

Abstract
In polyglutamine (polyQ) containing fragments of the Huntington’s disease protein huntingtin (htt), the N-terminal 17 amino acid httNT segment serves as the core of α-helical oligomers whose reversible assembly locally concentrates the polyQ segments, thereby facilitating polyQ amyloid nucleation. A variety of aggregation inhibitors have been described that achieve their effects by neutralizing this concentrating function of the httNT segment. In this paper we characterize the nature and limits of this inhibition for three means of suppressing httNT-mediated aggregation. We show that the previously described action of httNT peptide-based inhibitors is solely due to their ability to suppress the httNT-mediated aggregation pathway. That is, under httNT inhibition, nucleation of polyQ amyloid formation by a previously described alternative nucleation mechanism proceeds unabated and transiently dominates the aggregation process. Removal of the bulk of the httNT segment by proteolysis or mutagenesis also blocks the httNT-mediated pathway, allowing the alternative nucleation pathway to dominate. In contrast, the previously described immunoglobulin-based inhibitor, the anti-httNT VL 12.3 protein, effectively blocks both amyloid pathways, leading to stable accumulation of non-amyloid oligomers. These data show that the httNT-dependent and independent pathways of amyloid nucleation in polyQ-containing htt fragments are in direct kinetic competition. The results illustrate how amyloid polymorphism depends on assembly mechanism and kinetics, and have implications for how the intracellular environment can influence aggregation pathways.
The expanded CAG repeat diseases are a series of familial neurodegenerative conditions resulting from the expansion of a polyglutamine (polyQ) tract in a disease protein (1). While disease mechanisms remain obscure, the early cellular events are presumably triggered by pathogenic interactions between one or more cellular targets and either monomers or aggregates of the expanded polyQ protein (2). The polyQ repeat length dependence of both disease risk and disease onset (1) correlates with the repeat length dependence of aggregation onset (3, 4), and polyQ-containing aggregates are found in victims of each of these diseases (1, 5). The detailed structures of the toxic species and the identities of their cellular targets are not known.
As with many other aggregation disease proteins (6–11), polyQ proteins are able to form a number of polymorphic aggregates both in vitro and in vivo (2, 12, 13). Simple polyQ peptides, flanked by a few charged residues, undergo classical nucleated growth polymerization into amyloid fibrils without the intervention of the kind of non-amyloid oligomeric species often observed in amyloid formation by disease proteins like Aβ (2, 14, 15). In contrast, polyQ fused to some flanking sequences, such as the 17 amino acid N-terminus (httNT) of the Huntington’s disease (HD) protein huntingtin (htt), exhibit early formation of roughly spherical oligomers lacking β-structure, and these oligomers play critical roles in the formation of mature polyQ-core amyloid (2, 16–18). Aggregates of a wide range of morphologies have been isolated from a HD tg mouse model (13), and it is a reasonable possibility that only some of these aggregate polymorphs might be toxic. If so, it becomes particularly important to understand how the cellular environment interacts with the polyQ protein sequence to direct the formation of different aggregate morphologies.
Previous studies uncovered evidence that different growth conditions give rise to aggregates of polyQ-containing htt fragments that exhibit different morphologies and cytotoxicities (19). More recently, it was demonstrated that aggregates isolated from a number of HD tg mouse models are capable of seeding polyQ aggregation in vitro (20), suggesting an amyloid-like morphology and consistent with toxicity mechanisms postulating recruitment of normal polyQ proteins into aggregates (21, 22). Alternatively, non-amyloid oligomeric forms of htt fragments might be the toxic agents, consistent with the current view of aggregate toxicity in other disease states (8–10, 23). However, there is also some recent indirect evidence against a toxic role for such oligomers, in that analogs of polyQ-containing htt fragments that appear to be only capable of forming non-amyloid oligomers (18) are not toxic when expressed in mammalian cells (24, 25). The open question as to the nature of the toxic species in HD emphasizes the importance of better understanding the htt aggregation pathway and how it might be impacted by interactions with other molecules.
In this paper we describe how the aggregation reactions and products of polyQ-containing htt fragment model peptides are modified by certain effector molecules and protein modifications relevant to the cellular environment. We find that some events that target the key role of the httNT segment can delay httNT-mediated polyQ aggregation, providing a window for the operation of the normally underrepresented aggregation pathway analogous to that for simple polyQ peptides. Another httNT targeting inhibitor, in contrast, completely blocks nucleation of amyloid by any mechanism, leading to the stable accumulation of large non-amyloid oligomers. The results show that polyQ-containing htt fragments are capable of engaging at least three aggregation pathways leading to different aggregate products, and that the relative kinetics of these pathways can be influenced by other molecules and interactions. Since the different pathways generate aggregates of different structures and properties, the possibility exists for modulating toxicity not only by eliminating aggregation but also by its redirection towards more benign aggregated products.
MATERIALS AND METHODS
Materials
Synthetic peptides were obtained in unpurified form from the Keck Biotechnology Center at Yale (http://keck.med.yale.edu/ssps/), and were HPLC purified and disaggregated using a combination of aggregate dissociation and ultracentrifugation, as described (26). In some peptides an F17W mutation is present; previously we found that this mutation has a negligible effect on aggregation (16). Acetonitrile, hexafluoroisopropanol (99.5%, spectrophotometric grade), and formic acid were from Acros Organics, and trifluoroacetic acid (99.5%, Sequanal Grade) from Pierce.
General methods
The sedimentation assay (26), the ThT and 90° light scattering assays (27), and the nucleation kinetics analysis (14, 26), have been described. The isolation of aggregates for seeding analysis was conducted by centrifuging a reaction aliquot at 14800 rpm in an Eppendorf centrifuge at 4 ° C for 30 mins, washing the pellet 2–3 times with PBS, resuspending in buffer, and determining the aggregate concentration by an HPLC analysis of a formic acid dissolved aliquot, as described (26, 28). SEC was on a Superdex 75 10/200 column (GE Health Sciences) equilibrated in PBS on an Agilent 1200 isocratic HPLC system with a flow rate of 0.4 ml/min and elution monitored at 214 nm. The column was calibrated with aprotinin, cytochrome C, ribonuclease, ovalbumin and bovine serum albumin. In complex formation experiments, new peaks were characterized by analysis of 500 μL portions of the peak fractions on a reverse phase HPLC column with detection on an Agilent 1100 LC-MS system.
