


Abstract
Small RNAs (smRNAs) in plants, mainly microRNAs and small interfering RNAs, play important roles in both transcriptional and post-transcriptional gene regulation. The broad application of high-throughput sequencing technology has made routinely generation of bulk smRNA sequences in laboratories possible, thus has significantly increased the need for batch analysis tools. PsRobot is a web-based easy-to-use tool dedicated to the identification of smRNAs with stem-loop shaped precursors (such as microRNAs and short hairpin RNAs) and their target genes/transcripts. It performs fast analysis to identify smRNAs with stem-loop shaped precursors among batch input data and predicts their targets using a modified Smith–Waterman algorithm. PsRobot integrates the expression data of smRNAs in major plant smRNA biogenesis gene mutants and smRNA-associated protein complexes to give clues to the smRNA generation and functional processes. Besides improved specificity, the reliability of smRNA target prediction results can also be evaluated by mRNA cleavage (degradome) data. The cross species conservation statuses and the multiplicity of smRNA target sites are also provided. PsRobot is freely accessible at http://omicslab.genetics.ac.cn/psRobot/.
INTRODUCTION
MicroRNAs (miRNAs) and small interfering RNAs (siRNAs) are two major classes of endogenous regulatory small RNAs (smRNAs) in plants. They are usually 21–24 nucleotides (nt) long, and both function by pairing to targets via sequence complementarity (1). miRNAs are usually generated from limited genomic loci and mainly work post-transcriptionally to down-regulate target mRNAs, whereas siRNAs have much broader origins and can function at both transcriptional and post-transcriptional levels (2–6). Both miRNAs and siRNAs are typically identified by cloning and sequencing of small size RNAs (7). The development and application of the high-throughput sequencing technology have significantly advanced the studies on smRNAs, but also imposed increasing numbers of laboratories facing the tasks of data analysis.
Plant miRNAs are usually ∼21 nt long and processed from the pairing stem region of longer precursors with hairpin-shaped secondary structures (8). The presence of stem-loop precursor as the lowest energy folding form has been considered as one of the key criteria for the identification of new miRNAs (9,10). However, as many genomic loci giving rise to siRNAs can also be folded into hairpin-shaped structures (11,12), searching for miRNAs by their expression evidence and the presence of stem-loop precursors may yield many false-positive candidates. According to the community-agreed plant miRNA annotation criteria (9,13), the presence of dominantly expressed candidate sequence and the detection of the pairing sequence (miRNA*) are required for miRNAs (13). These constraints improved the prediction specificity, but the results are still far from ideal.
It has been shown that plant miRNAs mainly pair with their targets via nearly perfect sequence complementarity, very similar to the manner of siRNAs (9,13). This has made the predictions for plant miRNA targets relatively straightforward, yielding limited numbers of targets per miRNA rather than hundreds of ones in animals (9,13). Yet, it still requires large amount of work to identify real miRNA targets from the predicted candidate list. In addition, increasing lines of evidence have shown that gaps are also tolerable in the pairing between plant miRNAs and their targets (14), which will enlarge the candidate target list and produce more difficulties for experimental validation (15).
Most of the available smRNA target prediction software was designed for animal miRNAs and not ideal for plant data because of the common presence of large bulges in animal miRNA and target alignments as well as the different contributions of miRNA seed sequences or central sequences to the stability of miRNA: target pairs of animals and plants. Several recently developed plant miRNA target prediction tools, such as targetFinder (16,17), psRNATarget (18) and CleaveLand (19), have provided great help to researches. However, they are also limited by the requirement of local installation, lack of degradome data supports or dependence on the target prediction results of third-party software (Supplementary Table S1).
