


Abstract
Protein structures are necessary for understanding protein function at a molecular level. Dynamics and flexibility of protein structures are also key elements of protein function. So, we have proposed to look at protein flexibility using novel methods: (i) using a structural alphabet and (ii) combining classical X-ray B-factor data and molecular dynamics simulations. First, we established a library composed of structural prototypes (LSPs) to describe protein structure by a limited set of recurring local structures. We developed a prediction method that proposes structural candidates in terms of LSPs and predict protein flexibility along a given sequence. Second, we examine flexibility according to two different descriptors: X-ray B-factors considered as good indicators of flexibility and the root mean square fluctuations, based on molecular dynamics simulations. We then define three flexibility classes and propose a method based on the LSP prediction method for predicting flexibility along the sequence. This method does not resort to sophisticate learning of flexibility but predicts flexibility from average flexibility of predicted local structures. The method is implemented in PredyFlexy web server. Results are similar to those obtained with the most recent, cutting-edge methods based on direct learning of flexibility data conducted with sophisticated algorithms. PredyFlexy can be accessed at http://www.dsimb.inserm.fr/dsimb_tools/predyflexy/.
INTRODUCTION
X-ray experiments have been valuable tools to understand the intimate relation between protein structures and biological functions. They have revealed a large diversity of well-defined folds, each being adopted by members of a given functional family. However, recent studies have shown that conformational changes are required by numerous proteins in their folded state to accomplish their function [e.g. enzyme catalysis, activity modulation, macromolecular interactions, ligand binding and cell motility (1–4)]. This has led to revisit the importance of dynamics and to focus on regions with peculiar flexibility properties, supposed to participate in conformational changes. Hence, determining those regions would be extremely useful to decipher and eventually control biological function. Actually, few studies have focused on flexible regions in folded ordered proteins. Studies have mainly focused on (i) the analysis of specific protein structures to catch and/or simulate the flexible and rigid regions and (ii) the sole information of the sequence to predict flexibility.
In the first case, 3D structures are required all along. B-factors available with X-ray structures were first used as the main criteria to define protein rigidity and flexibility. Nowadays, the distinction between flexible and rigid regions takes advantage of dedicated approaches for exploring dynamics. The most popular approaches consist in atomistic molecular dynamics simulations which are available through different packages, such as Gromacs (5), Amber (6), NAMD (7) or Charmm (8). Principal component analyses of the resulting data allow identifying regions involved in the different type of motions and provide relevant information about the visited conformational space. Less time-consuming methods are also available, e.g. FlexServ (9), ElNemo (10) or Nomad (11), which perform normal mode analysis (NMA) of elastic network models. Data can also be gained with Brownian dynamics and discrete molecular dynamics (9) or more specialized approaches, e.g. to define hinges between domains, as StoneHinge (12), HingeProt (13) and tCONCOORD (14), which predict conformational flexibility based on geometrical considerations. All these methods give a large amount of data that bring quantitative information enabling precise ranking of flexible and rigid regions by highlighting local deformation as large domain motions.
In the second case, the prediction is based on the sole amino acid sequence. Historically, the flexibility was first predicted as Boolean, i.e. rigid or flexible, using simple statistical analyses of B-factor values (15,16). In the same spirit, Schlessinger et al. (17) developed more recently PROFBval, a method that improved the two-state flexibility prediction by using Artificial Neural Networks (ANNs) combined with evolutionary information. Instead of ANNs, Pan and Shen (18) used support vector regression coupled with random forest. Chen et al. (19) proposed an innovative development of logistic regressions and colocation-based representation with multiple features to predict flexible and rigid region. Nuclear magnetic resonance (NMR) data are alternative sources of information for protein dynamics. Zhang et al. (20) and Trott et al. (21) chose to exploit these data rather than X-ray B-factors. Zhang’s group used variation of backbone torsion angles from NMR structural models, whereas Trott et al. preferred order parameters to define the protein flexibility. Both groups performed prediction with neural networks. Galzitskaya et al. (22) extend the FoldUnfold methodology, which was originally designed to predict disorder, to the prediction of flexibility (23).
Interesting works related to protein flexibility prediction have focussed on more specific question. Hence, Hirose et al. used NMA to define specific motions in proteins. These motions were predicted using a random forest algorithm and were further used to explore protein–protein interaction (24). Hwang et al. (25) focused on prediction of flexible loops and combined B-factors, dihedral angles and accessibility. Kuznetsov et al. proposed a web server for predicting residue involved in conformational switches in proteins. Interestingly, it can use either protein sequence or structure. The prediction from the sequence is done with support vector machines (SVMs) (26,27).
