


Abstract
Seq2Logo is a web-based sequence logo generator. Sequence logos are a graphical representation of the information content stored in a multiple sequence alignment (MSA) and provide a compact and highly intuitive representation of the position-specific amino acid composition of binding motifs, active sites, etc. in biological sequences. Accurate generation of sequence logos is often compromised by sequence redundancy and low number of observations. Moreover, most methods available for sequence logo generation focus on displaying the position-specific enrichment of amino acids, discarding the equally valuable information related to amino acid depletion. Seq2logo aims at resolving these issues allowing the user to include sequence weighting to correct for data redundancy, pseudo counts to correct for low number of observations and different logotype representations each capturing different aspects related to amino acid enrichment and depletion. Besides allowing input in the format of peptides and MSA, Seq2Logo accepts input as Blast sequence profiles, providing easy access for non-expert end-users to characterize and identify functionally conserved/variable amino acids in any given protein of interest. The output from the server is a sequence logo and a PSSM. Seq2Logo is available at http://www.cbs.dtu.dk/biotools/Seq2Logo (14 May 2012, date last accessed).
INTRODUCTION
The idea of generating a logo from aligned sets of sequences was introduced in 1990 by Schneider and Stephens (1). The intention of a sequence logo is to concentrate into a single plot the general consensus, the order of predominance of residues at every position, the relative frequencies of every residue at every position, the amount of information present at every position and significant locations. This logo is then able to present all of the relevant information to the viewer in a fast and concise manner.
Several webservers exist to generate sequence logos from MSA’s (2–5). All these servers suffer from different limitations in the handling sequence redundancy and low number of observations. Moreover, to the best of our knowledge, all public sequence logo servers, with the exception of the Icelogo (4) and two-sample logo (5) methods, focus on displaying the position-specific enrichment of amino acids, discarding the equally valuable information related to amino acid depletion. Seq2logo aims at resolving these issues allowing the user to include sequence weighting to correct for data redundancy, pseudo counts to correct for low number of observations (6–8) and five different logotype representations each capturing different aspects related to amino acid enrichment and depletion. In addition to the usual Shannon logo (9), Seq2Logo includes the option to create Kullback–Leibler (KL) (10) logos where the depleted (under-represented) amino acids are represented on the negative y-axis. Besides the conventional KL logo, Seq2Logo can also display a weighted KL logo, where the relative height of each amino acid is proportional to the log-odds ratio and a probability weighted KL logo, where the relative height of each amino acid is proportional to the product of the probability and log-odds ratio. Finally, inspired by the work of Fujii et al. (11), Seq2Logo also includes an option to visualize PSSM (position-specific scoring matrix) logos, where the height of a bar is given by the sum of the absolute value of the PSSM weight matrix values and the height of a given amino acids is proportional to the absolute value of the weight matrix score. In particular, the weighted KL logo provides a visual and highly intuitive representation of both amino acid enrichment and depletion in for instance receptor binding motifs. Besides allowing input in the format of peptides and MSAs, the Seq2Logo server accepts inputs such as Blast sequence profiles, providing easy access for non-expert end-users to characterize and identify functionally conserved/variable amino acids in any given protein of interest.
MATERIALS AND METHODS
Seq2Logo implements two strategies to improve the accuracy of the estimated sequence logo. The first strategy is sequence weighting which corrects for data redundancy. The second strategy is pseudo counts which correct for a low number of observations. Sequence weighting is implemented as described in (6,8) and pseudo counts as described in (7). For details, see Supplementary Data.
