


Abstract
Circular dichroism (CD) spectroscopy is a widely used method for examining the structure, folding and conformational changes of proteins. A new online CD analysis server (DichroMatch) has been developed for identifying proteins with similar spectral characteristics by detecting possible structurally and functionally related proteins and homologues. DichroMatch includes six different methods for determining the spectral nearest neighbours to a query protein spectrum and provides metrics of how similar these spectra are and, if corresponding crystal structures are available for the closest matched proteins, information on their secondary structures and fold classifications. By default, DichroMatch uses all the entries in the Protein Circular Dichroism Data Bank (PCDDB) for its comparison set, providing the broadest range of publicly available protein spectra to match with the unknown protein. Alternatively, users can download or create their own specialized data sets, thereby enabling comparisons between the structures of related proteins such as wild-type versus mutants or homologues or a series of spectra of the same protein under different conditions. The DichroMatch server is freely available at http://dichromatch.cryst.bbk.ac.uk.
INTRODUCTION
Circular dichroism (CD) spectroscopy is a popular method used for characterising protein structures in solution. Protein CD spectra result in large part from peptide transitions associated with secondary structural components such as alpha, 310 and polyproline-II helices and antiparallel and parallel β-sheets. These produce characteristic CD spectral features in the ultraviolet (UV) wavelength regions through distinct interactions with the left and right circularly polarized light from a CD spectrophotometer or a synchrotron radiation circular dichroism (SRCD) beamline. To a first approximation, a CD spectrum is the net result of summing the spectra of each of the individual secondary structure components present in the protein weighted by their proportions present in the structure. A number of deconvolution methods have been developed to estimate secondary structural information from CD spectra (1). However, CD spectra of proteins are additionally influenced by their supersecondary structures, fold motifs and tertiary and quaternary structures, particularly in the lower vacuum ultraviolet (VUV) wavelength regions from around 190 to 160 nm (accessible by the associated technique of SRCD) (2). In addition, while the presence of aromatic amino acids and disulphide-bonded side chains mainly affect CD spectral shape in the near UV region above 240 nm, these side chain contributions, although usually of relatively low magnitude, can extend into the far UV and VUV regions.
The aim of the DichroMatch webserver is to provide a novel means of examining the degree of similarity between two proteins based on their CD spectral characteristics. This, in turn, may indicate the query and matched proteins share some structural (and possibly even functional) similarities. This tool should thus find use in identifying spectral nearest neighbours of an unknown query protein (especially if it has structural similarity but little sequence homology to known proteins), in examining similarities and differences between wild-type and mutant or orthologous proteins or in comparing a series of spectra collected from a single protein under different environmental conditions.
WEB SERVER DESCRIPTION
DichroMatch is a user-friendly webserver developed to enable the identification of closely related spectra to an input query spectrum, as the result of comparisons with a defined reference data set of protein spectra. By default, it uses the Protein Circular Dichroism Data Bank (PCDDB), a publicly accessible repository for the storage of validated CD/SRCD spectra together with their associated metadata, as its reference data set (3,4). Alternatively, the user can create a custom reference data set of selected protein spectra (either using an associated website http://createrds.cryst.bbk.ac.uk or by uploading a compressed collection of files) to perform a more focused matching of a query spectrum. DichroMatch employs six different methods for matching and provides a facility for determining and visualising spectra that share the closest characteristics to the query spectrum. In addition, it also provides metrics on the degree of similarity.
METHODS AVAILABLE
DichroMatch provides six different methods for comparing a query spectrum with spectra in the reference data set. One method is a simple comparison of the query spectrum to all reference spectra at each wavelength. The remaining methods modify the query, and sometimes the reference, spectrum in some way to account for other differences, including inaccuracies in protein concentration or cell pathlength measurements, or differences between CD machine calibrations, so that optimal matches are identified. The quality of each match is defined by a ‘goodness-of-fit’ parameter, the normalized root mean square deviation (NRMSD) (5), as follows:
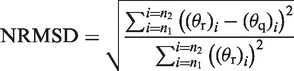 |
where n1 and n2 are the low and high wavelengths of the spectral range used for matching and θr and θq are the CD spectral values of the reference and query spectra, respectively, at the given wavelength i. A good quality match will be when the value of NRMSD is close to zero. For each of the six methods, the query spectrum is compared to each spectrum in the reference set in turn, and the three closest matching CD spectra from this set are displayed as the results, together with the NRMSD value, and where applicable, the relevant scaling term (see below).