Electron microscopy
Aliquots of aggregation reaction mixtures were taken at different time points and visualized by electron microscopy. A 3 μl sample was placed on a freshly glow-discharged carbon-coated grid, allowed to adsorb for 2 minutes, and washed with deionized water before staining with 2 μl of 1% uranyl acetate and blotting. Grids were imaged on a Tecnai T12 microscope (FEI Co., Hillsboro, Oregon) operating at 120kV and 30,000x magnification, and equipped with an UltraScan 1000 CCD camera (Gatan, Pleasanton, California) with post-column magnification of 1.4x.
Recombinant protein expression
The region encoding amino acids M1 to G115 of the αHtt-VL 12.3 domain was amplified by PCR using plasmid DNA (pNES-VL12.3, a gift from Dane Wittrup) as template. The resulting amplicon was then inserted into a pET15b expression vector (Novagen), using NdeI and XhoI restriction sites at the 5′ and 3′ ends, respectively, generating an N-terminal His-tagged αHtt-VL 12.3 domain construct. Following sequence verification, the VL12.3–15b plasmid was transformed into E. coli Rosetta2 (DE3) cells (Novagen). Cultures were grown at 37° C, induced with 1 mM IPTG at OD600 of ~0.8, and further grown at 16°C for 18 h for protein production. Cells were harvested by centrifugation, resuspended in TBS buffer (25 mM Tris.HCl, 150 mM NaCl, 3 mM NaN3, and pH 8.0) and lyzed by sonication. Protein was purified by metal-affinity chromatography on Ni2+-derivatized HisTrap columns (GE Healthcare) using a linear (20 – 1000 mM) imidazole gradient for elution. Purified protein was digested with thrombin in TBS buffer for removal of the His tag (leaving a Gly-Ser-His sequence at the N-terminus of the VL protein), followed by gel filtration on Superdex75 (GE Healthcare) in 20 mM sodium phosphate, 100 mM NaCl, and 3 mM NaN3 (pH 6.0). Purified proteins were concentrated using centriprep concentrators (Millipore) up to ~10 mg/ml and the buffer was simultaneously exchanged to 20 mM sodium phosphate, 3 mM NaN3, 90/10% H2O/D2O (pH 6.0). Structure of the purified protein was confirmed by mass spectrometry.
RESULTS
A variety of proteolytic cleavage events generating short polyQ-containing fragments of htt have been implicated in the HD disease mechanism (29), and the expression of expanded polyQ forms of such fragments, including one particular fragment, htt exon-1 (Fig. 1), has been widely shown to lead to aggregation and/or toxicity in cells and animals (30–35). Exon-1 consists of three well-defined sequence elements: the 17 amino acid httNT segment, the polyQ segment, and the Pro-rich segment (Fig. 1). Since chemically synthesized peptides containing the first two elements, plus polyPro segments as short as P6, exhibit aggregation properties very similar to those of full length exon-1 (B. Sahoo, D. Singer, T. Zuchner and R. Wetzel, Ms. in preparation), we have used such C-terminally truncated derivatives of htt exon1 to work out details of the aggregation mechanism (16–18, 36). Synthetic peptides studied in this paper are shown in Figure 1.
Figure 1.

Sequences of the exon1 N-terminal fragment of huntingtin and the various synthetic peptides studied.
Compromising the httNT role via inhibition with httNT peptides
Recently we reported that peptides related to the httNT sequence can be effective, if transitory, inhibitors of the aggregation of polyQ-containing htt fragment peptides (18). Thus, while 6 μM of httNTQ30P6K2 exhibits measureable aggregation within several hrs and is 50% aggregated by 18 hrs, the same concentration of peptide in the presence of a 1:1 ratio of httNT shows little aggregation after 18 hrs and requires ~ 80 hrs to reach 50% aggregation (Fig. 2A). Since we reckoned that the polyQ element of httNTQ30P6K2 might be capable of aggregating by several mechanisms, we inquired as to the nature of the residual aggregation that occurs in the presence of the httNT inhibitor. Electron micrographs of the non-inhibited aggregation of httNTQ30P6K2 show early formation of spherical oligomers (Fig. 3B, 0.25 hrs), followed by protofibrils (Fig. 3C, 2.5 hrs) and ultimately rough-edged fibrils (Fig. 3D, 144 hrs) (16). In contrast, in addition to the expected (18) mixed oligomer inhibition complexes, early amyloid-like aggregates are observed at 5.5 hrs when httNTQ30P6K2 is incubated in the presence of the httNT (Fig. S1H), and larger aggregates observed at 8.0 hrs (Fig. 3E; Fig. S1 I-P) exhibit the characteristic broad ribbons that are normally seen in amyloid aggregates of simple polyQ (Fig. 3J); such aggregates are never observed in uninhibited reactions of httNTQN peptides. EM images of the httNT inhibited reaction taken at subsequent times show aggregate morphologies more resembling the protofibrillar (Fig. 3F, 45 hrs) and fibrillar (Fig. 3G, 68 hrs) aggregates normally observed at earlier times in the non-inhibited aggregation of httNTQ30P6K2 (Fig 3B,C). The progression of aggregate morphologies suggest that at early times in the httNT inhibited reaction, there is a window within which httNTQ30P6K2 peptides can nucleate amyloid growth via the pathway normally observed for simple polyQ peptides, leading to characteristic ribbon-like aggregates. Eventually, however, inhibition of httNT-mediated amyloid nucleation is overcome, and the characteristic aggregate morphologies observed in htt N-terminal peptide aggregation emerge.
Figure 2.

Aspects of httNT inhibition. (A) Kinetics of httNTQ30P6K2 with (○) and without (●) httNT inhibition. (B) Aggregation of approximately 4 μM httNTQ30P6K2 either with no addition (○) or seeded with 7.5% by weight of: K2Q30K2 fibrils (△); fibrils from httNTQ30P6K2 aggregation with (▲) and without (●) added httNT inhibitor. (C) Aggregation of approximately 20 μM K2Q30K2 either with no addition (○) or seeded with 5 % by weight of: K2Q30K2 fibrils (△); fibrils from httNTQ30P6K2 aggregation with (▲) and without (●) added httNT inhibitor. For comparison purposes, all data were fit to straight lines; linear fits are expected for efficient seeded elongation reactions (37), but not for the spontaneous, unseeded reactions (14–16). In fact, some deviation from linearity is seen in data from both of the non-seeded reactions.
