. Clearly, our results open the door to further improvements such as the consideration of other covalent linking between side-chains and the modelling of ribosomal peptides at a genome scale can be considered.
FUNDING
The Institut National de la Santé et de la Recherche Medicale recurrent funding, the Centre National de la Recherche Scientifique recurrent funding, and the IA bioinformatics grant [BipBip]. Funding for open access charge: Institut National de la Santé et de la Recherche Medicale UMR-S 973.
Conflict of intertest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank the authors of the OSCAR-star program for useful help and adaptation of the program and also thank the referees for their useful criticism, Hervé Ménager for his help in Mobyle modifications and Julien Rey for this help for PEP-FOLD output specific formatting.
REFERENCES
- 1.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Systhetic therapeutic peptides: science and market. Drug Discov. Today. 2010;15:40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 2.Vanhee P, van der Sloot AM, Verschueren E, Serrano L, Rousseau F, Schymkowitz J. Computational design of peptide ligands. Trends Biotechnol. 29:231–239. doi: 10.1016/j.tibtech.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 3.Hobbs EC, Fontaine F, Yin X, Storz G. An expanding universe of small proteins. Curr. Opin. Microbiol. 2011;14:167–173. doi: 10.1016/j.mib.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vetter I, Davis JL, Rash LD, Anangi R, Mobli M, Alewood PF, Lewis RJ, King GF. Venomics: a new paradigm for natural products-based drug discovery. Amino Acids. 2011;40:15–28. doi: 10.1007/s00726-010-0516-4. [DOI] [PubMed] [Google Scholar]
- 5.Malavolta L, Cabral FR. Peptides: important tools for the treatment of central nervous system disorders. Neuropeptides. 2011;45:309–316. doi: 10.1016/j.npep.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Audie J, Boyd C. The synergistic use of computation, chemistry and biology to discover novel peptide-based drugs : the time is right. Curr. Pharm. Des. 2010;16:567–582. doi: 10.2174/138161210790361425. [DOI] [PubMed] [Google Scholar]
- 7.Deshmukh R, Purohit HJ. Peptide scaffolds: flexible molecular structures with diverse therapeutic potentials. Int. J. Pept. Res. Ther. 2012;18:125–143. [Google Scholar]
- 8.Vanhee P, Reumers J, Stricher F, Baeten L, Serrano L, Schymkowitz J, Rousseau F. PepX: a structural database of non-redundant protein-peptide complexes. Nucleic Acids Res. 2010;38:D545–D551. doi: 10.1093/nar/gkp893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.London N, Raveh B, Cohen E, Fathi G, Schueler-Furman O. Rosetta FlexPepDock web server—high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011;39:W249–W253. doi: 10.1093/nar/gkr431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Floris M, Masciocchi J, Fantom M, Moro S. Swimming into peptidomimetic chemical space using pepMMsMIMIC. Nucleic Acids Res. 2011;39:W261–W269. doi: 10.1093/nar/gkr287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maupetit J, Derreumaux P, Tufféry P. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009;37:W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jayaram B, Bhushan K, Shenoy SR, Narang P, Bose S, Agrawal P, Sahu D, Pandey V. Bhageerath: an energy based web enabled computer software suite for limiting the search space of tertiary structures of small globular proteins. Nucleic Acids Res. 2006;34:6195–6204. doi: 10.1093/nar/gkl789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaur H, Garg A, Raghava GP. PEPstr: a de novo method for tertiary structure predction of small bioactive peptides. Protein Pept. Lett. 2007;14:626–631. doi: 10.2174/092986607781483859. [DOI] [PubMed] [Google Scholar]
- 14.Beaufays J, Lins L, Thomas A, Brasseur R. In silico predictions of 3D structures of linear and cyclic peptides with natural and non-proteinogenic residues. J. Pept. Sci. 2012;18:17–24. doi: 10.1002/psc.1410. [DOI] [PubMed] [Google Scholar]