PsRobot is designed to partially solve the aforementioned problems in plant miRNA and target prediction. It incorporates the commonly agreed criteria to identify smRNAs with stem-loop shaped precursors from user-uploaded sequences and predict their targets. The multiple user adjustable parameters made the software able to meet different needs from users. To facilitate better classification and functional analysis of input sequences, psRobot integrates the expression information of input sequences in reported plant smRNA binding protein pull-down assay or mutants of major smRNA biogenesis pathway genes. For example, strong association with ARGONAUTE1 (AGO1) protein and expression depletion in dcl1 mutant will strengthen the confidence of a smRNA with stem-loop precursor as an miRNA (1). It also incorporates the available mRNA degradome data for users to evaluate the reliability of miRNA target prediction results. The multiplicity of miRNA binding sites on a single target as well as the cross species conservation status of the target sites are also analyzed and provided. PsRobot can be either used online or downloaded and installed locally. The local version offers a larger capacity for input data size and has the function to incorporate user-uploaded degradome data.
METHODS AND RESULTS
The Stem-loop smRNA Prediction Function
Input, algorithm and parameters
The stem-loop smRNA prediction function takes input smRNA sequences in FASTA or plain text format. For each query smRNA, the software finds its perfectly matched genomic origins, and extracts various lengths of upstream and downstream sequences as precursors, assuming that the smRNA may originate from either the 5′ or 3′ end of precursors, with 10 nt extension at one end of the precursor each time, till reaching the user defined precursor length. The secondary structures of the extracted precursor sequences are then evaluated by the MFOLD program (20). Precursor sequences with stem-loop structure as the minimal free energy folding form and the corresponding query smRNA will be selected and reported in the result pages (Fig. 1a), either in html or text format. If the conservation analysis function of smRNA sequences is selected, the cross species conservation status of smRNAs will be analyzed by aligning the query small sequences to eight selected plant genomes using the BLAST program (allowing up to two mismatches in the smRNA sequences), and the ClustalW (21) alignment of the identified smRNA homologous sequences will be included in the outputs (Fig 1b). The repetitive sequence regions of the 26 preloaded genomes were identified by the RepeatMasker program (http://repeatmasker.org) and stored in the background database. Every genomic locus of the query sequences will be searched against the database to identify repeat sequence originated smRNAs. Parameters for users to adjust include smRNA conservation analysis, the minimal and maximal numbers of mismatched nucleotides within the query smRNA sequences in the obtained precursor structures, the maximal lengths of extracted precursors, and the permission of large loop sequence in the qualified precursors. Although the precursor structures of most canonical plant miRNAs are very short and well paired, there are still some with large bulge or hairpin loops, such as ath-MIR393a and ath-MIR167d (22,23). Enable the “Retain large loop small RNA” function will include precursors with large loops in the prediction results.
Figure 1.

Output of stem-loop smRNA prediction. (A) The genomic mapping and stem-loop precursor prediction of the query smRNAs, detailed information of genomic location, precursor sequence, secondary structure of each predicted stem-loop smRNA loci and their folding energies are included. Sequence in red and capitalized letters is the query smRNA sequence; (B) shows the conservation status of the smRNA in 8 plant species; (C) shows the normalized reads of a given smRNA in smRNA biogenesis mutants and AGO-associated libraries.
Data collection
A preloaded species needs to be selected to define the origins of smRNAs. Up to 26 completed plant genomes are currently supported by psRobot, and future finished genomes will also be incorporated on their release. The Arabidopsis thaliana genome was downloaded from the Arabidopsis Information Resource (TAIR) (24) and the rice genome was from Rice Genome Annotation Project (RGAP) (25) and the Rice Annotation Project Database (RAP-DB) (26) databases. Other plant genomes were downloaded from the Phytozome genome database (27). SmRNA deep sequencing data in smRNA biogenesis mutants and AGO-associated libraries were collected from the NCBI Gene Expression Omnibus database with datasets GSE11094, GSE14695, GSE16959, GSE13605, GSE10036, GSE6682, GSE5343 and GSE6682 for Arabidopsis thaliana (28–35) and datasets GSE20748 and GSE18250 for rice (36,37).
