


Abstract
Three-dimensional protein structures provide invaluable information for understanding and regulating biological functions of proteins. The GalaxyWEB server predicts protein structure from sequence by template-based modeling and refines loop or terminus regions by ab initio modeling. This web server is based on the method tested in CASP9 (9th Critical Assessment of techniques for protein Structure Prediction) as ‘Seok-server’, which was assessed to be among top performing template-based modeling servers. The method generates reliable core structures from multiple templates and re-builds unreliable loops or termini by using an optimization-based refinement method. In addition to structure prediction, a user can also submit a refinement only job by providing a starting model structure and locations of loops or termini to refine. The web server can be freely accessed at http://galaxy.seoklab.org/.
INTRODUCTION
Three-dimensional protein structures provide essential information for atomic-level understanding of molecular functions designed by the nature and also for human design of new ligands regulating the protein functions. Computational methods for protein structure prediction have become complementary to experimental methods when close homologs of known experimental structures are available. With the ever-increasing sizes of both sequence and structure databases, the role of the structure prediction methods based on known structures of homologs (called template-based modeling, homology modeling or comparative modeling) is also increasing (1,2).
Traditionally, large emphasis has been placed on homolog detection and sequence alignment as essential elements of template-based modeling. More recently, obtaining model structures beyond the best available templates or improving models starting from the best available model structures have been discussed to be necessary for further advancement in the field (3–5). However, such improvement has proven to be very difficult, e.g. as demonstrated in the refinement category of recent CASP experiments. In the most recent CASP (CASP9), only three groups including us could achieve improvement in backbone structure quality, and the best improvement was only 0.37% (our own result) (5).
In this article, we introduce a new web server that provides two functions: protein structure prediction from sequence and refinement from user-provided model. The method is based on the ‘Seok-server’ tested in CASP9 and evaluated to be among top six servers (6). A lighter version of the original method with comparable performance is employed to provide more efficient service. In detail, lighter sampling is carried out both in the model-building and the refinement steps to reduce computation time. The template-based modeling method extensively uses multiple template information to construct reliable core regions and then refines up to three loops or termini detected to be unreliable. Two existing methods, HHsearch (7) and PROMALS3D (8), are used for template selection and sequence alignment, respectively. They are applied in such a way that reliable core structures are built by selecting templates of similar core structures and aligning core sequences. The remaining less conserved, unreliable regions are treated in the subsequent refinement stage. Better prediction of less conserved regions by an ab initio refinement method like the one introduced here would be invaluable for further functional or design studies because they often contribute to the specific functions of related proteins (9–11).
GALAXYWEB METHOD
A flowchart of the GalaxyWEB structure prediction (GalaxyTBM) and refinement (GalaxyREFINE) procedure is shown in Figure 1. First, candidates for templates are selected by rescoring HHsearch (7) results placing more weights on the secondary structure score for more difficult targets. The re-ranking score is a weighted sum of the Z-score of the HHsearch sequence score, Zseq, and that of the HHsearch secondary structure score, Zss,
where the weight w depends on the target difficulty estimated by the probability for the HHsearch top ranker, P, as
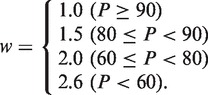 |
Figure 1.

Flowchart of the GalaxyWEB protein structure prediction pipeline which consists of protein structure prediction by GalaxyTBM and refinement by GalaxyREFINE.
Among the re-ranked top 20 homologs, multiple templates are selected by removing structural outliers based on mutual TM scores (12) for the aligned core regions. Average number of selected templates is 4.55 for the 68 single-domain CASP9 targets used as a test set. Multiple sequence alignment using PROMALS3D (8) is then performed for core regions deleting unaligned termini. Terminus sequence alignments are attached afterwards. Initial model structures are then built from the templates and the sequence alignment by a CSA (conformational space annealing) global optimization (13) of the restraints derived from templates by an in-house method (L. Heo, H. Park and C. Seok, unpublished data). The restraints are sum of approximately single-well potentials, similar to that developed by Thompson et al. (14). The range of restraint application between Cα pairs (up to 15 Å) is wider than Thompson et al. and similar to that in MODELLER (15). (In CASP9, more complex MODELLER restraints requiring more extensive sampling were used.) Unreliable local regions (ULRs) are then detected (16) from the initial model and a maximum of three ULRs are reconstructed ‘simultaneously’ by a CSA optimization of hybrid energy that consists of physics-based terms and knowledge-based terms (16,17). (In CASP9, ‘all’ ULRs were re-modeled individually, requiring more computation time than running a single optimization job.) During CSA optimization, the triaxial loop closure algorithm (18) is extensively used to generate geometrically proper backbone structures for loops (19). More details on the method and the effects of the strategy taken at each stage on the overall performance will be presented in a separate article (submitted). The modifications from the original Seok-server was made to provide the web service more efficiently, as the original method requires 2–3 times more computation power.
Performance of the method
Since the current web server employs a method lighter than the original Seok-server method tested in CASP9 both in the initial model building and refinement stages, the performance of the method was tested again on the 68 single-domain targets of CASP9. The backbone structure quality measured by average GDT-TS (20,21) is 68.5 by Seok-server and 67.6 by GalaxyWEB. The decreased performance of GalaxyWEB compared to the original Seok-server comes from the lighter optimization during model building and refinement. However, the result is still comparable to those of the top six server methods in CASP9. Initial model structures are improved in 65% of the cases in which refinement was performed when the local structure quality is measured by RMSD. The performance of the refinement method is more fully discussed in another article (17).
GALAXYWEB SERVER
Hardware and software
The GalaxyWEB server runs on a cluster of four Linux servers of 2.33 GHz Intel Xeon processors that consist of eight cores. The web application uses Python and the MySQL database. The structure prediction and refinement pipeline is implemented using Python by combining the two programs developed by other groups, HHsearch (7) and PROMALS3D (8), and our own program package for molecular modeling named GALAXY (16,17,19), which is written in Fortran 90. The JMol (http://www.jmol.org) is used for visualization of predicted structures.
Input and output
For structure prediction, a protein sequence must be provided in the FASTA format. For refinement only run, a user is required to provide a model structure to refine in the PDB format and to specify the residue number range for each region to refine. Expected run time for a structure prediction job is 7 h for a 500-residue protein and that for a refinement job is 2 h for a 26-residue loop or terminus. Five best models can be viewed and downloaded on the website, as shown in Figure 2. Full sets of models generated by the server can also be downloaded as a tar file.
Figure 2.