We take advantage of the method we previously elaborated to predict local protein structures. We have described global protein structures using a limited set of recurring local structures named long structural prototypes (LSPs) (28). These LSPs encompass all known local protein structures and ensure good quality 3D local approximation. We have proposed a prediction method based on evolutionary information coupled with SVMs. This method provides with a list of five possible structural candidates for a target sequence. The prediction rate reaches 63.1%, a rather high value given the high number of structural classes (29). We use the output of this structural prediction as the input for our prediction method of flexibility.
The originality of our method lies (i) in the use of a combination of two descriptors for quantifying protein dynamics, i.e. the X-ray B-factors and the root mean square fluctuation (RMSF) computed from molecular dynamics, (ii) in the prediction of flexibility through structural prediction of LSPs (see above) and (iii) by considering three classes of flexibility defined by the chosen descriptors and in which LSPs were distributed. This method turns out to be rather efficient compared to the most commonly used ones. The prediction rate is slightly better than the one of PROFbval (17) that was optimized for two classes. Importantly, we also propose a confidence index (CI) for assessing the quality of the prediction rate. The method is implemented in a useful web server PredyFlexy (http://www.dsimb.inserm.fr/dsimb_tools/predyflexy/) which is able to give different type of predictions as well a CI with outputs and flat file.
METHODS
The server can be used to predict protein flexibility as well as to predict local protein structure defined by LSPs. Figure 1 explains the two main steps of the prediction methodology. At first, LSPs are predicted and then using this prediction, protein flexibility is predicted. Prediction is defined using classical normalized B-factors (B-factorNorm) and normalized RMSF (RMSFNorm) from molecular dynamics.
Figure 1.

The framework of PredyFlexy and underlying methods. User must give a single sequence as input (a), a PSSM is computed using PSI-BLAST (b) and split into fragments of 11 residues. Prediction of LSPs is done using trained SVMs (c); scores are ranked and the best five are kept (d). Using these LSPs, prediction of flexibility is done in three states (rigid, intermediate and flexible) (e); predicted B-factorNorm, RMSFNorm and a confidence index are also provided (f).
LSP prediction
We have proposed a library consisting of 120 overlapping structural classes of 11-residue long fragments (28). This library was constructed with an original unsupervised structural clustering method called the Hybrid Protein Model (HPM) (30). The hybrid protein principle is similar to a self-organizing neural network (31,32). It was constructed as a ring of N neurons (here N = 120), each representing a cluster of structurally similar 3D fragments encoded into series of Protein Blocks (PBs). PBs are a structural alphabet (33), i.e. a set of local protein fragments, able to provide correct approximation of protein structure. Its training strategy consisted in learning the similarities between protein structural fragments deduced from the alignment of their series of PBs (34,35). Once the HPM was trained, each neuron or cluster was associated with a set of fragments representing a structural class using root mean square deviation (RMSD) (28). For each class, a mean representative fragment, or a ‘local structure prototype’ (LSP), was chosen. The 120 LSPs correctly approximated the local structure ensembles. The major advantage of this library is its capacity to capture the continuity between the identified recurrent local structures (29). Relevant sequence–structure relationships were also observed and further used for prediction. Briefly, LSP prediction is based on SVM training. With the LSP prediction, a CI that is based on the discriminative power of the SVMs is provided. The higher the CI, the better the prediction rate. For more details on LSPs and their prediction, please see (36).
Protein structure datasets
A dataset of 172 X-ray high-resolution (≤1.5 Å) globular protein structures was extracted from the Protein Data Bank (PDB) using the PDB-REPREDB database web service (37) that provides the user with different choices of thresholds for selecting chains of given sequence and structural similarity. The method is detailed in (38). We chose chains sharing <10% sequence identity and for which the Cα RMSD between aligned residues differ by at least 10 Å. Proteins composed of a single domain, not involved in a protein complex, and that did not have extensive number of contacts with ligands were only considered. A final dataset of 43 protein structures was obtained. This dataset 1 was used to calibrate thresholds for RMSF computed from molecular dynamics simulations using Gromacs (5). Parameters and conditions defined in (39) were used for the simulations. A larger, non-redundant databank composed of 1421 X-ray structures with resolution higher than 1.5 Å, sequence identity smaller than 30% and Cα RMSDs larger than 10 Å [selected using PDB-REPRDB (37)] was used for the prediction itself (data set 2).