smRNA biogenesis and functional data
PsRobot incorporates published smRNA sequencing data from major plant smRNA biogenesis and function associated protein complexes or gene mutants and returns this information together with the stem-loop prediction results. If a query sequence presents in any of the preloaded database, its sequence reads in each database will be listed (Fig. 1c). As it has been shown that smRNAs of different origins are processed by different Dicer-like (DCL) family proteins and associate with different AGO protein complexes, such information will facilitate users to evaluate the types and functions of the query smRNAs. For example, decreased expression in dcl1 mutant together with enrichment in the AGO1 association libraries provide strong evidence for authentic miRNAs (1), whereas decreased expression in dcl3 mutant together with enrichment in the AGO4 association libraries indicate the smRNAs as siRNAs (38).
Performance
To evaluate the accuracy of stem-loop smRNA prediction function, all miRBase (release v17) (39) recorded Arabidopsis thaliana miRNAs were collected and used as the test dataset. Among the 213 nonredundant sequences, 202 (∼94%) miRNAs were successfully identified as stem-loop smRNAs.
The smRNA Target Prediction Function
Input and parameters
To use the smRNA target prediction function, users can either select the known miRNAs from a plant species or submit their own sequences. The software will search for target sites among the pre-loaded genes/transcripts of the corresponding genome or the user-uploaded target library. The uploaded query smRNA sequences and target library should be either in FASTA or plain text format. The parameters for users to adjust include the following: (1) penalty score for the alignment between smRNAs and targets, which is defined by the formulas below; (2) the boundaries of essential sequence region, within which mismatches or gaps will receive double penalty scores than other regions; (3) the threshold for the total number of gaps within the smRNA and target alignment region; and (4) the region within which gaps are permitted. Degradome sequences mapped within the target sites will be analyzed and presented. Only preloaded degradome data are available for the online version of psRobot, yet users can incorporate their own degradome data via the psRobot_deg program once psRobot is installed locally.
Algorithms
As the pairings between smRNAs and target mRNAs involve global sequence alignment of smRNAs and local sequence alignment of targets, we applied a modified Smith–Waterman algorithm (Formula 1) (40) with the defined scoring system (Formula 2) to calculate the alignment scores between the query smRNAs and targets. The penalty score of each candidate alignment is obtained by subtracting the actual alignment score from the ideal perfect global pairing score (Formula 3). Alignments meet the penalty score cutoff will be backtraced and reported in the result page (Fig. 2). Exhaustive search will be performed on each mRNA to search for the potential presence of multiple target sites (target multiplicity). Parallel computing method is used to accelerate the speed. A standalone local version of this function is also available for download to facilitate analysis on large datasets.
Figure 2.

Result summary table of smRNA target prediction function.
Formula 1:
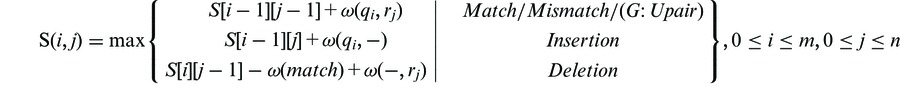 |
Formula 2:
Formula 3:
Output of the smRNA target prediction function
The primary output of the smRNA target prediction function is summarized in a sortable and searchable table (Fig. 2), with the query sequence, target alignment, alignment penalty score, target annotation, multiplicity of target sites and other supporting information. The contents of the expandable links in the result table are summarized in the next sections.
Target site conservation
Homolog gene groups of eight plant species (Arabidopsis thaliana, Brachypodium distachyon, Carica papaya, Oryza sativa, Populus trichocarpa, Sorghum bicolor, Vitis vinifera, Zea mays) are generated using OrthoMCL (41) with the default parameters, and serve as the source data for the conservation analysis of target sites. Predicted targets are searched against this source data for both paralogous and orthologous sequences with conserved target sites (Fig. 2). The multiple alignments of the conserved target sites and the alignments between smRNA and targets for homolog genes can be viewed via the hyperlink in the “Conservation” column (Fig. 3b and c).
Figure 3.