Figure 3.

Electron micrographs of various polyglutamine aggregates. Empty grid (A); aggregation reaction of httNTQ30P6K2 alone at 15 mins (B), 2.5 hrs (C), and 100 hrs (D); aggregation of httNTQ30P6K2 in the presence of a 1:1 molar ratio of httNT collected at 8 hrs (E), 45 hrs (F) and 68 hrs (G); aggregation of httNTQ30P6K2 plus a 1:1 molar ratio of the anti-httNT VL protein collected at 120 hrs (H); aggregation of MF-Q37P10K2, 74 hrs (I); final aggregates of K2Q30K2 (J). The scale bar (A) of 50 nm is applicable to all panels.
Further kinetics studies add additional support to this view. An examination of the concentration dependence of the initial aggregation rates (Fig. 4) of wild type httNTQ30P6K2 (■) and its F17W mutant (▲) in the absence of httNT inhibition yields log-log slopes of 1.20 and 0.95 as previously reported (16). In contrast, the corresponding slope for the same peptide in the presence of inhibitor (△) is 2.65, which is very similar to the slope of 2.57 for aggregation of a simple K2Q30K2 peptide (●) (Fig. 4). This slope change is consistent with the EM data in suggesting that the early aggregation of httNTQ30P6K2 in the presence of httNT occurs via the classical, nucleated growth mechanism previously determined for simple polyQ peptides (14, 15).
Figure 4.
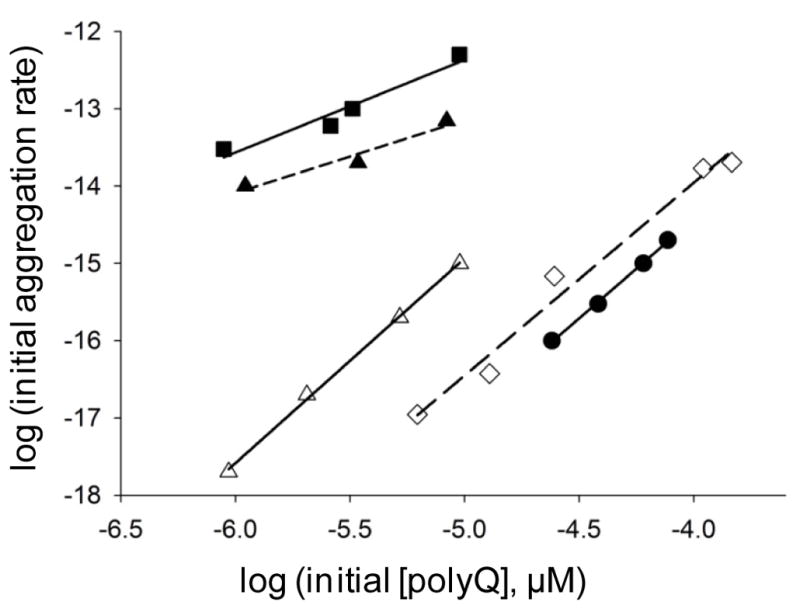
Concentration dependence of initial aggregation rates of polyQ peptides. Slopes of time-squared plots of initial aggregation kinetic data are plotted vs. concentration in a log-log plot (14). The slope of the log-log plot minus 2 is n*, the critical nucleus (14). Determination for K2Q30K2 (●, slope = 2.57, n* = 0.57), httNTQ30P6 (■, slope = 1.20, n* = − 0.80), httNTQ30P6K2 (F17W) (▲, slope = 0.95, n* = − 1.05), httNTQ30P6K2 (F17W) in the presence of httNT (△, slope = 2.65, n* = 0.65), and MF-Q37P10K2 (◇, slope = 2.57, n* = 0.57). Previously we found that the F17W mutation (Methods) has a negligible effect on aggregation kinetics (16). Data for K2Q30K2 and httNTQ30P6K2 peptides alone are from reference (16).
As a further test, the mature aggregates from the inhibited and non-inhibited aggregation reactions of httNTQ30P6K2, along with control amyloid-like aggregates of K2Q30K2, were collected and studied for their seeding properties, as a measure of structural relatedness (37, 38). With httNTQ30P6K2 monomers, only the product from the non-inhibited aggregation of httNTQ30P6K2 (●) exhibited significant enhancement compared with the non-seeded control (○), while K2Q30K2 aggregates (△) and the products of httNT-inhibited httNTQ30P6K2 aggregation (▲) were equally ineffective (Fig. 2B). With K2Q30K2 monomers, the product of httNT-inhibited aggregation of httNTQ30P6K2 (▲) likewise behaved like simple K2Q30K2 aggregates (△), with both aggregates significantly enhancing aggregation compared to the unseeded control (○) (Fig. 2C). In contrast, the product of non-inhibited httNTQ30P6K2 aggregation (●) was an ineffective seed, producing no change from the unseeded rate (Fig. 2C). Together the results show that (a) the seeding properties of non-inhibited httNTQ30P6K2 aggregation and K2Q30K2 aggregation are different, and (b) the seeding potential of the product of inhibited aggregation of httNTQ30P6K2 is identical to that of K2Q30K2 aggregates. These seeding results imply underlying structural similarities and differences. It is interesting that the final aggregates of the httNT-inhibited reaction behave like K2Q30K2 aggregates, even though they are quite similar in appearance to the aggregates from the non-inhibited httNTQ30P6K2 reaction (Fig. 3D,G). This shows that aggregate properties cannot always be correlated with EM morphologies.
Compromising the httNT role via inhibition with an anti-httNT intrabody
Previously, Wittrup and colleagues described a novel immunoglobulin-related molecule capable of binding the httNT segment of htt, obtained by a combination of mutagenesis and yeast surface display selection (39). This light chain variable domain-related protein, αHtt-VL 12.3, which lacks both a heavy chain partner and the normal, native Ig fold intra-domain disulfide bond, exhibited a binding constant for httNT peptides in the low nM range (39, 40), inhibited htt N-terminal fragment aggregation in vitro, and was a good inhibitor of htt aggregation and toxicity in mammalian cells in culture (39). Interestingly, the X-ray crystal structure of a 1:1 complex of this VL 12.3 with an httNT fragment shows that httNT is bound in an α-helical conformation (40), similar to the structure induced upon httNT self-assembly into tetramers and larger oligomers (36) (17). We used a cDNA encoding this protein to engineer a vector for expression in E. coli (see Methods). After purification and proteolytic removal of the His tag, the 12 kDa protein was obtained and examined for its mechanism of aggregation inhibition.