In a sequence logo, the height of the bar is equal to the information content at each amino acid position. The information content is calculated using the relation 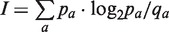 , where pa and qa are the observed probability (calculated from the data) and background probability, respectively, of the amino acid a. If an equiprobable background amino acid distribution is applied, a conventional Shannon sequence logo is displayed. If a background amino acid distribution reflecting the prevalence of the different amino acids is applied, a Kullback–Leibler sequence logo is displayed. The choice of the Kullback–Leibler logotype in Seq2Logo not only provides correction for the uneven distribution of amino acids, but also expresses the depleted amino acids (where pa < qa) on the negative side of the y-axis. This enables the user to quickly identify enriched and depleted (under-represented) amino acids. To enhance the identification and information of the depleted amino acids, Seq2Logo includes another logotype called weighted Kullback–Leibler. This logo type presents each individual amino acid proportional to its relative log-odds score [log2(pa/qa)]. Another logotype is included called probability weighted Kullback–Leibler, where the relative height of each individual amino acid is proportional to pa · log2(pa/qa). Finally, Seq2Logo includes an option to display PSSM-logos (11), where the height of a bar is equal to the sum of the absolute value of the PSSM weight matrix values and the height of each amino acid is proportional to the absolute value of the weight matrix score (with negative values displayed on the negative y-axis).
, where pa and qa are the observed probability (calculated from the data) and background probability, respectively, of the amino acid a. If an equiprobable background amino acid distribution is applied, a conventional Shannon sequence logo is displayed. If a background amino acid distribution reflecting the prevalence of the different amino acids is applied, a Kullback–Leibler sequence logo is displayed. The choice of the Kullback–Leibler logotype in Seq2Logo not only provides correction for the uneven distribution of amino acids, but also expresses the depleted amino acids (where pa < qa) on the negative side of the y-axis. This enables the user to quickly identify enriched and depleted (under-represented) amino acids. To enhance the identification and information of the depleted amino acids, Seq2Logo includes another logotype called weighted Kullback–Leibler. This logo type presents each individual amino acid proportional to its relative log-odds score [log2(pa/qa)]. Another logotype is included called probability weighted Kullback–Leibler, where the relative height of each individual amino acid is proportional to pa · log2(pa/qa). Finally, Seq2Logo includes an option to display PSSM-logos (11), where the height of a bar is equal to the sum of the absolute value of the PSSM weight matrix values and the height of each amino acid is proportional to the absolute value of the weight matrix score (with negative values displayed on the negative y-axis).
THE WEB SERVER
The Seq2Logo server has a simple interface that allows non-expert users to generate and customize accurate logos from any amino acid sequence data of interest.
Input
The interface is split in two parts for easy overview. The first and the most important part is submission (Figure 1, left panel). Here, the user can upload or paste in the input data in addition to specifying the logotype (Shannon, Kullback–Leibler, Weighted Kullback–Leibler, probability weighted Kullback–Leibler or PSSM-logo) and conditions for handling the input data (sequence weighting and pseudo counts). Seq2Logo can read sequence data in the following formats: Fasta, ClustalW, Raw peptide sequences and Weight/Blast matrix (for details on each format refer to Supplementary Data). The detection of the format happens automatically through the identification of key elements from each format. In the submission part, the user further specifies which output files should be created. In the graphical layout (Figure 1, right panel), the user can customize the graphical layout of the logo plot. Page size sets the resolution of the image and stacks per line and lines per page determine how the logo should look. Assigning each amino acid symbol to a color defines the amino acid colors. There are six colors to choose from: Red, green, blue, yellow, purple or orange. All amino acids left out will be black. Several predefined color-schemes are available. The user can also rotate the position numbers on the x-axis and hide various features of the graph.
Figure 1.

The submission (left) and graphical layout (right) part of the web interface. In the submission part the user specifies the input file, the format of output files, the logotype and the conditions for the handling of the input data. In the Graphical Layout part, the user customizes the graphical layout of the logo plot; page size, stacks per line, lines per page, colours, bars, rotation of position numbers and title.
Output
An example of the output from Seq2Logo generated using the input specifications from Figure 1 is shown in Figure 2. The figure shows on the positive y-axis, the amino acids enriched at each peptide position and on the negative y-axis the corresponding depleted amino acids. In this case, the logo is calculated from a set of 13 artificial peptide sequences proposed to bind the HLA-A*02:01 class I major histocompatibility complex (MHC) molecule. This molecule has a binding motif with strong interactions at P2 and P9 both positions with prevalence for hydrophobic amino acids (12).