Illustration of expandable links in the target prediction result table. (A) A sample line of the target prediction result table; (B) target site conservation analysis results. Homolog genes with the same smRNA target site, both within and cross-species, are shown in the ClustalW multiple alignment format; (C) the alignments between miRNA and targets provoked by mousing over the hyperlinks of gene IDs listed in (B); (D) Degradome data of the predicted smRNA target. Positions listed in the table are orders of nucleotides in the smRNA sequences, plus upstream and downstream 5 nt each; ‘Loci number’ represents the number of perfectly mapped loci of each degradome sequence, marked at the start nucleotide position of each sequence. Normalized reads starting at each nucleotide position in the degradome data are listed in following rows. The histogram shows the distribution of the degradome reads. In the example, the target should be cleaved after the 12th nucleotide according to the degradome data; (E) Expression change of target gene in smRNA biogenesis pathway gene mutants compared to wild-type plants. Fold changes of the normalized expression values between mutants and wildtype are shown in the table and the Y axis of the plot. Data from different datasets are distinguished by colors in the plot.
Degradome data
It has been shown that the miRNA cleavage products of targets can be cloned and detected by high-throughput sequencing technology, generating mRNA degradome data among which the 5′ ends of sequences mark the cleavage sites of miRNAs (28,37,42–44). PsRobot integrates well-produced datasets of Arabidopsis thaliana (GSE11094) and rice (GSE18248) in the target prediction results (Fig. 2) (28,37). The abundance of degradome sequences (after normalized to reads per million, RPM) is marked at the starting genomic loci of each sequence (Fig. 3d). Candidate target sites with abundant degradome sequences are more likely to be authentic miRNA targets. The position with the most abundant degradome sequences should represent the miRNA cleavage site.
Target expression in smRNA biogenesis mutants
As the production of smRNAs will be significantly impaired in the mutants of genes involved in plant smRNA biogenesis pathways, such as the dcl, hyl1, hen1 and rdr family genes (38), expression increment of genes in these mutants may indicate an inhibitory target effect by smRNAs. To facilitate inspections from this perspective, psRobot collected the published microarray data and integrated them in the target prediction results (Fig. 3e). Currently, this function is only available for Arabidopsis thaliana (GSE2473, GSE3011, GSE24887) (45,46) and will be expanded to other species on the availability of required data.
Performance
To test the reliability of the smRNA target prediction results, we selected 75 Arabidopsis thaliana miRNAs with at least one reported target in the ASRP database (Supplementary Table S2) (47). A total of 995 genes were predicted as the targets of the 75 miRNAs using the default parameters, of which 306 (31%) targets were reported by the ASRP database, representing 89% of the 344 validated Arabidopsis miRNA targets (Supplementary Table S1). Significant improvement of the prediction results was achieved by combining the information of target site conservation, detectability in degradome data and expression change in smRNA biogenesis mutants as filters, as demonstrated by the reduction of total predicted targets to 542, of which 292 (54%) were confirmed targets from ASRP (85% of validated targets) (Supplementary Table S1).
CONCLUSIONS
Computational prediction of miRNAs and their targets have suffered high false-positive rate because of limited constraints to apply. To generate more specific prediction results, psRobot integrated the biogenesis and protein association information of plant smRNAs, as well as conservation, cleavage and smRNA dependency information for mRNAs. These information can facilitate users to quickly identify bona fide miRNAs or other functional stem-loop smRNAs and their candidate targets. The ability to handle both single or batch sequence input and the availability of online and local version of the software renders it high flexibility in application.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2.
FUNDING
National Natural Science Foundation of China [90917016 and 30921061 to X.W.]; Chinese Academy of Sciences [20100322 to X.W.]; Ministry of Agriculture [2011ZX08010-002-002 to X.W.]. Funding for open access charge: National Natural Science Foundation of China [90917016].
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr. Guan-Zheng Luo for the discussion of web structure. The authors also thank Guihai Feng, Yong-Xin Liu and Xin-Min Zhang for testing the local software.