GalaxyWEB output page (A). Five top-ranking models are shown in static images (B). They can also be viewed using the Jmol structure viewer. The residue ranges of the refined ULRs are summarized in the table (C) and also indicated in the secondary structure figure (D) in which secondary structure of the first model is compared with the prediction obtained from sequence using PSIPRED.
CONCLUSIONS
GalaxyWEB is a web server for protein structure prediction and refinement. A distinct feature of the server from other protein structure servers is that unreliable regions for which template information is not available or inconsistent are detected and refined by an ab initio method. Model structures obtained by other methods may also be refined by specifying the regions to refine. The ab initio loop and terminus modeling method is one of few refinement methods that can actually improve on the starting models, as demonstrated in CASP9.
FUNDING
National Research Foundation of Korea funded by the Ministry of Education, Science and Technology [2011-0012456]; Center for Marine Natural Products and Drug Discovery (CMDD), one of the MarineBio21 programs funded by the Ministry of Land, Transport and Maritime Affairs of Korea. Funding for open access charge: Seoul National University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhang Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008;18:342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 3.Keedy DA, Williams CJ, Headd JJ, Arendall WB, III, Chen VB, Kapral GJ, Gillespie RA, Block JN, Zemla A, Richardson DC, et al. The other 90% of the protein: assessment beyond the Calphas for CASP8 template-based and high-accuracy models. Proteins. 2009;77(Suppl. 9):29–49. doi: 10.1002/prot.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kopp J, Bordoli L, Battey JN, Kiefer F, Schwede T. Assessment of CASP7 predictions for template-based modeling targets. Proteins. 2007;69(Suppl. 8):38–56. doi: 10.1002/prot.21753. [DOI] [PubMed] [Google Scholar]
- 5.MacCallum JL, Perez A, Schnieders MJ, Hua L, Jacobson MP, Dill KA. Assessment of protein structure refinement in CASP9. Proteins. 2011;79(Suppl. 10):74–90. doi: 10.1002/prot.23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mariani V, Kiefer F, Schmidt T, Haas J, Schwede T. Assessment of template based protein structure predictions in CASP9. Proteins. 2011;79(Suppl. 10):37–58. doi: 10.1002/prot.23177. [DOI] [PubMed] [Google Scholar]
- 7.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21:951–960. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 8.Pei J, Kim BH, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. Molecular Biology of the Cell. New York: Garland Publishing Inc.; 1994. [Google Scholar]
- 10.Shi L, Javitch JA. The second extracellular loop of the dopamine D2 receptor lines the binding-site crevice. Proc. Natl Acad. Sci. USA. 2004;101:440–445. doi: 10.1073/pnas.2237265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aparicio R, Ferreira ST, Polikarpov I. Closed conformation of the active site loop of rabbit muscle triosephosphate isomerase in the absence of substrate: evidence of conformational heterogeneity. J. Mol. Biol. 2003;334:1023–1041. doi: 10.1016/j.jmb.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joo K, Lee J, Seo JH, Lee K, Kim BG. All-atom chain-building by optimizing MODELLER energy function using conformational space annealing. Proteins. 2009;75:1010–1023. doi: 10.1002/prot.22312. [DOI] [PubMed] [Google Scholar]
- 14.Thompson J, Baker D. Incorporation of evolutionary information into Rosetta comparative modeling. Proteins. 2011;79:2380–2388. doi: 10.1002/prot.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 16.Park H, Ko J, Joo K, Lee J, Seok C. Refinement of protein termini in template-based modeling using conformational space annealing. Proteins. 2011;79:2725–2734. doi: 10.1002/prot.23101. [DOI] [PubMed] [Google Scholar]
- 17.Park H, Seok C. Refinement of unreliable local regions in template-based protein models. Proteins. 2012 doi: 10.1002/prot.24086. April 10 (doi: 10.1002/prot.24086; epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 18.Coutsias EA, Seok C, Jacobson MP, Dill KA. A kinematic view of loop closure. J. Comput. Chem. 2004;25:510–528. doi: 10.1002/jcc.10416. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Lee D, Park H, Coutsias EA, Seok C. Protein loop modeling by using fragment assembly and analytical loop closure. Proteins. 2010;78:3428–3436. doi: 10.1002/prot.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zemla A. LGA: a method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zemla A, Venclovas C, Moult J, Fidelis K. Processing and analysis of CASP3 protein structure predictions. Proteins. 1999;(Suppl. 3):22–29. doi: 10.1002/(sici)1097-0134(1999)37:3+<22::aid-prot5>3.3.co;2-n. [DOI] [PubMed] [Google Scholar]