Figure 2.

Output from Seq2Logo. The upper panel shows the sequence logo calculated from a set of 13 artificial peptide sequences using the specification defined in Figure 1 (sequence weighting using clustering, pseudo count with a weight of 200 and logotype as Kullback–Leibler). Enriched amino acids are shown on the positive y-axis and depleted amino acids on the negative y-axis. The lower panel gives the position-specific (log-odds) scoring matrix (PSSM) calculated by Seq2Logo. Each line corresponds to a position and gives the consensus amino acid and the log-odds scores for the 20 amino acids.
One of the distinct powers of Seq2Logo is its ability to deal with data redundancy and low number of observations. To the best of our knowledge, no other public sequence logo servers share this ability. In Figure 3, the cruciality of these features for the generation of accurate sequence logos describing a binding motif is illustrated. The figure displays Shannon sequence logos generated by Seq2Logo, using different option to improve the accuracy, as well as sequence logos generated by Weblogo (2) and EnoLOGOS (3). When comparing the logos calculated from the small sample data set with the logo obtained from the larger data set, it is apparent that the inclusion of sequence weighting and pseudo counts have a significant positive impact on the overall accuracy of the binding motif description.
Figure 3.

Sequence logos generated from small sequence samples. All logos except the right logo in the lower row were calculated from a set of 13 artificial peptide sequences proposed to bind HLA-A*02:01 (see Figure 1). The upper row shows logos calculated by Seq2Logo using: (i) without sequence weighting and pseudo count correction, (ii) sequence weighting by clustering and no pseudo count correction and (iii) sequence weighting by clustering and pseudo count correction with a weight on prior of 200. The lower row shows logos calculated using: (i) Weblogo with ‘small sample correction’, (ii) EnoLOGOS and (iii) Seq2Logo from a set of 229 HLA-A*02:01 9mer ligands downloaded from the SYFPEITHI database (12) with sequence weighting by clustering and pseudo count correction with a weight on prior of 200.
The other distinct feature of Seq2Logo compared to most other public sequence logo server is the display of depleted amino acids on the negative y-axis in Kullback–Leibler logos. Most sequence logo servers display the relative height of the different amino acids in a manner proportional to their frequency, thus displaying only the position-specific enrichment of amino acids, discarding the equally valuable information related to amino acid depletion. To improve on this issue, Seq2Logo includes a series of distinct logotypes (see Figure 4). In addition to the usual Shannon logo, Seq2Logo includes the option to create Kullback–Leibler (KL) logos where depleted amino acids are represented on the negative y-axis. Besides the conventional KL logo, Seq2Logo can also display a weighted KL logo, where the relative height of each amino acid is proportional to the log-odds ratio and a probability weighted KL logo, where the relative height of each amino acid is proportional to the product of the probability and log-odds ratio. In particular, the weighted KL logo provides a visual and highly intuitive representation of both amino acid enrichment and depletion in for instance receptor binding motifs. Besides these information-based logotypes, Seq2Logo offers the possibility of displaying PSSM-logos calculated either from a log-odds weight matrix derived by Seq2Logo from a multiple sequence alignment or from a user-defined PSSM. In the PSSM-logo, the height of the bar and amino acid at each position is proportional to the absolute value of the PSSM weight matrix values. This logotype is particularly powerful when illustrating depletion of a small set of amino acids form otherwise variable positions in a sequence motif. One such example is N-linked glycosylation sites that are known to have the motif N-X-S/T where X can be any amino acid but P. Visualizing this motif as an information-based sequence logo will not capture the depletion of P at the position between N and S/T as all amino acids except P are found at this position, hence making the overall information content very small. On the other hand, visualizing the motif as a PSSM-logo, the strong depletion of P at the position between N and S/T becomes apparent (see Figure 5).
Figure 4.