In our standard sedimentation aggregation assay, this protein proved a very effective inhibitor of httNTQ30P6K2 aggregation at a 1:1 molar ratio (Fig. 5A). After a slow drop in soluble httNTQ30P6K2 in the inhibited reaction to about 70% the starting value, little or no further loss of monomer occurs up to over 150 hrs (Fig. 5A, -○-). A small but significant amount of the αHtt-VL 12.3 domain protein also becomes pelletable over the same time frame (Fig. 5A, -△-). In parallel with the generation of pelletable material, right angle light scattering confirmed the generation of protein particles (Fig. 5B). At the same time, there is no detectible ThT signal throughout the incubation of httNTQ30P6K2 with the αHtt-VL 12.3 domain protein (Fig. 5C), suggesting that the aggregates formed are not amyloid. Thus, in contrast to the inhibition by 1:1 ratios of httNT peptides, the αHtt-VL 12.3 domain protein under the same conditions provides strong inhibition of httNT-mediated amyloid nucleation in polyQ-containing htt fragments. We conducted further experiments to explore this difference.
Figure 5.

Kinetics of aggregation of a 1:1 molar ratio of VL 12.3 and httNTQ30P6K2 by the sedimentation (A), 90° light scattering (B) and ThT (C) assays. HttNTQ30P6K2 with (○) and without (●) the VL protein; VL protein monomer concentration in mixture with httNTQ30P6K2 (△); VL protein incubated alone (▲).
We first examined complex formation under native conditions between VL 12.3 and httNTQ30P6K2, using SEC. As previously described (40), we found that the αHtt-VL 12.3 domain protein behaves aberrantly in SEC, eluting well after the position normally occupied by small solute molecules (Fig. 6A, peak c). In contrast, the httNTQ30P6K2 peptide elutes at 31.6 mins (Fig. 6A), corresponding to a predicted MW of 7.7 KDa. The slight discrepancy between the SEC determined MW and the actual (6.7 KDa) is most likely due to the non-globular nature of the htt N-terminal fragment (SEC standards used are all globular proteins; see Methods). A mixture of excess αHtt-VL 12.3 domain protein with httNTQ30P6K2 generates a new peak eluting at 29.2 mins (Fig. 6A, peak b), corresponding to an SEC determined MW of 12.6 KDa. This is somewhat lower than expected (19.0 kDa) for a 1:1 complex, perhaps due to some residual interaction of the αHtt-VL 12.3 domain component with the solid support. Interestingly, the SEC profile of the mixture also exhibits several higher MW forms, including a small peak corresponding to ~ 43 kDa (Fig. 6A). The main 29.2 min SEC peak was collected and analyzed by LC-MS (see Methods). In the RP-HPLC of this SEC peak, two peaks were observed, giving MS signals corresponding to the known masses of httNTQ30P6K2 and the αHtt-VL 12.3 domain protein (Fig. 6B). Thus, the major product of the initial interaction of httNTQ30P6K2 and the αHtt-VL 12.3 domain protein in solution is, as expected from other analyses (40), a 1:1 complex.
Figure 6.

Size exclusion chromatography of the anti-httNT VL 12.3 protein. (A) Chromatograms of anti-httNT VL 12.3 protein alone (dotted line), httNTQ30P6K2 alone (solid line) and a mixture of httNTQ30P6K2 with excess anti-httNT VL 12.3 protein (dashed line). Arrows indicate void volume (a), httNTQ30P6K2: anti-httNT VL 12.3 complex (b, 29.2 mins, 12.6 KDa from standard curve; 19.0 KDa actual), httNTQ30P6K2 alone (c, 31.6 mins, 7.7 KDa from standard curve, 6.7 KDa actual), position of salt elution (d), anti-httNT VL 12.3 protein alone (e); (B) LC-MS of the SEC peak “b” above identifying two components with the m/z of httNTQ30P6K2 eluting at ~ 16.5 mins and for the anti-httNT VL 12.3 protein eluting at ~ 19.5 mins.
Further examination of the incubated mixture of these two proteins by EM revealed additional higher assembly forms. EM images collected at 120 hrs consistently show a field of spherical aggregates with aggregates in the 8 –25 nm range (Fig. 3H). There is a curious ‘beads-on-a-string’ appearance to these images, the significance of which is not clear. These large oligomers presumably are the source of the ThT-negative (Fig. 5C), light scattering positive (Fig. 5B) material as well as the incomplete recovery of monomer in the sedimentation assay from solutions of httNTQ30P6K2 and the αHtt-VL 12.3 domain protein (Fig. 5A). It is tempting to assign the observed strong inhibition of amyloid formation to the existence of these oligomers, in analogy to other peptide-based inhibitors of the aggregation of polyQ-containing htt fragments (18). This interpretation does not hold up under scrutiny, however. Since oligomer formation does not nearly go to completion (Fig. 5A), it would be expected that the residual (~70%) concentration of low MW material should be capable of supporting the alternative amyloid formation pathway, as in the case of httNT inhibition described above. There is no evidence of this, however, by EM or other analysis. Based on the SEC analysis (Fig. 6A), it is likely that the bulk of the httNTQ30P6K2 in this solution is engaged in complex formation with the VL 12.3 protein to generate a heterodimer (Fig. 6A). It would therefore appear that this heterodimer formation is sufficient to very effectively block nucleation of polyQ amyloid formation. This suggests an ability of the 1:1 complex to interfere with the interactions of the polyQ chains that are required to engage the classical nucleated growth mechanism seen for simple polyQ peptides and for httNT-inhibited httNTQ30P6K2 (see above). It is not clear how complexation of the VL protein with the httNT segment alone might achieve this.