- 15.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Method Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 17.Duvignaud JB, Leclerc D, Gagné SM. Structure and dynamics changes induced by 2,2,2-trifluoro-ethanol (TFE) on the N-terminal half of hepatitis C virus core protein. Biochem. Cell Biol. 2010;88:315–323. doi: 10.1139/o09-155. [DOI] [PubMed] [Google Scholar]
- 18.Ragunathan P, Ponnuraj K. Expression, purification and structural analysis of a fibrinogen receptor FbsA from Streptococcus agalactiae. Protein J. 2011;30:159–166. doi: 10.1007/s10930-011-9317-1. [DOI] [PubMed] [Google Scholar]
- 19.Chopra N, Agarwal S, Verma S, Bhatnagar S, Bhatnagar R. Modeling of the structure and interactions of the B. anthracis antitoxin, MoxX: deletion mutant studies highlight its modular structure and repressor function. J. Comput. Aided Mol. Des. 2011;25:275–291. doi: 10.1007/s10822-011-9419-z. [DOI] [PubMed] [Google Scholar]
- 20.Gupta SK, Singh A, Srivastava M, Gupta SK, Akhoon BA. In silico DNA vaccine designing against human papillomavirus (HPV) causing cervical cancer. Vaccine. 2009;28:120–131. doi: 10.1016/j.vaccine.2009.09.095. [DOI] [PubMed] [Google Scholar]
- 21.Clark RJ, Craik DJ. Native chemical ligation applied to the synthesis and bioengineering of circular peptides and proteins. Biopolymers. 2010;94:414–422. doi: 10.1002/bip.21372. [DOI] [PubMed] [Google Scholar]
- 22.Chakraborty K, Thakurela S, Prajapati RS, Indu S, Ali PS, Ramakrishnan C, Varadarajan R. Protein stabilization by introduction of cross-strand disulfides. Biochemistry. 2005;44:14638–14646. doi: 10.1021/bi050921s. [DOI] [PubMed] [Google Scholar]
- 23.Maupetit J, Derreumaux P, Tufféry P. A fast method for large-scale de novo peptide and miniprotein structure prediction. J. Comput. Chem. 2010;31:726–738. doi: 10.1002/jcc.21365. [DOI] [PubMed] [Google Scholar]
- 24.Camproux AC, Gautier R, Tufféry P. A hidden Markov model derived structural alphabet for proteins. J. Mol. Biol. 2004;339:591–605. doi: 10.1016/j.jmb.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 25.Maupetit J, Tufféry P, Derreumaux P. A coarse-grained protein force field for folding and structure prediction. Proteins. 2007;69:394–408. doi: 10.1002/prot.21505. [DOI] [PubMed] [Google Scholar]
- 26.Tufféry P, Guyon F, Derreumaux P. Improved greedy algorithm for protein structure reconstruction. J. Comput. Chem. 2005;26:506–513. doi: 10.1002/jcc.20181. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57:702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Eickholt J, Cheng J. APOLLO: a quality assessment service for single and multiple protein models. Bioinformatics. 2011;27:1715–1716. doi: 10.1093/bioinformatics/btr268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Tegge AN, Cheng J. Evaluating the absolute quality of a single protein model using structural features and support vector machines. Proteins. 2009;75:638–647. doi: 10.1002/prot.22275. [DOI] [PubMed] [Google Scholar]
- 30.Liand S, Zheng D, Zhang C, Standley DM. Fast and accurante prediction of protein side-chain conformations. Bioinformatics. 2011;27:2913–2914. doi: 10.1093/bioinformatics/btr482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 32.Néron B, Ménager H, Maufrais C, Joly N, Maupetit J, Letort S, Carrere S, Tufféry P, Letondal C. Mobyle: a new full web bioinformatics framework. Bioinformatics. 2009;25:3005–3011. doi: 10.1093/bioinformatics/btp493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartshorn MJ. AstexViewer: a visualisation aid for structure-based drug design. J. Comput. Aided Mol. Des. 2002;16:871–881. doi: 10.1023/a:1023813504011. [DOI] [PubMed] [Google Scholar]
- 34.Hanson RM. Jmol: a paradigm shift in crystallographic visualization. J. Appl. Crystallogr. 2010;43:1250–1260. [Google Scholar]
- 35.Chebaro Y, Dong X, Laghaei R, Derreumaux P, Mousseau N. Replica exchange molecular dynamics simulations of coarse-grained proteins in implicit solvent. J. Phys. Chem. B. 2009;113:267–274. doi: 10.1021/jp805309e. [DOI] [PubMed] [Google Scholar]