Method 1: simple fit
In the simple fit method, the query spectrum is compared to each of the reference spectra and the NRMSD is used as the metric to determine the three closest matching spectra. No spectral modifications of any kind are used, as this method makes the assumption that all input experimental parameters have generated a correctly scaled query spectrum. The same assumption is made for the correctness of the PCDDB spectra, but as these spectra have already been subjected to significant validation procedures before deposition (3), this is likely the case.
Method 2: factor scaling
Factor scaling is designed to eliminate issues associated with errors in the magnitude of the query spectrum as well as accounting for magnitude differences resulting from, for instance, structural flexibility or the presence of intrinsically disordered regions. Since magnitude errors can arise from several common sources (such as protein concentration determinations, sample cell pathlength or the value of the mean residue weight used to convert CD spectra into delta epsilon values), this can produce large (but invalid) variations between spectra (6,7). The factor scaling method obviates these differences in a manner parallel to that used to determine the magnitude dependence of CD secondary structure analyses (8). The factor scaling method systematically scales the query CD spectrum, multiplying it by a factor between +3.0 and −3.0 in increments of 0.01. The ‘best fits’ produced are those with the lowest NRMSD results and with scale factors closest to 1.0. The rescaling process retains the overall shape of the input spectrum while altering all the peak magnitudes by a single value, thus retaining the largest amount of information content in the spectra: the peak positions and relative peak magnitudes.
Method 3: rotational strength
This method uses the fact that the area under the curve of a CD spectrum is proportional to the rotational strength of the chromophores which generated the spectrum. It assumes that two proteins of similar secondary structure will have similar overall rotational strengths (i.e. the area under the spectrum of a mostly helical protein is greater than that for a mostly sheet protein). This method therefore puts the greatest emphasis on the magnitudes and less so on the shapes. It most effectively discriminates between proteins of different classes, i.e. those composed predominantly of alpha helices or β-sheets or even irregularly structured proteins. It is good at corroborating results from other methods. Again, the closest three matches are generated as output.
Method 4: normalized comparison
This method was developed primarily to obviate issues arising from incorrect positioning of the zero value of the Y-axis which can arise from mismatching baselines, but the method could also be of value when there is a significant signal at the high wavelength end of the spectrum (for instance, due to strong aromatic amino acid signals). By scaling both query and reference to minimum and maximum magnitude values between 0 and 1, this method retains the relative peak positions and magnitudes and hence the spectral shape but not where the spectra cross the Y-axis.
Method 5: ratio comparison
The percentages of different types of secondary structure content present in a protein are highly correlated with the CD magnitudes of three spectral peak positions: at 195, 208 and 222 nm and in particular the ratios of these peaks (9). For example, a spectrum with a high 195/208 ratio is much more likely to contain a high helix content than one with a low 195/208 ratio, which more likely arises from proteins with high sheet contents. This method removes much of the spectral information, relying solely on finding the minimal difference in the NRMSDs calculated between the ratio values at only these three wavelength values for the query spectrum and each of the reference spectra in turn. A full NRMSD over the matching wavelength range is used to order the three closest results and is also the final value displayed. This ‘minimalistic’ approach to matching enables potential results to be obtained where an interfering signal exists at other wavelengths due to components such as aromatic residues or disulfide links. Since it only includes a minimal amount of spectral information, it is likely to be the least accurate method but can be used as a corroboration of other methods. The modified plot also shows the match with the query and reference spectra normalized, but this is only for ease of viewing similarities and is not used in the calculation algorithms.
Method 6: wavelength shift
This method obviates differences that can arise from variabilities within the calibration of the CD instruments used to collect the query and reference spectra; these can result in erroneous apparent shifts in wavelengths. Although there are well-established methods for calibrations (6,7), they are often not applied on a regular basis and can have significant effects on spectral analyses (7). Wavelength shifts can also result from proteins being in different solvent environments. This method therefore shifts the query spectrum by values between +3.0 and −3.0 nm in 1 nm steps, with the three best fits being those with the lowest calculated NRMSD values, regardless of the extent of the shift.