Definition of protein structure flexibility classes
We extracted Cα B-factors from the PDB files of the protein structures dataset 1. For comparison purposes, the raw values were normalized for each protein using the method in (40). After removing outliers detected statistically with a median-based approach, the normalized B-factors were calculated as B-factorNorm = (B-factorRaw − µ)/σ, where µ and σ stand for the mean and the standard deviation of the Cα B-factor, respectively. Flexibility of each 11-residue long overlapping fragment in the dataset was characterized by the B-factorNorm associated with its central Cα.
Similarly, we extracted flexibility measurements from molecular dynamics simulations. Cα RMSF was calculated using g_rmsf GROMACS tool (5) after superimposing each snapshot structure on the initial conformation. Cα RMSF gives the mean amplitude of each Cα movement compared to a mean reference position:
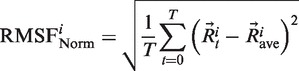 |
where T is the production time expressed in snapshot number, 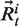 the coordinates of Cα atom i of structure at time t and
the coordinates of Cα atom i of structure at time t and 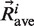 the average coordinates of Cα atom i over production time. Raw RMSF values were normalized for each protein. The RMSFNorm associated with the central Cα of each 11-residue fragment characterized the flexibility using molecular dynamics.
the average coordinates of Cα atom i over production time. Raw RMSF values were normalized for each protein. The RMSFNorm associated with the central Cα of each 11-residue fragment characterized the flexibility using molecular dynamics.
Hence, to each fragment is associated a couple of values B-factorNorm and RMSFNorm. The three flexibility classes, rigid, intermediate and flexible, were then defined from a fine calibration of thresholds combining Cα RMSF (noted τF) and B-factors (noted τB). The calibration was based on a backward–forward procedure targeting the optimal flexibility prediction rate. Fragments for which the couple (Cα B-factors, Cα RMSF) is (i) smaller than τB1, τF1 are rigid, (ii) larger than τB1, τF1 but smaller τB2, τF2 are intermediate and (iii) larger τB2, τF2 are flexible.
Finally, a detailed analysis of RMSF and Cα B-factors couples for each LSP allowed attributing a well-defined flexibility class to each of them as well as a mean B-factorNorm and a mean RMSFNorm. This was obtained by (i) computing the propensity of fragments belonging to a LSP to be associated with a given flexibility class and (ii) selecting as the unique assigned class for each LSP, the class that maximizes the propensity (see (39) for details).
Flexibility prediction
For a target sequence, the local structure prediction is first performed and yields the five best LSP candidates. Then, the predicted flexibility class is obtained by simply calculating the rounded average of the flexibility classes of the five candidates. In the same way, the B-factorNorm and RMSFNorm are predicted by averaging the mean B-factorNorm and RMSFNorm of the five structural candidates. At this stage, no training on the data was performed. The prediction reflects the informativity of structural prediction from sequence for flexibility.
DISCUSSION
The PredyFlexy method is based on the flexibility analysis of local protein structures through an appropriate combination of the B-factor of X-ray experiment and the fluctuation of residues during molecular dynamics simulations. A correlation (r2 = 0.68) was obtained between Cα-B-factorNorm and Cα-RMSFNorm. This value confirms that even though related, both descriptors bring different information justifying the interest to combine both measures of the flexibility. The PredyFlexy method led to an average, well-balanced prediction rate of 49.4% for the three defined flexibility classes, a value considerably higher than a random prediction rate. The correlation r2 between observed and predicted values for B-factorNorm and RMSFNorm reached 0.71 and 0.69, respectively. When outliers (5% of the values), detected by the median-based approach proposed by Smith et al. (40), were excluded, correlations r2 climbed to 0.94 and 0.96, respectively. This correlation is slightly better than the best correlation value obtained by the PONDR VSL1 prediction methods (41).
For comparison purpose, we regrouped our three flexibility classes into two classes to assess a two-class prediction. Depending on the grouping, we obtained prediction rates comparable and even better than the current methods available (17,18). Details are given in Table IV of (39). This confirms that LSP description is truly useful for addressing flexibility prediction.
Web server
PredyFlexy provides a user-friendly web interface that combines predictions for local structure and flexibility. The homepage contains a short summary of the two aspects of the method. In this page, the sole input, the protein sequence, must be provided. Two possibilities are offered: the sequence, in FASTA format, may be pasted in a first window frame or downloaded from a file, the filename being given in a second window frame. This page contains additional links: ‘Contacts’ which refers to authors’ homepage, ‘About Method’ which details the methodology and its flowchart, ‘Download’ which allows to obtain a local version of the program by sending an email for registration, ‘Example’ which illustrates with a concrete case, the input and output of the server (see below) and ‘DSIMB’ which connects to team’s homepage. In Figure 1, the different steps that led from a protein sequence to the output results of the prediction are described.