Compromising the httNT role via partial excission
Previously we showed that the polyQ containing fragment of trypsin cleavage of httNTQ37P10K2, namely, SF-Q37P10K2, undergoes amyloid nucleation at a slower rate than the uncleaved molecule, and exhibits a nucleation mechanism and aggregate morphologies identical to those from simple polyQ peptides (15). Consistent with the suggestion that similar cleavage fragments occur in cells (41), tg mouse experiments have been initiated investigating the effect of expression of a full length htt molecule in which the 17 amino acid httNT sequence is replaced by the Met-Phe sequence (William Yang, personal communication). Here we investigate the impact of this sequence change in aggregation experiments in vitro.
As expected, the peptide MF-Q37P10K2 (○) aggregates appreciably more slowly than a matched concentration of httNTQ37P10K2 (●) (Fig. 7A). At the same time, however, MF-Q37P10K2 at a higher concentration (●) aggregates somewhat faster than the same concentration of Q37P10K2 (⋄) (Fig. 7B). Analysis of the concentration dependence of the initial aggregation of MF-Q37P10K2 yields a log-log plot (Fig. 4, ⋄) slope of 2.5, almost identical to the slopes for K2Q30K2 (●) and the httNT-inhibited aggregation of httNTQ30P6K2 (△). Finally, the morphology of the MF-Q37P10K2 aggregates (Fig. 3I) is identical to typical amyloid-like aggregates of simple polyQ peptides (Fig. 3J). Thus, aggregation kinetics and aggregate morphologies are consistent in indicating that this analog of a trypsin-cleaved htt N-terminal fragment follows an aggregation pathway essentially identical to that of simple polyQ peptides.
Figure 7.

Kinetics of MF-Q37P10K2 aggregation by the sedimentation assay. (A) Aggregation of 6 μM MF-Q37P10K2 (○) compared to 6 μM httNTQ37P10K2 (●); (B) aggregation of 31 μM MF-Q37P10K2 (○;) compared to 48 μM Q37P10K2 (●).
DISCUSSION
The phenomenon of aggregate polymorphism has emerged as a major area of interest in amyloid assembly and properties (11, 42, 43). The ability to make multiple morphologies of stable products is nearly ubiquitous in amyloid systems and distinguishes them from native proteins. For example, not only do amyloidogenic peptides like Aβ exhibit a variety of aggregate non-amyloid morphologies, some of which may be particularly toxic (44), but even mature amyloid fibrils of the same wild type Aβ peptide can exhibit a surprising number of distinct structural types (45–47). The polymorphisms exhibited by mature amyloid fibrils are almost certainly linked to the phenomena of strain effects in prion biology, at least in yeast prion systems (11, 43). While the ability of protein aggregates to exhibit multiple, stable structural types is accounted for by the polymeric nature of protein assemblies (48), the mechanistic basis for how growth conditions (6, 19, 49–51), mutations (7, 52–54), and/or other, sporadic, factors (55) control the kinetic accessibility that ultimately favors particular aggregate polymorphs has received less attention. This general problem may be particularly well-defined in the huntingtin system. While simple polyQ peptides are almost unique in the amyloid world in being able to assemble into amyloid fibrils without the mediation of non-amyloid intermediates (15), certain flanking sequences to polyQ, such as the httNT segment of huntingtin (16, 17), mediate amyloid nucleation by undergoing an initial, independent assembly pathway (2). At the same time, in contrast to the current wisdom that such oligomers tend to be the primary toxic agents in aggregation-based neurodegenerative diseases (42), the situation may be different in polyQ diseases. We have argued that the reported low toxicity (24) of a htt exon1 analog containing a particular type of proline-mutated polyQ segment (56) is evidence against a toxic role for non-amyloid oligomers of htt fragments (18). Consistent with this view, a well-supported mechanism for aggregate toxicity in polyQ diseases, the recruitment/sequestration hypothesis (22), is only likely to operate via mature amyloid-like polyQ aggregates, and not with oligomers, due to the relative abilities of these two aggregate classes to seed aggregate elongation (16) and hence recruit monomers. Perhaps related to such functional diversity, different morphologies of polyQ aggregates have been shown to exhibit different cytotoxicities (19).
Our working model for the aggregation options open to polyQ-containing htt fragments is shown in Figure 8. Monomers (a) of such fragments are in principle capable of entering two distinct aggregation pathways. Pathway A involves the efficient formation of α-helix rich oligomers according to the pathway described previously (16, 17). In this pathway, the httNT segment of these peptides, which is disordered in the monomer state (16), mediates α-helix bundle formation (b,c,d) that collects the disordered polyQ elements at a high local concentration and thereby facilitates the formation of polyQ amyloid nuclei (e) that can then elongate into βrich amyloid like aggregates by monomer addition (f). (It should be noted that alternative models have also been described for how flanking sequences interact with polyQ sequences to effect aggregate formation (57–59)). If, for any of a number of reasons, httNT mediated α-helix bundle formation is not feasible or efficient, polyQ-containing htt fragments can undergo the previously described (14, 15) classical nucleation pathway. In this B Pathway, for relatively long (15) polyQ peptides, the critical nucleus is a special, rare conformation of the monomer (g) that is capable of serving as the template for the initiation of amyloid elongation (h). This paper concerns cases in which the role of the httNT sequence is compromised by virtue of its removal, or by the action of certain httNT-complexation agents. Elsewhere we will describe what happens when httNT function is compromised due to sequence modifications within httNT, such as post-translational modifications (52) (R. Mishra and R. Wetzel, manuscripts in preparation).
Figure 8.

Kinetically competing pathways for nucleation of amyloid formation in polyQ-containing htt fragments. Monomeric fragment “a” (httNT = green, polyQ = orange, proline-rich = black) can enter both Pathway A or Pathway B to nucleate amyloid growth, with relative rates depending on structures and conditions. In Pathway A, “a” can reversibly assemble into tetramer “b”, which can further assemble into octomer “c”, dodecamer “d”, and higher order oligomers (not shown). These oligomers are held together primarily via α-helical packing of their httNT moieties while the polyQ and polyP segments are relatively solvent exposed with no defined structure. Each of “b”, “c” and “d”, as well as higher order oligomers, has a certain propensity to undergo amyloid nucleus (“e”) formation in which some of the attached polyQ sequences assemble into amyloid. Once formed, nucleus “e” can initiate amyloid growth by monomer addition (“f”). In Pathway B, “a” rarely and reversibly undergoes a highly energetically unfavorable folding event to generate a defined conformation or conformations (“g”, the critical nucleus) capable of interacting with the polyQ of another htt fragment to initiate amyloid elongation (“h”). This is a schematic drawing meant to indicate general features of these pathways and structural details, such as the orientation of helices in the tetramer and the packing interfaces in higher oligomers, are yet to be worked out. Likewise, many structural features of the final aggregates of the B pathway and how they relate to the products of the A pathway have not been investigated.
