


Abstract
High-throughput technologies including transcriptional profiling, proteomics and reverse genetics screens provide detailed molecular descriptions of cellular responses to perturbations. However, it is difficult to integrate these diverse data to reconstruct biologically meaningful signaling networks. Previously, we have established a framework for integrating transcriptional, proteomic and interactome data by searching for the solution to the prize-collecting Steiner tree problem. Here, we present a web server, SteinerNet, to make this method available in a user-friendly format for a broad range of users with data from any species. At a minimum, a user only needs to provide a set of experimentally detected proteins and/or genes and the server will search for connections among these data from the provided interactomes for yeast, human, mouse, Drosophila melanogaster and Caenorhabditis elegans. More advanced users can upload their own interactome data as well. The server provides interactive visualization of the resulting optimal network and downloadable files detailing the analysis and results. We believe that SteinerNet will be useful for researchers who would like to integrate their high-throughput data for a specific condition or cellular response and to find biologically meaningful pathways. SteinerNet is accessible at http://fraenkel.mit.edu/steinernet.
INTRODUCTION
Monitoring the changes in gene expression, protein levels and post-translational modifications of proteins is crucial to better understand signaling pathways, the mechanisms of action of drugs and the changes that occur during disease. Many approaches are available for the analysis of individual types of data. However, integration and curation of multiple ‘omic’ data sources are more challenging (1). Typically, there is little overlap among the proteins and genes identified by different methods (2), and the overlap between experimental findings and known pathways is very low. Previous publications have shown that a more coherent view of the underlying biological processes can be obtained by using a network approach in which the hits from ‘omic’ experiments are mapped onto a network of protein–protein interactions.
Given the high rates of false positives and negatives in these data, the resulting networks are frequently too large and noisy to interpret directly, leading to a number of algorithmic approaches to the problem (3–16). Flow optimization has been used to reconstruct pathways by inferring genetic hits and transcriptional data (16), and this method is available as a web server (9). In another approach, transcriptional data obtained from gene knockout experiments are integrated with interactome and causal paths are identified by linear programming (11). Bayesian networks have been used to integrate siRNA data in insulin signaling (13) and copy number and gene expression data for finding drivers in diseases (3). A maximum-likelihood-based approach has been applied to reveal causal paths in transcriptional regulation by integrating gene knockout experiments (15). Other approaches include network inference from gene expression (6,17), electric circuits (8,10,12) and network propagation (14).
In our previous work, we have shown that one successful approach to this problem is constrained optimization to identify a subset of the ‘omic’ hits that are connected directly or indirectly by high probability interactions (7). This was achieved by searching for the solution to the prize-collecting Steiner tree (PCST) problem (4,5,7). In this approach, the detected proteins/genes in experiments are defined as ‘terminal nodes’ and we seek to connect them to each other either directly or through other undetected proteins (Steiner nodes) using protein–protein and protein–gene interactions. A critical feature of the algorithm is that we do not require it to connect all the terminal nodes. Rather, we seek a network composed of high-confidence edges that ultimately link a subset of the termini. To identify this network, we assign costs to each interaction reflecting our confidence that the interaction is real. In addition, we assign penalties to the terminal nodes based on our confidence in the proteomic or transcriptional data. The PCST algorithm identifies a relevant subnetwork by simultaneously minimizing the cost of edges included in the tree and the penalties of terminals that are excluded.
The PCST approach has been evaluated on the phosphoproteomic and transcriptional data representing the well-characterized yeast pheromone response. The benefits of this approach are that (i) it is robust to noise in experiments, (ii) it integrates both transcriptional and proteomics data in a single pipeline, (iii) the resulting optimum tree contains functionally correlated components and (iv) it contains relevant signaling proteins and transcription factors that were undetected in experiments.
Here, we present the web application of this approach in SteinerNet to make this method (7) available in a user-friendly way. SteinerNet takes a set of proteins/genes for processing and returns an optimum and compact network from these data. It contains a visualization panel that provides the user a basic display of the optimum Steiner tree and a download panel that provides links to download all the analyses and results. We believe that SteinerNet will help researchers in integrating their high-throughput data to reveal hidden components in their specific system and to obtain biologically meaningful compact networks.