Input
A single FASTA sequence must be provided (Figure 1A). A check is performed to ensure that only natural amino acids are used.
Background step: ‘PredyFlexy running’
For the given sequence, a Position Substitution Sequence Matrix (PSSM) is first computed with PSI-BLAST v. 2.2.09 (42) using default parameters and SWISS-PROT databank (43) (Figure 1B). The sequence is then divided into overlapping fragments of 11 residues long (Figure 1B), corresponding to the LSP size. In a third step, LSP prediction is done using 120 independent SVMs (libsvm-2.81) that was previously optimized for each LSP (36). This method yields for a target sequence a list of five structural candidates associated with the highest scores (Figure 1D). The prediction rate reaches 63.1%, a rather high value given the high number of structural classes (36). From this prediction, the corresponding flexibility class of the LSP is attributed. Hence, at this stage, each sequence fragment is characterized by five flexibility states, one per structural LSP in the list. Finally, the predicted flexibility state of an 11-residue sequence is a simple mean of the flexibility states for the five predicted LSP candidates (Figure 1E). Using a similar approach, local B-factorNorm and RMSFNorm are predicted (Figure 1F).
Output
Once the job is finished, a window opens with the results. Results are given as a text file that can be downloaded. Results may also be visualized through different graphical outputs. The first graphs represent the values, along the sequence, of the B-factorNorm (green), the RMSFNorm (yellow) on the left y-axis and on the right y-axis and the CI (gray line). For clarity, the results are represented by blocks of 50 residues. The sequence is reported in the same graph above the x-axis. These combined representations allow the user to focus on the regions with a high CI, i.e. larger than 15 (representing >25% of residues), frequently associated with regions with low flexibility. In the second part of the page, a table summarizes the results of the local structure prediction, the CI and the flexibility class. The lines correspond to each position along the sequence. In the two first columns, the position and the corresponding amino acid (one letter) are indicated. The five following columns contain the five best LSP candidates represented by their 3D structure and their corresponding number in the HPM (for details, see (36)). The two last columns correspond to the CI value and the predicted flexibility class (0 for rigid, 1 for intermediate and 2 for flexible). The CI is represented by 19 discrete values ranked from 1 to 19, with the prediction confidence increasing. For a rapid visualization inspection, values for CI and flexibility classes are also represented with colors. Note that due to the LSP size, the first 10 and last 10 residues are not predicted.
The text file brings the same information (except the 3D representation) and two additional columns for the predicted B-factorNorm and RMSFNorm.
Implementation
Implementation of this tool is done in Python and HTML, while the graphical plots are done using R software (44,45). The front-end use is based on html and php. Perl/cgi programs control the input while python and other programs carry out the processing behind the database search and pairwise comparisons.
Figure 2 illustrates the results of the prediction of isomerase in a region ranging from residue 100 to 150. As the CI is higher than 15, the regions from (a) to (d) are very well predicted, while the region (e) is not reliable with a very low CI (=3). So, the user can be quite sure of a succession from flexible (a) to rigid (b) with an intermediate to flexible zone (c) and then come back to rigid zone (d). By looking at the distribution of predicted LSPs, the user can analyze more deeply what could be the local conformations adopted by this region, i.e. a succession of short helical regions alternated by short loops going to an extended conformation.
Figure 2.

Protein prediction example. The prediction highlights the regions from residue 100 to 150. In regions (a) to (d) are located residues predicted with a high accuracy (confidence index of 15 or better): it represents flexible (a) to rigid (b) with an intermediate to a flexible zone (c) and then coming back to rigid zone (d). Following region (e) is predicted as flexible, but the low confidence index (=3) makes the prediction not reliable.
CONCLUSION
Very few web servers are dedicated to the prediction of flexibility from the sole information of the sequence. We propose an original tool that combines in one run the prediction of the local structures and the associated flexibility. We also chose to predict flexibility in three classes compared with two in most studies. We also provide B-factor and RMSF prediction. In addition, very useful and important information is provided by the CI. This value allows the user to assess the predictability of its sequence or region of interest. We hope that the availability of our method through PredyFlexy web server will help researchers to better understand the properties of their protein and design new experiments focusing on appropriate regions depending on their goal.