As previously described (18), httNT and related sequences appear to inhibit pathway A by readily forming mixed α-helical oligomers with the polyQ-containing htt N-terminal fragment. This dilutes the local concentration of the disordered polyQ segments and hence retards nucleation. At the same time, inhibition is incomplete, so that nucleation and growth of amyloid do finally occur (18). As we shown here, when pathway A is transiently restricted, pathway B remains open, resulting in a concentration dependence characteristic of the B (and not the A) pathway, and in the formation of polyQ amyloid aggregates that resemble aggregates of simple polyQ peptides both in the EM (Fig. 3E; Fig. S1, I-P) and in their cross-seeding properties (Fig. 2B,C). These data show that the lack of evidence for the simple polyQ aggregation pathway in httNTQN peptides under most circumstances is not because this pathway is inactivated by the httNT moiety, but rather because it is outstripped kinetically by the alternate, httNT-mediated pathway. When the kinetic advantage of the httNT-mediated pathway is compromised, the simple polyQ nucleation and aggregation pathway effectively competes. The kinetic partitioning between these two well-characterized pathways of amyloid nucleation and growth provides a molecular basis for how certain chaperones can influence the course of huntingtin fragment aggregation (57, 60, 61). In Figure 8, the products of pathway B are drawn to indicate the absence of the fully formed httNT α-helix that is known to exist in the final product of pathway A (17, 36). The fate of the httNT segment in these aggregates is unknown, however, and the altered structure is indicated schematically only as a reminder that the final aggregates must be structurally different at some level, since they are functionally different. In fact, the basis for the difference in seeding abilities of the final aggregates of the inhibited and non-inhibited reactions is not clear. This may be a consequence of the amount of httNT inhibitor that appears to be bound to the fibrils (18). Alternatively, it may reflect some very subtle structural difference of the polyQ β-sheet networks of these two fibrils.
Although the VL 12.3 protein also likely inhibits pathway A by specifically complexing httNT and preventing its self-association, there is no evidence for the formation of pathway B products, in contrast to the results of httNT inhibition. Our data suggest that, even after long incubation, the bulk of a mixture of VL 12.3 and htt N-terminal fragment consists of heterodimers of these two proteins, with only a minor portion of the proteins involved in the formation of large oligomers. It might have been expected that the polyQ segment of httNTQ30P6K2 in these heterodimers might be as free to engage the B pathway (Fig. 8) as are httNTQ30P6K2 monomers under httNT inhibition, but there is no evidence that this pathway is open to the VL 12.3/httNTQN heterodimers. This implies that, in spite of the focus of VL 12.3 on the httNT segment, the polyQ sequence in the heterodimer exists in a conformational state that resists the nucleation and/or elongation steps shown in the B pathway.
Since we previously showed that polyQ peptides containing a C-terminal polyPro extension undergo amyloid nucleation by the same mechanism as simple polyQ peptides (62), it is not surprising that the peptide MF-Q37P10K2 exhibits a log-log slope similar to that of K2Q30K2 (Fig. 4). The more rapid aggregation in the 40 μM range of MF-Q37P10K2 compared with Q37P10K2, however, shows that the MF sequence does play a role in aggregation. Just as the Ser-Phe sequence of httNT proximal to the polyQ has been shown to take part in β-sheet formation in the mature amyloid fibrils of htt N-terminal fragments (36), it is possible that the polyQ proximal Met-Phe in MF-Q37P10K2 is included in amyloid structure and by virtue of that might affect nucleation or elongation in a positive way. Regardless of this slight enhancing effect, however, it is also clear that MF-Q37P10K2 aggregates much less rapidly than the corresponding httNTQ37P10K2 (Fig. 7A), and this rate difference is expected to be greatly enhanced at the low concentrations that obtain in the cell, based on a projection to low, cellular concentrations from their disparate concentration dependencies (Fig. 4).
Although the httNT segment of htt has a number of important possible targeting and trafficking roles (63–67), it is clear from these and other studies (16–18, 36, 52, 57) that this flanking sequence of the polyQ also greatly contributes to biophysical properties. The data presented in this paper show that different fates of the httNT segment of polyQ-containing htt fragments can have different consequences, not only for aggregation rates, but also for the mechanism of aggregation and hence for the structure and properties of the aggregate products. Thus, httNT inhibition of httNTQN aggregation produces amyloid aggregates that are much more capable of recruiting other polyQ proteins into the growing aggregate than are the amyloid products of non-inhibited aggregation reactions (Fig. 2C). This observation is relevant to the recent description of the seeding ability of aggregates isolated from various HD tg mice (20), and to recruitment-sequestration models of polyQ toxicity (21, 22). It remains to be seen whether there exist molecules in the cell capable of producing httNT-like inhibitory effects, but certain molecular chaperones are attractive candidates for such molecules (57, 60, 61). In contrast to httNT, the VL 12.3 protein completely blocks amyloid formation in vitro, instead generating a mixture of heterodimers and non-amyloid oligomers. Given this result, the observation that this protein in cell culture inhibits both inclusion formation and cytotoxicity (68) is most consistent with the hypothesis, discussed above, that it is the amyloid fibril form of polyQ-containing htt fragments that are toxic, while non-amyloid oligomers are benign (18). Given that the removal of the httNT segment from htt N-terminal fragments slows but does not abrogate polyQ amyloid formation, while completely eliminating the ability of such fragments to make non-amyloid oligomers, it will be interesting to consider the effects of this modification on disease course and neuronal pathology observed in ongoing tg mouse experiments (Gu et al., Ms. in preparation).
Supplementary Material
Acknowledgments
Funding support from the National Institutes of Health, R01 AG019322 (to R.W.) and SAP #4100026429 from the Commonwealth of Pennsylvania (to A.M.G.).
