

Abstract
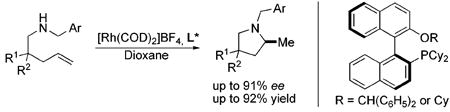
Metal-catalyzed enantioselective intramolecular hydroamination of olefins1 is one of the most conceptually simple and atom-economical approaches to the construction of enantioenriched nitrogen heterocycles,2 which are valuable synthons in the preparation of natural products and biologically active molecules.3 Despite significant advances,4,5,6 no general solution for this challenging transformation has yet been realized. Some of the most reactive catalysts are based on lanthanides5 and group 4 transition metals,6 but only a small number of highly enantioselective (>90% ee) reactions have been described.5a, 6b, 6c Noteworthy is Schafer’s6c elegant report of chiral zirconium amidate complexes which provide good enantioselectivity for a range of terminal alkenes, but require a geminal dialkyl group7 to bias the substrates toward cyclization. Furthermore, the sensitivity of rare earth and group 4 metal catalysts toward air and moisture and their low functional group tolerance have limited their synthetic utility and motivated efforts to develop alternative systems. Some late transition metal complexes have been investigated for enantioselective hydroamination, with good results for alkynes,8 allenes,9 dienes,10 and vinylarenes.11 Very recently one example of an intermolecular hydroamination of unactivated 1-alkenes with an ee up to 78% was reported.12,13 To the best of our knowledge, however, there are no examples of late transition metal-catalyzed enantioselective intramolecular hydroamination of unactivated olefins. Herein we disclose the first example of a rhodium-catalyzed14 asymmetric intramolecular addition of amines to olefins for the synthesis of a variety of enantioenriched 2-methylpyrrolidines with good enantioselectivity (up to 91% ee).
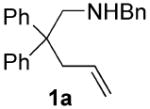
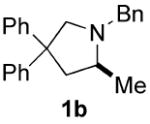
Recent reports of the successful application of the dialkylbiaryl phosphines DavePhos15a, t-BuDavePhos15b and JohnPhos15c in rhodium-, platinum- and gold-catalyzed intramolecular hydroamination by Hartwig14a and Widenhoefer16,17 respectively, stimulated us to develop a late transition metal-catalyzed enantioselective version of this process using chiral ligands of the type developed in our laboratory.15 We began by examining the cyclization of N-benzyl-2,2-diphenyl-4-pentenamine 1a to 1-benzyl-2-methyl-4,4-diphenylpyrrolidine 1b as a test reaction (eq. 1). Following the seminal work of Hartwig,14a we exposed 1a to 5 mol% [Rh(COD)2]BF4 and 6 mol% KenPhos L1,18 providing pyrrolidine 1b in 95% yield (GC) and 38% ee (entry 1). Varying the size of the substituent on nitrogen or phosphorus provided ligands19,20 that gave the product in good yield but poor ee (entries 2 and 4). Control experiments in which the metal was omitted or replaced by a protic acid resulted in no product formation. No improvement in yield or enantioselectivity was seen if alternative Rh sources were employed (see supporting information).
We next turned our attention to MOP21 derivatives, the use of Cy-MOP L6,22 afforded 1b in 96% yield with 53% ee (entry 6). Given this promising result, several Cy-MOP-type ligands were prepared using a slight modification of the literature procedure.22 The selectivity of the hydroamination reaction improved with increasing size of the oxygen substituent (entries 6-8). We found that the dialkylphosphino group was essential for high activity; attempts to employ the diphenylphosphine L10 furnished <10% yield of product (entry 10). Among the ligands evaluated, L9 with a (diphenyl)methyl-substituted alkoxy group provided the best combination of reactivity and enantioselectivity, affording 1b in 95% yield and 80% ee (entry 9).
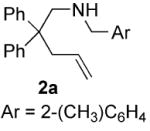
The scope of the reaction is summarized in Table 2. Cyclization of substrates with geminal substitution in the homoallylic position proved to be the most facile, the reactions proceeding in the highest yields and with good enantioselectivities. Typically these reactions could be carried out at 70 °C with 5 mol% Rh. However, for the easiest substrates the reaction still proceeded at an acceptable rate at 50 °C, affording a slight increase in enantioselectivity (entry 2 and 5). In the case of substrates lacking substitution that facilitate cyclization, the use of the less sterically demanding ligand L8 was more efficient. Using a catalyst system based on L8, these substrates could be cyclized with comparable enantioselectivity, although in somewhat lower yield (entries 10-13). These results are some of the best reported for the enantioselective cyclization of unbiased substrates using a transition metal-based catalyst.5a,5d In the case of racemic N-2-methylbenzyl-2-phenyl-4-pentenamine 5a no kinetic resolution was observed, the diastereomeric products were obtained in a 1.1:1 ratio with high enantiomeric excess (87% and 91% ee respectively, entry 8).
Table 2.
Rhodium-Catalyzed Enantioselective Intramolecular Hydroamination of Aminoalkenes[a]
| entry | alkenyl amine | product | T(°C) | t(h) | yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1 |
|
|
70 | 15 | 91 | 80(S) |
| 2 | 50 | 24 | 90 | 83(S) | ||
| 3 |
|
|
70 | 15 | 92 | 84(S) |
| 4d | 70 | 20 | 88 | 84(S) | ||
| 5 | 50 | 24 | 91 | 88(S) | ||
| 6 |
|
|
70 | 20 | 75 | 62(S) |
| 7 |
|