The different logotype representations covered by Seq2Logo. Sequence logos generated from at set of 13 artificial peptide sequences proposed to bind HLA-A*02:01 (see Figure 1). All logos were calculated using clustering and pseudo counts with a weight on prior at 200. Upper row, left panel: Shannon, right panel: Kullback–Leibler. Lower row left panel: weighted Kullback–Leibler, right panel: probability weighted Kullback–Leibler.
Figure 5.

PSSM-logo for the N-linked glycosylation motif. The motif was calculated from a set of 2128 unique experimentally verify N-glycosylation sites downloaded from the UniprotKB protein database. Only peptide fragments of length 11 (5 before and 5 after the N) were included in the analysis.
A powerful way to characterize sequence conservation/variation within a protein family is by use of sequence profiles. Such sequence profiles can be obtained using Psi-Blast (7). Seq2Logo accepts input of such sequence profile in the Blast profile format allowing easy access for non-expert end-users to characterize and identify functionally conserved/variable amino acids in any given protein of interest. Blast sequence profile can be generated either in-house using a command like ‘blastpgp −d db −e 0.00001 −j 4 −Q blastprofile −i fasta −o out’, where db is the sequence database used to search by Blast, −e defines the e-value cut-off for significant hits, −j defines the number of Psi-blast iterations, −i is the input file in FASTA format, −Q is the output file for the blast profile (the file to be used by Seq2Logo to visualized the sequence profile) and −o is the file for the blast output. Alternatively, the Blast2logo webserver (www.cbs.dtu.dk/biotools/Blast2logo (14 May 2012, date last accessed)) can be used to obtain the sequence profile. Figure 6 demonstrates the use of Seq2Logo to display a sequence profile for Rhamnogalacturonan acetylesterase (PDBid 1K7C, chain A). The active site of 1K7C.A is defined by the residues S9, G42, N74, D192 and H195 (13). All these residues are highly conserved in the sequence logo (in fact they are among the 10 residues with the highest information content, data not shown). Another striking observation from the logo is the lack of sequence information in the area between positions 75 and 105, suggesting that this part of the protein is highly variable (most likely an insertion) within the protein family. Both these observations illustrate the power of sequence profiles combined with Seq2Logo as a simple tool to identify functionally important residues and insertions in protein sequences.
Figure 6.

Seq2Logo visualization of a Blast sequence profile for 1K7C chain A. The Blast profile was obtained using Blast2logo (www.cbs.dtu.dk/biotools/Blast2logo (14 May 2012, date last accessed)) searching against the nr70 sequence database with default options. The active site of 1K7C:A is defined by the residues S9, G42, N74, D192 and H195 (13). All these residues show up as highly conserved in the sequence logo.
INTEGRATING SEQ2LOGO WITH OTHER PREDICTION SERVERS
To improve the usability and make Seq2Logo able to cooperate with other programs and servers, a form-handler was implemented on the server that makes it possible to send input data directly to Seq2Logo. This simple form-handler allows a quick and easy transfer of data to Seq2Logo and defines a platform for using Seq2Logo as a visualization tool for other programs. The form data sent to Seq2Logo is inserted directly into the input field. An instruction of how to implement this transfer can be found at: http://www.cbs.dtu.dk/biotools/Seq2Logo-1.0/bin/easytransferbutton.html (14 May 2012, date last accessed).
DISCUSSION AND CONCLUSION
Sequence logos provide a powerful way to visualize amino acid preferences in a receptor binding motif, as well as sequence conservation/variation and the location of functionally essential residues in multiple sequence alignments. Accurate estimation of a sequence motif is often compromised by data redundancy and low number of observations. Inappropriate handling of these issues can lead to inaccurate estimation of the sequence motif and subsequent poor sequence logo representation. Moreover, the majority of sequence logo webservers have a poor visualization of the information related to amino acid depletion since they focus on displaying the position-specific enrichment of amino acids.