We gratefully acknowledge Dane Wittrup (MIT) for the gift of the cDNA encoding the αHtt-VL 12.3 domain protein, and discussions with William Yang (UCLA). We thank Bart Roland for providing some of the aggregation kinetics data, and Drs. James Conway and Alexander Makhov for helpful discussions and access to the Structural Biology Department’s cryo-EM facility.
ABBREVIATIONS AND TEXTUAL FOOTNOTES
- polyQ
polyglutamine
- htt
huntingtin
- httNT
17 amino acid N-terminus of huntingtin preceding the polyQ sequence
- anti-httNT VL 12.3 protein
immunoglobulin light chain variable domain-derived httNT binding protein
- HD
Huntington’s disease
Footnotes
SUPPORTING INFORMATION AVAILABLE
A figure containing additional EM images of aggregates, as well as a brief discussion of the provenance and significance of these aggregates, is included in a single pdf file as supporting information. These data characterize in more detail the appearance of “simple polyQ-type” aggregates as intermediates in httNT-inhibited reactions of httNTQ30P6K2 peptides. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bates GP, Benn C. The polyglutamine diseases. In: Bates GP, Harper PS, Jones L, editors. Huntington’s Disease. Oxford University Press; Oxford, U.K: 2002. pp. 429–472. [Google Scholar]
- 2.Wetzel R. Physical chemistry of polyglutamine: Intriguing tales of a monotonous sequence. J Mol Biol. 2012 doi: 10.1016/j.jmb.2012.01.030. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci U S A. 1999;96:4604–4609. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–182. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 5.Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu XF, Greiner E, Park CS, Wang N, Sopher BL, La Spada AR, Osmand A, Margolis RL, Sun YE, Yang XW. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron. 2011;70:427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer’s peptide Ab. J Mol Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- 7.Helms LR, Wetzel R. Specificity of abnormal assembly in immunoglobulin light chain deposition disease and amyloidosis. J Mol Biol. 1996;257:77–86. doi: 10.1006/jmbi.1996.0148. [DOI] [PubMed] [Google Scholar]
- 8.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- 9.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 10.Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- 11.Kodali R, Wetzel R. Polymorphism in the intermediates and products of amyloid assembly. Curr Opin Struct Biol. 2007;17:48–57. doi: 10.1016/j.sbi.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 12.Ross CA, Poirier MA. Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- 13.Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, Bates GP. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen S, Ferrone F, Wetzel R. Huntington’s Disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci USA. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328–336. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, Anjum DH, Kodali R, Creamer TP, Conway JF, Gronenborn AM, Wetzel R. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–389. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R. Slow amyloid nucleation via α-helix-rich oligomeric intermediates in short polyglutamine-containing Huntingtin fragments. J Mol Biol. 2012;415:881–899. doi: 10.1016/j.jmb.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mishra R, Jayaraman M, Roland BP, Landrum E, Fullam R, Kodali R, Thakur AK, Arduini I, Wetzel R. Inhibiting nucleation of amyloid structure in a huntingtin fragment by targeting α-helix rich oligomeric intermediates. J Mol Biol. 2012;415:900–917. doi: 10.1016/j.jmb.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc Natl Acad Sci U S A. 2009;106:9679–9684. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta S, Jie S, Colby DW. Protein misfolding detected early in the pathogenesis of a transgenic mouse model of Huntington’s disease using an amyloid seeding assay. J Biol Chem. 2011 doi: 10.1074/jbc.M111.305417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 22.McCampbell A, Fischbeck KH. Polyglutamine and CBP: fatal attraction? Nat Med. 2001;7:528–530. doi: 10.1038/87842. [DOI] [PubMed] [Google Scholar]
- 23.Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 24.Poirier MA, Jiang H, Ross CA. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum Mol Genet. 2005;14:765–774. doi: 10.1093/hmg/ddi071. [DOI] [PubMed] [Google Scholar]
- 25.Zhang QC, Yeh TL, Leyva A, Frank LG, Miller J, Kim YE, Langen R, Finkbeiner S, Amzel ML, Ross CA, Poirier MA. A compact beta model of huntingtin toxicity. J Biol Chem. 2011;286:8188–8196. doi: 10.1074/jbc.M110.192013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, Thiagarajan G, Wetzel R. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Berthelier V, Hamilton JB, O’Nuallain B, Wetzel R. Amyloid-like features of polyglutamine aggregates and their assembly kinetics. Biochemistry. 2002;41:7391–7399. doi: 10.1021/bi011772q. [DOI] [PubMed] [Google Scholar]
- 28.Jayaraman M, Thakur AK, Kar K, Kodali R, Wetzel R. Assays for studying nucleated aggregation of polyglutamine proteins. Methods. 2011;53:246–254. doi: 10.1016/j.ymeth.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zuccato C, Valenza M, Cattaneo E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol Rev. 2010;90:905–981. doi: 10.1152/physrev.00041.2009. [DOI] [PubMed] [Google Scholar]
- 30.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 31.Faber PW, Alter JR, MacDonald ME, Hart AC. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci U S A. 1999;96:179–184. doi: 10.1073/pnas.96.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci U S A. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Apostol BL, Kazantsev A, Raffioni S, Illes K, Pallos J, Bodai L, Slepko N, Bear JE, Gertler FB, Hersch S, Housman DE, Marsh JL, Thompson LM. A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc Natl Acad Sci U S A. 2003;100:5950–5955. doi: 10.1073/pnas.2628045100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington’s disease. Neurobiol Dis. 2004;16:546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 35.Wang CE, Tydlacka S, Orr AL, Yang SH, Graham RK, Hayden MR, Li S, Chan AW, Li XJ. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington’s disease. Hum Mol Genet. 2008;17:2738–2751. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PC. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J Am Chem Soc. 2011;133:4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 38.Krebs MR, Morozova-Roche LA, Daniel K, Robinson CV, Dobson CM. Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci. 2004;13:1933–1938. doi: 10.1110/ps.04707004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colby DW, Garg P, Holden T, Chao G, Webster JM, Messer A, Ingram VM, Wittrup KD. Development of a human light chain variable domain (V(L)) intracellular antibody specific for the amino terminus of huntingtin via yeast surface display. J Mol Biol. 2004;342:901–912. doi: 10.1016/j.jmb.2004.07.054. [DOI] [PubMed] [Google Scholar]