INPUT
File formats
Spectral data input is via text files in one of three commonly used CD formats selected from a pull-down menu: .gen [generated by the processing program CDTool (10) and available as a PCDDB output format], .pcd [the standard output format of downloads from the PCDDB (3)] and a simple two-column (wavelength, value) text format. The last of these is a general format that can be constructed from any other file type (including all commercial CD instruments and SRCD beamline ASCII file outputs) using a text editor or spreadsheet software and is most commonly used as the input format to the popular DichroWeb CD analysis server (1,11). The default units are delta epsilon; however, the user can also provide their spectra in other common units used in CD spectroscopy, such as millidegrees or mean residue ellipticity. These units can be converted by the program to delta epsilon units following input of the appropriate additional experimental parameters such as protein concentration, cell pathlength and mean residue weight. The user may chose the wavelength range over which the matching is performed, but the query spectrum must have wavelength coverage of at least between 192 and 222 nm to generate a meaningful match. Ideally, the range should include the lowest wavelength data possible to optimize the information content and produce the best matches (12–14). The maximum wavelength used for the matching is 240 nm because above this sequence peculiarities of proteins such as aromatic and disulphide content tend to dominate the spectrum, as opposed to the backbone fold of the protein.
Reference data sets
The default reference data set for DichroMatch includes all spectral entries in the PCDDB for which a crystal structure is available. This can be altered (by ticking a box) to include all validated PCDDB entries, whether or not crystal structure information is available for them. Note that the PCDDB entries for which crystal structures are available include all the proteins in the SP175 (soluble protein) (15) and MP180 (membrane protein) (16) reference data sets specifically designed to cover fold and secondary structure space. Alternatively, it is possible to employ a user-defined reference data set. This might be desirable, for instance, if the user had a series of spectra of related proteins produced by their own laboratory and not yet deposited in the PCDDB or if the user only wanted to consider a subset of all proteins present in the PCDDB. User-defined reference data sets can be produced using the CreateRDS tool (http://createrds.cryst.bbk.ac.uk). In that case, input formats for individual entries may be in any of the three above-defined formats (.gen, .pcd and text). Alternatively, the user may use a reference data set consisting of any number of files which they have created in a .zip or .tar.gz compression format from a directory they created named ‘reference_set’ which contains files in any one of these formats, but mixed formatting is not permitted.
OUTPUT
DichroMatch provides the closest three matched spectra to the query spectrum produced by the six methods, as indicated by the lowest NRMSD parameter, with the second ranking parameter being a scale factor, if used, closest to 1.0. A graphical output is produced for each method, which for the default reference data set will have a descriptor banner listing the PCDDB identifier code and the name of the protein for that entry. In all cases, the ‘original data’ for the query spectrum (Figure 1, left) is overlaid with the best-matched reference spectrum. For the ‘Simple Fit’ method, there is only one plot for each result and no modifications to the data are made. For the five other methods, a second graphical output (Figure 1, right) is of ‘modified data’ overlaid on that of the matched reference protein (however, the Ratio Comparison method does not use this modified data, it is there for ease of viewing similarities). Note that in the ‘Normalized Comparison’ method, both query and reference spectral data are modified, whereas in the other methods where modification is made, it is only of the query spectrum. For each match, the result can be downloaded via clickable links below the spectra as a .png graphic or as a three-column (wavelength, query and reference) or six-column (wavelength, query, reference, modified wavelength, modified query and modified reference) text file, and if the PCDDB entries were used as the reference data set, a link is provided to that entry using the clickable box provided. In addition, if a PCDDB entry with a known crystal structure has been identified as a match, a link to the secondary structure of the matched reference protein is included, based on a DSSP calculation (17) from the crystal coordinates available, and a link is also available to other information such as the protein sequence, the corresponding Protein Data Bank files (18), and to the cited publication for that PCDDB entry.
Figure 1.