FUNDING
Ministry of Research (France); University of Paris Diderot; Sorbonne Paris Cité; National Institute for Blood Transfusion (INTS, France); Institute for Health and Medical Research (INSERM, France) (to A.G.d.B., C.E. and J.C.G.); French Ministry of Research (to A.B. and P.C.). Funding for open access charge: INSERM.
Conflict of interest statement. None declared.
REFERENCES
- 1.Beckett D. Regulating transcription regulators via allostery and flexibility. Proc. Natl Acad. Sci. USA. 2009;106:22035–22036. doi: 10.1073/pnas.0912300107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammes GG, Benkovic SJ, Hammes-Schiffer S. Flexibility, diversity, and cooperativity: pillars of enzyme catalysis. Biochemistry. 2011;50:10422–10430. doi: 10.1021/bi201486f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lill MA. Efficient incorporation of protein flexibility and dynamics into molecular docking simulations. Biochemistry. 2011;50:6157–6169. doi: 10.1021/bi2004558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin JH. Accommodating protein flexibility for structure-based drug design. Curr. Top. Med. Chem. 2011;11:171–178. doi: 10.2174/156802611794863580. [DOI] [PubMed] [Google Scholar]
- 5.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 6.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Camps J, Carrillo O, Emperador A, Orellana L, Hospital A, Rueda M, Cicin-Sain D, D’Abramo M, Gelpi JL, Orozco M. FlexServ: an integrated tool for the analysis of protein flexibility. Bioinformatics. 2009;25:1709–1710. doi: 10.1093/bioinformatics/btp304. [DOI] [PubMed] [Google Scholar]
- 10.Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindahl E, Azuara C, Koehl P, Delarue M. NOMAD-Ref: visualization, deformation and refinement of macromolecular structures based on all-atom normal mode analysis. Nucleic Acids Res. 2006;34:W52–W56. doi: 10.1093/nar/gkl082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keating KS, Flores SC, Gerstein MB, Kuhn LA. StoneHinge: hinge prediction by network analysis of individual protein structures. Protein Sci. 2009;18:359–371. doi: 10.1002/pro.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emekli U, Schneidman-Duhovny D, Wolfson HJ, Nussinov R, Haliloglu T. HingeProt: automated prediction of hinges in protein structures. Proteins. 2008;70:1219–1227. doi: 10.1002/prot.21613. [DOI] [PubMed] [Google Scholar]
- 14.Seeliger D, De Groot BL. tCONCOORD-GUI: visually supported conformational sampling of bioactive molecules. J. Comput. Chem. 2009;30:1160–1166. doi: 10.1002/jcc.21127. [DOI] [PubMed] [Google Scholar]
- 15.Karplus P, Schulz G. Prediction of chain flexibility in proteins. A tool for the selection of peptide antigens. Naturwissenschaften. 72:212–213. [Google Scholar]
- 16.Vihinen M, Torkkila E, Riikonen P. Accuracy of protein flexibility predictions. Proteins. 1994;19:141–149. doi: 10.1002/prot.340190207. [DOI] [PubMed] [Google Scholar]
- 17.Schlessinger A, Yachdav G, Rost B. PROFbval: predict flexible and rigid residues in proteins. Bioinformatics. 2006;22:891–893. doi: 10.1093/bioinformatics/btl032. [DOI] [PubMed] [Google Scholar]
- 18.Pan XY, Shen HB. Robust prediction of B-factor profile from sequence using two-stage SVR based on random forest feature selection. Protein Pept. Lett. 2009;16:1447–1454. doi: 10.2174/092986609789839250. [DOI] [PubMed] [Google Scholar]
- 19.Chen K, Kurgan LA, Ruan J. Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs. BMC Struct. Biol. 2007;7:25. doi: 10.1186/1472-6807-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang T, Faraggi E, Zhou Y. Fluctuations of backbone torsion angles obtained from NMR-determined structures and their prediction. Proteins. 2010;78:3353–3362. doi: 10.1002/prot.22842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trott O, Siggers K, Rost B, Palmer AG., III Protein conformational flexibility prediction using machine learning. J. Magn. Reson. 2008;192:37–47. doi: 10.1016/j.jmr.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galzitskaya OV, Garbuzynskiy SO, Lobanov MY. FoldUnfold: web server for the prediction of disordered regions in protein chain. Bioinformatics. 2006;22:2948–2949. doi: 10.1093/bioinformatics/btl504. [DOI] [PubMed] [Google Scholar]