SteinerNet WEB SERVER
Inputs
SteinerNet is the user-friendly way to run the framework published in our previous work (7). Users working with yeast, human, mouse, Caenorhabditis elegans or Drosophila melanogaster data need only provide a terminal set consisting of proteins and/or genes of interest with associated penalties for each node and the web server will identify a network containing a subset of these nodes connected by high-confidence interactions. Terminal nodes can be obtained from experimentally detected proteins/genes. Each terminal should have a ‘penalty’ associated with it. The higher the penalty, the more the algorithm will attempt to keep the terminal in the final tree. If there is no reason to prefer one node to another, all the penalties can be set to a uniform value. The networks discovered by SteinerNet have a structure known as a rooted tree, and the algorithm will attempt to connect all the termini to one node that is designated as the root. The user has the option of specifying a root node together with the terminal set. If the user does not specify, the algorithm selects the node having highest penalty value as the root.
SteinerNet provides interactome databases for five species. The yeast interactome, which is the one used in our prior publication (7), is available in ORF names or in gene symbols. The human, mouse, C. elegans and D. melanogaster interactomes are available in Ensembl IDs or gene symbols (retrieved from STRING Database v8.3 (18) where experimental and database options are combined to calculate interaction probabilities). An interaction probability is assigned to each edge. This probability represents the reliability of a given interaction. For the yeast interactome, the evidence for interactions is collected from public databases and probabilities are obtained by applying Bayes rule to compute the individual probability for each edge in the interactome. For other species, we used the scoring scheme of the STRING database by considering only experiment and database evidence channels [for more details, see (7,18)]. More advanced users and those working in other species can upload their own interactomes. The identifiers of the nodes in the interactome and in the terminal list must be consistent. User can convert identifiers to a standard format using external servers such as DAVID or HUGO. Instructions about the input format of the terminal set are detailed on the tutorial pages.
In our formulation of the PCST problem, we use gene expression changes as evidence of upstream changes in signaling pathways. Therefore, all gene expression changes are mapped to nodes representing mRNAs, which are only connected to transcriptional regulatory proteins (Figure 1). If the user has transcriptional data, transcription factor to DNA interactions are also required to connect mRNAs to the interactome. Transcription factor to DNA interactions are provided only for yeast and can be uploaded for other species.
Figure 1.

The concept figure and the flowchart of SteinerNet. Once a job is submitted through SteinerNet, it is added to the queue and waits to be processed. During processing a job, three consecutive steps are performed in the background. First, input data (interactome and terminal set) are integrated. Then, an optimum solution tree is identified for the given dataset using constrained optimization. At the final step, outputs are post-processed to provide easy visualization and links for downloading the results.
The algorithm has only one parameter which is called β. This parameter controls the trade-off between the cost of excluding termini from the solution and the cost of including edges. Separate β values can be used for the protein termini and the mRNA termini. Once all the data have been entered, the user will be directed to a unique URL where the output will be posted when it is ready. In addition, the user may provide an e-mail address to receive a notification when the results are ready.
To illustrate the use of SteinerNet, we applied it to phosphoproteomic data from the EGF-stimulated ERBB pathway. The data were retrieved from the work by Naegle et al. (19) where 77 phosphopeptides were identified at four different time points. When we mapped these phosphopeptides to their corresponding proteins, we obtained 57 proteins. In this case study, we assigned uniform penalties to the nodes in the terminal set to illustrate the web server in a simple way and we used the default human interactome in the web server. The root node was not specified and β was set to 4.
Outputs
The output page is composed of two panels. The left panel is designed for visualization and the right panel is designed for data download. The left panel displays the optimum Steiner tree using the Cytoscape Web plug-in (20). This visualization is provided to give users a quick look before they download output files. Selecting the ‘Go to the augmented network’ tab gives the visualization of the nodes from the PCST with all the edges among these nodes that are present in the interactome, regardless of whether they were included in the tree. At the bottom of the right panel, the interactive pie chart illustrates statistics of the included terminals. On the right panel, resulting networks (optimum PCST and the augmented network) can be downloaded as Cytoscape-compatible SIF files. In addition, users may download several properties of nodes and edges included in the solution, such as node betweenness in the solution and in the augmented network, node degree, edge betweenness in the solution and in augmented network and results from edge-betweenness clustering.
The details of the optimization stages may be downloaded. These contain the value of the objective function during each iteration step. The results of the jobs will be kept for 15 days on the web server and then removed.
Figure 2 shows the output when SteinerNet was run on the phosphoproteins from the ERBB pathway identified by Naegle et al. (19) as described above. In the optimum tree of this example, 35 of 57 terminals are included. These 35 proteins are connected via 18 Steiner nodes. Although these 18 proteins are not detected in experimental data, these proteins are enriched especially in signal transduction, phosphorylation and receptor protein tyrosine kinase signaling pathway. The output page of this example and other examples can be browsed interactively using the tab ‘Sample Output’ on the main page of SteinerNet.
Figure 2.