Sample output of the DichroMatch server using the factor scaling method. The query spectrum is coloured in blue, the closest matched protein spectrum from the reference data set is in red and the difference at each wavelength is shown by the green bars on the graph. Left panel: the original unscaled query spectrum overlaid with the reference spectrum. Right panel: the closest fit query spectrum after factor scaling overlaid with the reference spectrum. In the grey descriptor banner at the top is the PCDDB ID code and name of the matched protein (if the default reference data set is used) or the name of the matching protein file (if a user-defined reference data set is used). The NRMSD value is listed in the coloured box below the plots, which is ‘traffic light colour-coded’ to indicate good (green), medium (orange) or poor (red) fits. The scale factor used to modify the query protein to obtain this match is listed. Below that are a series of clickable boxes that enable the user to download the original and modified query spectral plots, a three-column (or six-column) text file containing the wavelength, query and reference protein spectral data (and the additional modified spectral data if appropriate), the full PCDDB entry file of the matched protein (if the default reference data set is used) and, if available, the secondary structure of the matched protein, calculated from the protein crystal coordinates, and other information such as the protein sequence, PDB files. and cited publication for the matched PCDDB entry.
The ‘traffic light’ system: aid to interpretation
The match between the query and reference spectra, as indicated by the NRMSD parameter, is given a degree of quality assessment by a traffic light colour system. A green background suggests that there is a good match and that the sample and reference protein may share structural similarities. A yellow/orange background suggests a medium fit where the extent of structural similarity needs to be investigated further. A red background suggests even the ‘best’ fits are not good and that there is no corresponding protein spectrum in the reference data set. Red fits should not be considered correct. However, these traffic lights are only meant as general guidance regarding match quality. As with all CD analyses methods, the interpretation of the results rely on the user’s judgement based on the context and type of sample examined.
VALIDATION
The aim of the DichroMatch website is to provide a novel means of investigating CD spectral similarities between proteins. The comparisons therefore are not based on a single structural characteristic, as is the case for methods that aim to extract secondary structural compositions (1). Since the DichroMatch website provides a unique means for identifying novel features in proteins with spectral similarities, it is not possible to strictly ‘validate’ the accuracy of the method against a known parameter or set of parameters. However, as an indication that the results can correlate with the one contributor (secondary structure) to the spectral features for which there is comparison data, each method was tested against a data set of 71 proteins of known structure (14) and found to produce good correlations (Supplementary Figure S1). However, it is very important to note that because DichroMatch was specifically devised to identify similarities present which are not fully explained by secondary structure, very high correlations were not expected with this test. Indeed, had the correlations been too good, they would have been an indication that this method was not providing any novel information.
HELP PAGES
The DichroMatch website includes a help facility at http://dichromatch.cryst.bbk.ac.uk/help (which can be accessed by clicking on ‘Help Me’ on the upper right hand corner of the home page). This describes the input and output formats and methods available on the server, information for interpreting the results and how to create reference data sets using the file compression formats, as well as a link to the CreateRDS server that can be used to create reference data sets. It also includes links to related structural and sequence data bases, a contact email link for any user queries and information about the authorship and support of the site. The Help Pages will be updated upon acceptance of this manuscript to include the citation for the DichroMatch server.
AVAILABILITY
DichroMatch is freely accessible to all users at http://dichromatch.cryst.bbk.ac.uk. Likewise, the reference data set formatting server located at http://createrds.cryst.bbk.ac.uk is freely accessible to all users.
Users who identify spectral matches using the PCDDB (3) as their reference data set are expected to cite it as the source of the information. Furthermore, specific spectral matches should cite the original producers of the matched spectral file (available in the cognate .pcd file output from the PCDDB).
CONCLUSIONS
The DichroMatch website provides a means of identifying spectral nearest neighbours for an input query CD spectrum based on a reference data set of CD spectra. The default reference data set consists of the complete validated holdings of the PCDDB for which crystal structures are available. User-defined specialist reference data sets can also be used to generate results for specialized analyses, including for example, focusing on variations within the chosen protein group.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figure S1.