- 23.Mamonova TB, Glyakina AV, Kurnikova MG, Galzitskaya OV. Flexibility and mobility in mesophilic and thermophilic homologous proteins from molecular dynamics and FoldUnfold method. J. Bioinform. Comput. Biol. 2010;8:377–394. doi: 10.1142/s0219720010004690. [DOI] [PubMed] [Google Scholar]
- 24.Hirose S, Yokota K, Kuroda Y, Wako H, Endo S, Kanai S, Noguchi T. Prediction of protein motions from amino acid sequence and its application to protein-protein interaction. BMC Struct. Biol. 2010;10:20. doi: 10.1186/1472-6807-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang H, Vreven T, Whitfield TW, Wiehe K, Weng Z. A machine learning approach for the prediction of protein surface loop flexibility. Proteins. 2011;79:2467–2474. doi: 10.1002/prot.23070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuznetsov IB. Ordered conformational change in the protein backbone: prediction of conformationally variable positions from sequence and low-resolution structural data. Proteins. 2008;72:74–87. doi: 10.1002/prot.21899. [DOI] [PubMed] [Google Scholar]
- 27.Kuznetsov IB, McDuffie M. FlexPred: a web-server for predicting residue positions involved in conformational switches in proteins. Bioinformation. 2008;3:134–136. doi: 10.6026/97320630003134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benros C, de Brevern AG, Etchebest C, Hazout S. Assessing a novel approach for predicting local 3D protein structures from sequence. Proteins. 2006;62:865–880. doi: 10.1002/prot.20815. [DOI] [PubMed] [Google Scholar]
- 29.Benros C, de Brevern AG, Hazout S. Analyzing the sequence-structure relationship of a library of local structural prototypes. J. Theor. Biol. 2009;256:215–226. doi: 10.1016/j.jtbi.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 30.de Brevern AG, Hazout S. ‘Hybrid protein model’ for optimally defining 3D protein structure fragments. Bioinformatics. 2003;19:345–353. doi: 10.1093/bioinformatics/btf859. [DOI] [PubMed] [Google Scholar]
- 31.Kohonen T. Self-organized formation of topologically correct feature maps. Biol. Cybern. 1982;43:59–69. [Google Scholar]
- 32.Kohonen T. Self-Organizing Maps. 3rd edn. Berlin: Springer; 2001. [Google Scholar]
- 33.Offmann B, Tyagi M, de Brevern AG. Local protein structures. Curr. Bioinform. 2007;3:165–202. [Google Scholar]
- 34.Joseph AP, Agarwal G, Mahajan S, Gelly JC, Swapna LS, Offmann B, Cadet F, Bornot A, Tyagi M, Valadie H, et al. A short survey on protein blocks. Biophys. Rev. 2010;2:137–147. doi: 10.1007/s12551-010-0036-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Brevern AG, Etchebest C, Hazout S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins. 2000;41:271–287. doi: 10.1002/1097-0134(20001115)41:3<271::aid-prot10>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 36.Bornot A, Etchebest C, de Brevern AG. A new prediction strategy for long local protein structures using an original description. Proteins. 2009;76:570–587. doi: 10.1002/prot.22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noguchi T, Akiyama Y. PDB-REPRDB: a database of representative protein chains from the Protein Data Bank (PDB) in 2003. Nucleic Acids Res. 2003;31:492–493. doi: 10.1093/nar/gkg022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noguchi T, Matsuda H, Akiyama Y. PDB-REPRDB: a database of representative protein chains from the Protein Data Bank (PDB) Nucleic Acids Res. 2001;29:219–220. doi: 10.1093/nar/29.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bornot A, Etchebest C, de Brevern AG. Predicting protein flexibility through the prediction of local structures. Proteins. 2011;79:839–852. doi: 10.1002/prot.22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith DK, Radivojac P, Obradovic Z, Dunker AK, Zhu G. Improved amino acid flexibility parameters. Protein Sci. 2003;12:1060–1072. doi: 10.1110/ps.0236203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Obradovic Z, Peng K, Vucetic S, Radivojac P, Dunker AK. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005;61(Suppl. 7):176–182. doi: 10.1002/prot.20735. [DOI] [PubMed] [Google Scholar]
- 42.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.UniProt Consortium. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–D219. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 1996;5:299–314. [Google Scholar]
- 45.R Development Core Team. Computing, R. F. f. S. 2011. R: a language and environment for statistical computing. Vienna, Austria. [Google Scholar]