Here, we have proposed a novel sequence logo generator, Seq2Logo that aims at addressing these shortcomings and allow non-expert end-users, via an easy to use web-interface, to generate accurate sequence logos from protein sequence data. We have demonstrated that Seq2Logo can deal with sequence redundancy and low number of observations in a manner superior to that of other public available sequence logo generators like Weblogo and ENOlogos. Besides the conventional Shannon sequence logo, Seq2Logo also incorporates distinct logotypes where depleted amino acids are displayed on the negative y-axis. These logotypes offer a unique possibility for Seq2Logo to display for instance receptor-binding motifs in a format that highlights both favored and disfavored amino acids at the different positions in the motif.
A sequence profile is a powerful way to capture position-specific information about sequence conservation/variation within a protein family. Seq2Logo accepts sequence profiles in the Blast format as input and can in a very simple and intuitive manner be used in combination with Blast as a tool to visualize sequence profiles and identify functionally conserved/variable amino acids in any given protein of interest.
Finally, to allow other servers dealing with multiple sequence alignments and binding motifs to directly cooperate with Seq2Logo and benefit from its improved features, the server includes a form-handler that enables communication with Seq2Logo via a simple html form. This feature has allowed for a simple and effective improvement to two of our own webservers NNAlign (14) and Blast2logo (www.cbs.dtu.dk/biotools/Blast2logo (14 May 2012, date last accessed)), and we believe this to be an additional feature that will become very useful for other webserver developers within the field of for instance receptor-binding motif characterization.
In its current form, Seq2Logo can only handle amino acid input data. The reason for this limitation is that most of its unique features like pseudo count estimates from Blosum substitution matrices and sequence weighting of are specific for amino acid data. The ability to also handle nucleic acids will be a part of a future update for the method.
In conclusion, we believe Seq2Logo to be an important and novel tool for non-expert users to construct accurate sequence logos describing receptor binding motifs and sequence variations in multiple sequence alignments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Methods and Supplementary References [6–8,15,16].
FUNDING
Funding for open access charge: National Institutes of Health (NIH) [contract nos HHSN272200900045C and HHSNN26600400006C].
Conflict of interest statement. None declared.
REFERENCES
- 1.Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Workman CT, Yin Y, Corcoran DL, Ideker T, Stormo GD, Benos PV. enoLOGOS: a versatile web tool for energy normalized sequence logos. Nucleic Acids Res. 2005;33:W389–W392. doi: 10.1093/nar/gki439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colaert N, Helsens K, Martens L, Vandekerckhove J, Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- 5.Vacic V, Iakoucheva LM, Radivojac P. Two Sample Logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics. 2006;22:1536–1537. doi: 10.1093/bioinformatics/btl151. [DOI] [PubMed] [Google Scholar]
- 6.Henikoff S, Henikoff JG. Position-based sequence weights. J. Mol. Biol. 1994;243:574–578. doi: 10.1016/0022-2836(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 7.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O. Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics. 2004;20:1388–1397. doi: 10.1093/bioinformatics/bth100. [DOI] [PubMed] [Google Scholar]
- 9.Shannon CE. A mathematical theory of communication. Bell Syst. Tech. J. 1948;27:379–423, 623–656. [Google Scholar]
- 10.Kullback S, Leibler RA. On Information and Sufficiency. Ann. Math. Stat. 1951;22:79–86. [Google Scholar]
- 11.Fujii K, Zhu G, Liu Y, Hallam J, Chen L, Herrero J, Shaw S. Kinase peptide specificity: improved determination and relevance to protein phosphorylation. Proc. Natl Acad. Sci. USA. 2004;101:13744–13749. doi: 10.1073/pnas.0401881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–219. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 13.Porter CT, Bartlett GJ, Thornton JM. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:D129–D133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Andreatta M, Schafer-Nielsen C, Lund O, Buus S, Nielsen M. NNAlign: a web-based prediction method allowing non-expert end-user discovery of sequence motifs in quantitative peptide data. PLoS One. 2011;6:e26781. doi: 10.1371/journal.pone.0026781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc. Natl Acad. Sci. USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hobohm U, Scharf M, Schneider R, Sander C. Selection of representative protein data sets. Protein Sci. 1992;1:409–417. doi: 10.1002/pro.5560010313. [DOI] [PMC free article] [PubMed] [Google Scholar]