REFERENCES
- 1.Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant. Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- 2.Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, Hilbert JL, Bartel DP, Crete P. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell. 2004;16:69–79. doi: 10.1016/j.molcel.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 3.Borsani O, Zhu J, Verslues PE, Sunkar R, Zhu JK. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005;123:1279–1291. doi: 10.1016/j.cell.2005.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang XJ, Gaasterland T, Chua NH. Genome-wide prediction and identification of cis-natural antisense transcripts in Arabidopsis thaliana. Genome biology. 2005;6:R30. doi: 10.1186/gb-2005-6-4-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Liu XS, Liu QR, Wei L. Genome-wide in silico identification and analysis of cis natural antisense transcripts (cis-NATs) in ten species. Nucleic Acids Res. 2006;34:3465–3475. doi: 10.1093/nar/gkl473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang H, Chua NH, Wang XJ. Prediction of trans-antisense transcripts in Arabidopsis thaliana. Genome Biol. 2006;7:R92. doi: 10.1186/gb-2006-7-10-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reinhart BJ, Weinstein EG, Rhoades MW, Bartel B, Bartel DP. MicroRNAs in plants. Genes Dev. 2002;16:1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006;38 Suppl:S31–S36. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- 9.Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/s0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- 10.Meyers BC, Simon SA, Zhai J. MicroRNA processing: battle of the bulge. Curr. Biol.: CB. 2010;20:R68–R70. doi: 10.1016/j.cub.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Zilberman D, Cao X, Johansen LK, Xie Z, Carrington JC, Jacobsen SE. Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr. Biol. 2004;14:1214–1220. doi: 10.1016/j.cub.2004.06.055. [DOI] [PubMed] [Google Scholar]
- 12.Dunoyer P, Brosnan CA, Schott G, Wang Y, Jay F, Alioua A, Himber C, Voinnet O. An endogenous, systemic RNAi pathway in plants. EMBO J. 2010;29:1699–1712. doi: 10.1038/emboj.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20:3186–3190. doi: 10.1105/tpc.108.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell. 2004;14:787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 15.Dai X, Zhuang Z, Zhao PX. Computational analysis of miRNA targets in plants: current status and challenges. Brief. Bioinform. 2010;12:115–121. doi: 10.1093/bib/bbq065. [DOI] [PubMed] [Google Scholar]
- 16.Allen E, Xie Z, Gustafson AM, Carrington JC. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 17.Fahlgren N, Howell MD, Kasschau KD, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Law TF, Grant SR, Dangl JL, et al. High-throughput sequencing of Arabidopsis microRNAs: evidence for frequent birth and death of MIRNA genes. PloS one. 2007;2:e219. doi: 10.1371/journal.pone.0000219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai X, Zhao PX. psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res. 2011;39:W155–W159. doi: 10.1093/nar/gkr319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Addo-Quaye C, Miller W, Axtell MJ. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25:130–131. doi: 10.1093/bioinformatics/btn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 22.Wang XJ, Reyes JL, Chua NH, Gaasterland T. Prediction and identification of Arabidopsis thaliana microRNAs and their mRNA targets. Genome Biol. 2004;5:R65. doi: 10.1186/gb-2004-5-9-r65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001–2019. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, Sasidharan R, Muller R, Dreher K, Alexander DL, Garcia-Hernandez M, et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2011;40:D1202–D1210. doi: 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ouyang S, Zhu W, Hamilton J, Lin H, Campbell M, Childs K, Thibaud-Nissen F, Malek RL, Lee Y, Zheng L, et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 2007;35:D883–D887. doi: 10.1093/nar/gkl976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka T, Antonio BA, Kikuchi S, Matsumoto T, Nagamura Y, Numa H, Sakai H, Wu J, Itoh T, Sasaki T, et al. The Rice Annotation Project Database (RAP-DB): 2008 update. Nucleic Acids Res. 2008;36:D1028–D1033. doi: 10.1093/nar/gkm978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2011;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nat Biotechnol. 2008;26:941–946. doi: 10.1038/nbt1417. [DOI] [PubMed] [Google Scholar]