- 40.Schiefner A, Chatwell L, Korner J, Neumaier I, Colby DW, Volkmer R, Wittrup KD, Skerra A. A Disulfide-Free Single-Domain V(L) Intrabody with Blocking Activity towards Huntingtin Reveals a Novel Mode of Epitope Recognition. J Mol Biol. 2011;414:337–355. doi: 10.1016/j.jmb.2011.09.034. [DOI] [PubMed] [Google Scholar]
- 41.Southwell AL, Bugg CW, Kaltenbach LS, Dunn D, Butland S, Weiss A, Paganetti P, Lo DC, Patterson PH. Perturbation with intrabodies reveals that calpain cleavage is required for degradation of huntingtin exon 1. PLoS ONE. 2011;6:e16676. doi: 10.1371/journal.pone.0016676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stefani M. Structural polymorphism of amyloid oligomers and fibrils underlies different fibrillization pathways: immunogenicity and cytotoxicity. Curr Protein Pept Sci. 2010;11:343–354. doi: 10.2174/138920310791330631. [DOI] [PubMed] [Google Scholar]
- 43.Toyama BH, Weissman JS. Annual Review of Biochemistry. Vol. 80. Annual Reviews; Palo Alto: 2011. Amyloid Structure: Conformational Diversity and Consequences; pp. 557–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldsbury CS, Wirtz S, Muller SA, Sunderji S, Wicki P, Aebi U, Frey P. Studies on the in vitro assembly of a beta 1–40: implications for the search for a beta fibril formation inhibitors. J Struct Biol. 2000;130:217–231. doi: 10.1006/jsbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 46.Paravastua AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci U S A. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kodali R, Williams AD, Chemuru S, Wetzel R. A beta(1–40) Forms Five Distinct Amyloid Structures whose beta-Sheet Contents and Fibril Stabilities Are Correlated. Journal of Molecular Biology. 2010;401:503–517. doi: 10.1016/j.jmb.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wetzel R, Shivaprasad S, Williams AD. Plasticity of amyloid fibrils. Biochemistry. 2007;46:1–10. doi: 10.1021/bi0620959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 50.Kumar S, Udgaonkar JB. Conformational conversion may precede or follow aggregate elongation on alternative pathways of amyloid protofibril formation. J Mol Biol. 2009;385:1266–1276. doi: 10.1016/j.jmb.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 51.Kumar S, Udgaonkar JB. Structurally distinct amyloid protofibrils form on separate pathways of aggregation of a small protein. Biochemistry. 2009;48:6441–6449. doi: 10.1021/bi900682w. [DOI] [PubMed] [Google Scholar]
- 52.Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S, Steffan JS, Thompson LM, Wetzel R, Yang XW. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron. 2009;64:828–840. doi: 10.1016/j.neuron.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Furukawa Y, Kaneko K, Yamanaka K, Nukina N. Mutation-dependent polymorphism of Cu,Zn-superoxide dismutase aggregates in the familial form of amyotrophic lateral sclerosis. J Biol Chem. 2010;285:22221–22231. doi: 10.1074/jbc.M110.113597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ding F, Furukawa Y, Nukina N, Dokholyan NV. Local Unfolding of Cu, Zn Superoxide Dismutase Monomer Determines the Morphology of Fibrillar Aggregates. J Mol Biol. 2011 doi: 10.1016/j.jmb.2011.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldsbury C, Frey P, Olivieri V, Aebi U, Muller SA. Multiple assembly pathways underlie amyloid-beta fibril polymorphisms. J Mol Biol. 2005;352:282–298. doi: 10.1016/j.jmb.2005.07.029. [DOI] [PubMed] [Google Scholar]
- 56.Thakur A, Wetzel R. Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci U S A. 2002;99:17014–17019. doi: 10.1073/pnas.252523899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tam S, Spiess C, Auyeung W, Joachimiak L, Chen B, Poirier MA, Frydman J. The chaperonin TRiC blocks a huntingtin sequence element that promotes the conformational switch to aggregation. Nat Struct Mol Biol. 2009;16:1279–1285. doi: 10.1038/nsmb.1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williamson TE, Vitalis A, Crick SL, Pappu RV. Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin. J Mol Biol. 2010;396:1295–1309. doi: 10.1016/j.jmb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fiumara F, Fioriti L, Kandel ER, Hendrickson WA. Essential role of coiled coils for aggregation and activity of Q/N-rich prions and PolyQ proteins. Cell. 2010;143:1121–1135. doi: 10.1016/j.cell.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- 61.Tam S, Geller R, Spiess C, Frydman J. The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat Cell Biol. 2006;8:1155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, Creamer TP, Wetzel R. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–535. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 63.Steffan JS, Agrawal N, Pallos J, Rockabrand E, Trotman LC, Slepko N, Illes K, Lukacsovich T, Zhu YZ, Cattaneo E, Pandolfi PP, Thompson LM, Marsh JL. SUMO modification of Huntingtin and Huntington’s disease pathology. Science. 2004;304:100–104. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 64.Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 65.Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, Sullivan PG, Steffan JS, Sensi SL, Thompson LM. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16:61–77. doi: 10.1093/hmg/ddl440. [DOI] [PubMed] [Google Scholar]
- 66.Thompson LM, Aiken CT, Kaltenbach LS, Agrawal N, Illes K, Khoshnan A, Martinez-Vincente M, Arrasate M, JGOS-R, Khashwji H, Lukacsovich T, Zhu YZ, Lau AL, Massey A, Hayden MR, Zeitlin SO, Finkbeiner S, Green KN, Laferla FM, Bates G, Huang L, Patterson PH, Lo DC, Cuervo AM, Marsh JL, Steffan JS. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J Cell Biol. 2009;187:1083–1099. doi: 10.1083/jcb.200909067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Atwal RS, Desmond CR, Caron N, Maiuri T, Xia JR, Sipione S, Truant R. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol. 2011;7:453–460. doi: 10.1038/nchembio.582. [DOI] [PubMed] [Google Scholar]
- 68.Colby DW, Chu Y, Cassady JP, Duennwald M, Zazulak H, Webster JM, Messer A, Lindquist S, Ingram VM, Wittrup KD. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc Natl Acad Sci U S A. 2004;101:17616–17621. doi: 10.1073/pnas.0408134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.