A snapshot from the SteinerNet output page. The left panel provides the visualization of the optimum Steiner tree. The ‘Go to the Augmented Network’ tab shows the same set of nodes with all the edges between them that are present in the interactome. The right panel is designed as a download page. Here, a user can download all input and output data, analyses and results. At the bottom, an interactive pie chart is provided to check the number of terminals included in the optimum tree.
Due to the fact that the underlying PCST problem is NP-hard, running the web server can take some time, depending on both the interactome size and terminal set size. In our work, this optimization step takes 237 s on the yeast data set, containing 5957 nodes, 34 712 edges and 224 protein/gene terminal nodes. In addition, the input human interactome in Figure 2 contains 4241 nodes and 12 980 edges, and the terminal set contains 57 proteins. The optimization step takes 6 s in this case. Depending on the size of the input sets, running SteinerNet takes from a few minutes up to a few hours. For more information on run-time, see Ljubic et al. (21) who present a very rigorous analysis on non-biological benchmark sets of the external tool (dhea-code) we use to solve the PCST problem.
Implementation
SteinerNet uses CGI scripting with Perl for the web interface and job submission and Python for performing data integration and post-processing in the background. It then calls the dhea-code (21) as an external tool to perform the branch-and-cut algorithm for solving the PCST problem. SteinerNet website is free and open to all non-commercial users and there is no login requirement. SteinerNet is accessible at http://fraenkel.mit.edu/steinernet.
OVERVIEW OF THE STEINERNET METHODOLOGY
We used the Goemans–Williamson formulation of the PCST problem. In a given undirected graph, G(V, E), penalties (p(v) ≥ 0) are assigned to nodes (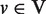 ) and costs (c(e) ≥ 0) are assigned to the edges (
) and costs (c(e) ≥ 0) are assigned to the edges (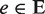 ). Nodes having non-zero penalty values are called ‘terminal nodes’. Intermediate nodes included in the tree to connect terminals are called ‘Steiner nodes’. By minimizing the sum of the total cost of all edges in the tree and the total penalties of all nodes not contained in the tree, we are able to obtain compact networks. The objective function to be minimized is
). Nodes having non-zero penalty values are called ‘terminal nodes’. Intermediate nodes included in the tree to connect terminals are called ‘Steiner nodes’. By minimizing the sum of the total cost of all edges in the tree and the total penalties of all nodes not contained in the tree, we are able to obtain compact networks. The objective function to be minimized is
| (1) |
where VT is the set of vertices and ET is the set of edges in the solution tree. As defined in Eq. (1), the algorithm pays cost, c(e), for included edges (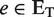 ) and pays penalty p(v) for excluded terminal nodes (
) and pays penalty p(v) for excluded terminal nodes (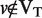 ). The branch-and-cut algorithm, implemented in dhea-code (21), is used to find the exact solution to the PCST problem. The default parameter set of dhea-code is used in calculations. Analysis of suboptimal solutions has illustrated that the branch-and-cut algorithm is robust to the noise in the interactome (7).
). The branch-and-cut algorithm, implemented in dhea-code (21), is used to find the exact solution to the PCST problem. The default parameter set of dhea-code is used in calculations. Analysis of suboptimal solutions has illustrated that the branch-and-cut algorithm is robust to the noise in the interactome (7).
In SteinerNet, the undirected graph, G, is composed of edges representing physical protein–protein and transcription factor-to-gene interactions and nodes representing either proteins or genes. The edge costs are defined as negative logarithm of the interaction probability. In this way, interactions with higher probability have lower cost values. Terminal nodes are defined as the set of proteins/genes having proteomics or transcriptional data. The size of the resulting tree is controlled by the parameter beta (β) which changes the amplitude of the penalty to exclude a terminal node. Larger values of β lead to including more terminal nodes. This parameter is stable for a wide range of values (7).
CONCLUSION
SteinerNet provides a convenient tool for using a powerful constrained optimization method to reconstruct signaling and response pathways by integrating multiple ‘omic’ data. The SteinerNet method seeks a network composed of high-confidence interactions that ultimately link a subset of the omic hits either directly or through intermediate proteins. This is achieved by solving the PCST problem. Previously, we have shown that the solution of the PCST problem in yeast pheromone response data reveals functionally coherent pathways and hidden components that are not detected in experiments. The SteinerNet web server makes this approach available to a larger community for reconstructing response pathways for data from any species. The user-friendly interface of SteinerNet and its built-in visualization feature provide an extensive analysis of the results, identifying functionally relevant proteins. We believe that SteinerNet will serve a diverse range of researchers who would like to integrate multiple ‘omic’ data sources to reconstruct biologically meaningful pathways.