- 29.Fahlgren N, Sullivan CM, Kasschau KD, Chapman EJ, Cumbie JS, Montgomery TA, Gilbert SD, Dasenko M, Backman TW, Givan SA, et al. Computational and analytical framework for small RNA profiling by high-throughput sequencing. RNA. 2009;15:992–1002. doi: 10.1261/rna.1473809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardcastle TJ, Kelly KA. baySeq: empirical Bayesian methods for identifying differential expression in sequence count data. BMC Bioinformatics. 2010;11:422. doi: 10.1186/1471-2105-11-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery TA, Yoo SJ, Fahlgren N, Gilbert SD, Howell MD, Sullivan CM, Alexander A, Nguyen G, Allen E, Ahn JH, et al. AGO1-miR173 complex initiates phased siRNA formation in plants. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20055–20062. doi: 10.1073/pnas.0810241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, et al. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5' terminal nucleotide. Cell. 2008;133:116–127. doi: 10.1016/j.cell.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, Carrington JC. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS. Biol. 2007;5:e57. doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu C, Kulkarni K, Souret FF, MuthuValliappan R, Tej SS, Poethig RS, Henderson IR, Jacobsen SE, Wang W, Green PJ, et al. MicroRNAs and other small RNAs enriched in the Arabidopsis RNA-dependent RNA polymerase-2 mutant. Genome Res. 2006;16:1276–1288. doi: 10.1101/gr.5530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howell MD, Fahlgren N, Chapman EJ, Cumbie JS, Sullivan CM, Givan SA, Kasschau KD, Carrington JC. Genome-wide analysis of the RNA-DEPENDENT RNA POLYMERASE6/DICER-LIKE4 pathway in Arabidopsis reveals dependency on miRNA- and tasiRNA-directed targeting. Plant Cell. 2007;19:926–942. doi: 10.1105/tpc.107.050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. DNA methylation mediated by a microRNA pathway. Mol. Cell. 2010;38:465–475. doi: 10.1016/j.molcel.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Wu L, Zhang Q, Zhou H, Ni F, Wu X, Qi Y. Rice MicroRNA effector complexes and targets. Plant Cell. 2009;21:3421–3435. doi: 10.1105/tpc.109.070938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vazquez F. Arabidopsis endogenous small RNAs: highways and byways. Trends Plant Sci. 2006;11:460–468. doi: 10.1016/j.tplants.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 39.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith TF, Waterman MS. Identification of common molecular subsequences. J. Mol. Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 41.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008;18:758–762. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pantaleo V, Szittya G, Moxon S, Miozzi L, Moulton V, Dalmay T, Burgyan J. Identification of grapevine microRNAs and their targets using high-throughput sequencing and degradome analysis. Plant J. 2010;62:960–976. doi: 10.1111/j.0960-7412.2010.04208.x. [DOI] [PubMed] [Google Scholar]
- 44. Zhou M, G.L., Li P, Song X, Wei L, Chen Z, Cao X. (2010) Degradome sequencing reveals endogenous small RNA targets in rice (Oryza sativa L. ssp. indica). Front. Biol., 5,67–90.
- 45.Xie Z, Allen E, Wilken A, Carrington JC. DICER-LIKE 4 functions in trans-acting small interfering RNA biogenesis and vegetative phase change in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 2005;102:12984–12989. doi: 10.1073/pnas.0506426102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willmann MR, Mehalick AJ, Packer RL, Jenik PD. MicroRNAs regulate the timing of embryo maturation in Arabidopsis. Plant Physiol. 2011;155:1871–1884. doi: 10.1104/pp.110.171355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gustafson AM, Allen E, Givan S, Smith D, Carrington JC, Kasschau KD. ASRP: the Arabidopsis Small RNA Project Database. Nucleic Acids Res. 2005;33:D637–D640. doi: 10.1093/nar/gki127. [DOI] [PMC free article] [PubMed] [Google Scholar]