FUNDING
U.K. Biotechnology and Biological Sciences Research Council: Bioinformatics and Biological Resources [F010346 to B.A.W. and F010362 to R.W.J.]. Funding for open access charge: U.K. Biotechnology and Biological Science Research Council [F010346 to B.A.W. and F010362 to R.W.J.].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank members of the Wallace Group at Birkbeck College, University of London for advice, helpful discussions and testing of the software.
REFERENCES
- 1.Whitmore L, Wallace BA. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 2.Wallace BA. Protein characterisation by synchrotron radiation circular dichroism spectroscopy. Q. Rev. Biophys. 2009;42:317–370. doi: 10.1017/S003358351000003X. [DOI] [PubMed] [Google Scholar]
- 3.Whitmore L, Woollett B, Miles AJ, Klose DP, Janes RW, Wallace BA. PCDDB: the Protein Circular Dichroism Data Bank, a repository for circular dichroism spectral and metadata. Nucleic Acids Res. 2011;39:D480–D486. doi: 10.1093/nar/gkq1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitmore L, Woollett B, Miles AJ, Janes RW, Wallace BA. The protein circular dichroism data bank, a Web-based site for access to circular dichroism spectroscopic data. Structure. 2010;18:1267–1269. doi: 10.1016/j.str.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 5.Mao D, Wachter E, Wallace BA. Folding of the mitochondrial proton adenosinetriphosphatase proteolipid in phospholipid vesicles. Biochemistry. 1982;21:4960–4968. doi: 10.1021/bi00263a020. [DOI] [PubMed] [Google Scholar]
- 6.Miles AJ, Wien F, Lees JG, Rodger A, Janes RW, Wallace BA. Calibration and standardisation of synchrotron radiation circular dichroism and conventional circular dichroism spectrophotometers. Spectroscopy. 2003;17:653–661. [Google Scholar]
- 7.Miles AJ, Wien F, Lees JG, Wallace BA. Calibration and standardisation of synchrotron radiation and conventional circular dichroism spectrometers. Part 2: factors affecting magnitude and wavelength. Spectroscopy. 2005;19:43–51. [Google Scholar]
- 8.Miles AJ, Whitmore L, Wallace BA. Spectral magnitude effects on the analyses of secondary structure from circular dichroism spectroscopic data. Protein Sci. 2005;14:368–374. doi: 10.1110/ps.041019905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stone T. University of London; 2009. The analysis of protein circular dichroism spectra. PhD thesis. [Google Scholar]
- 10.Lees JG, Smith BR, Wien F, Miles AJ, Wallace BA. CDtool—an integrated software package for circular dichroism spectroscopic data processing, analysis and archiving. Anal. Biochem. 2004;332:285–289. doi: 10.1016/j.ab.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Whitmore L, Wallace BA. DichroWeb, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004;32:W668–W673. doi: 10.1093/nar/gkh371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hennessey JP, Johnson WC. Information content in the circular dichroism of proteins. Biochemistry. 1981;20:1085–1094. doi: 10.1021/bi00508a007. [DOI] [PubMed] [Google Scholar]
- 13.Wallace BA, Janes RW. Synchrotron radiation circular dichroism spectroscopy of proteins: secondary structure, fold recognition, and structural genomics. Curr. Opin. Chem. Biol. 2001;5:567–571. doi: 10.1016/s1367-5931(00)00243-x. [DOI] [PubMed] [Google Scholar]
- 14.Lees JG, Miles AJ, Janes RW, Wallace BA. Novel methods for secondary structure determination using low wavelength (VUV) circular dichroism spectroscopic data. BMC Bioinformatics. 2006;7:507. doi: 10.1186/1471-2105-7-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lees JG, Miles AJ, Wien F, Wallace BA. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics. 2006;22:1955–1962. doi: 10.1093/bioinformatics/btl327. [DOI] [PubMed] [Google Scholar]
- 16.Abdul-Gader A, Miles AJ, Wallace BA. A reference dataset for the analyses of membrane protein secondary structures and transmembrane residues using circular dichroism spectroscopy. Bioinformatics. 2011;27:1630–1636. doi: 10.1093/bioinformatics/btr234. [DOI] [PubMed] [Google Scholar]
- 17.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 18.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]