FUNDING
National Institutes of Health [U54-CA112967 and R01-GM089903]; National Science Foundation [Award No. DB1-0821391]. Funding for open access charge: NIH [U54-CA112967].
Conflict of interest statement. None declared.
REFERENCES
- 1.Palsson B, Zengler K. The challenges of integrating multi-omic data sets. Nat. Chem. Biol. 2010;6:787–789. doi: 10.1038/nchembio.462. [DOI] [PubMed] [Google Scholar]
- 2.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 3.Akavia UD, Litvin O, Kim J, Sanchez-Garcia F, Kotliar D, Causton HC, Pochanard P, Mozes E, Garraway LA, Pe'er D. An integrated approach to uncover drivers of cancer. Cell. 2010;143:1005–1017. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailly-Bechet M, Borgs C, Braunstein A, Chayes J, Dagkessamanskaia A, Francois JM, Zecchina R. Finding undetected protein associations in cell signaling by belief propagation. Proc. Natl Acad. Sci. USA. 2010;108:882–887. doi: 10.1073/pnas.1004751108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dittrich MT, Klau GW, Rosenwald A, Dandekar T, Muller T. Identifying functional modules in protein-protein interaction networks: an integrated exact approach. Bioinformatics. 2008;24:i223–i231. doi: 10.1093/bioinformatics/btn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- 7.Huang SS, Fraenkel E. Integrating proteomic, transcriptional, and interactome data reveals hidden components of signaling and regulatory networks. Sci. Signal. 2009;2:ra40. doi: 10.1126/scisignal.2000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YA, Wuchty S, Przytycka TM. Identifying causal genes and dysregulated pathways in complex diseases. PLoS Comput. Biol. 2011;7:e1001095. doi: 10.1371/journal.pcbi.1001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lan A, Smoly IY, Rapaport G, Lindquist S, Fraenkel E, Yeger-Lotem E. ResponseNet: revealing signaling and regulatory networks linking genetic and transcriptomic screening data. Nucleic Acids Res. 2011;39:W424–W429. doi: 10.1093/nar/gkr359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Missiuro PV, Liu K, Zou L, Ross BC, Zhao G, Liu JS, Ge H. Information flow analysis of interactome networks. PLoS Comput. Biol. 2009;5:e1000350. doi: 10.1371/journal.pcbi.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ourfali O, Shlomi T, Ideker T, Ruppin E, Sharan R. SPINE: a framework for signaling-regulatory pathway inference from cause-effect experiments. Bioinformatics. 2007;23:i359–i366. doi: 10.1093/bioinformatics/btm170. [DOI] [PubMed] [Google Scholar]
- 12.Suthram S, Beyer A, Karp RM, Eldar Y, Ideker T. eQED: an efficient method for interpreting eQTL associations using protein networks. Mol. Syst. Biol. 2008;4:162. doi: 10.1038/msb.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tu Z, Argmann C, Wong KK, Mitnaul LJ, Edwards S, Sach IC, Zhu J, Schadt EE. Integrating siRNA and protein-protein interaction data to identify an expanded insulin signaling network. Genome Res. 2009;19:1057–1067. doi: 10.1101/gr.087890.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput. Biol. 2010;6:e1000641. doi: 10.1371/journal.pcbi.1000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeang CH, Ideker T, Jaakkola T. Physical network models. J. Comput. Biol. 2004;11:243–262. doi: 10.1089/1066527041410382. [DOI] [PubMed] [Google Scholar]
- 16.Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, et al. Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity. Nat. Genet. 2009;41:316–323. doi: 10.1038/ng.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bailly-Bechet M, Bradde S, Braunstein A, Flaxman A, Foini F, Zecchina R. Clustering with shallow trees. J. Stat. Mech. 2009 P12010. [Google Scholar]
- 18.Jensen LJ, Kuhn M, Stark M, Chaffron S, Creevey C, Muller J, Doerks T, Julien P, Roth A, Simonovic M, et al. STRING 8—a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37:D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naegle KM, Welsch RE, Yaffe MB, White FM, Lauffenburger DA. MCAM: multiple clustering analysis methodology for deriving hypotheses and insights from high-throughput proteomic datasets. PLoS Comput. Biol. 2011;7:e1002119. doi: 10.1371/journal.pcbi.1002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ljubic I, Weiskircher R, Pferschy U, Klau GW, Mutzel P, Fischetti M. An algorithmic framework for the exact solution of the Prize-Collecting Steiner Tree Problem. Math. Program. 2006;105:427–449. [Google Scholar]