|
70 | 20 | 80 | 63(S) |
| 8e |
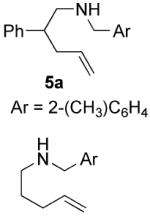
|
|
70 | 20 | 80 (1.1:1) | 87(2S, 4S) |
| 91(2S, 4S) | ||||||
| 9f | Ar = C6H5(6a) | 6b | 70 | 20 | 48 | 90(S) |
| 10g | Ar = 2-(CH3)C6H4(7a) | 7b | 70 | 20 | 50 | 86(S) |
| 11g | Ar = 4-(Cl)C6H4 (8a) | 8b | 70 | 20 | 63 | 85(S) |
| 12g | Ar = 4-(MeO)C6H4(9a) | 9b | 70 | 30 | 35 | 85(S) |
| 13g | Ar = 4-(CO2Me)C6H4 (10a) | 10b | 70 | 20 | 61 | 83(S) |
| 14h |

|

|
100 | 10 | 85 | 64(S) |
Reaction conditions: 0.5 mmol alkenyl amine, 5 mol% of [Rh(COD)2]BF4, 6 mol% of ligand L9, dioxane (0.5 mL).
Isolated yields (average of two runs).
The ee values were determined by chiral HPLC, GC or 1H NMR analysis of its derivatives. The absolute configuration of 1b was determined by converting it to the known (S)-MTPA amide. The configuration of 6b, 7b, 8b, 9b and 10b were determined by converting them to the known N-α-naphthyl amide. The configuration of 11b was determined by converting it to the known N-acetyl-2-methylindoline. See supporting information for details. The configurations of other amines were assigned by analogy.
2.5 mol% Rh and 3 mol% ligand L9.
Diastereomeric ratio given in parentheses.
10 mol% Rh and 12 mol% ligand L9.
5 mol% Rh and 6 mol% ligand L8.
Yield of isolated product following derivatization with acetic anhydride.
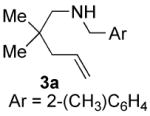
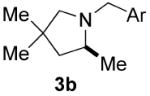
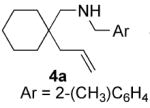
The nature of the protecting group on nitrogen had a pronounced influence on the outcome of the reaction. We found that N-(2-methyl)benzyl aminoolefins gave higher enantioselectivity than N-benzyl aminoolefins in some cases, without adversely effecting the yield (entries 3-8). Presumably the substituent at the 2-position results in a more ordered transition state. Increasing the size of this substituent or using a 2,6-dimethylbenzyl protecting group, however, elicited a profound decrease in reactivity, perhaps as a result of inhibition of metal binding. Varying the para-substituent on the aryl ring of the nitrogen protecting group had little effect on the enantioselectivity, but electron-donating substituents retarded the reaction and resulted in a lower yield. 23 Typically, unprotected aminoolefins exhibited poor reactivity, however, 2-allylaniline did undergo hydroamination to yield 2-methylindoline with moderate enantioselectivity (entry 14).
Functional groups including esters and aryl chlorides were tolerated under the reaction conditions rendering the products amenable to further synthetic manipulation (entry 11 and 13). The absolute configuration of the products was determined to be (S) by conversion of 1b to the known (S)-MPTA amide,6c 6b-10b to the known N-naphthyl amides5e and 11b to the previously reported N-acetyl-2-methylindoline. 24
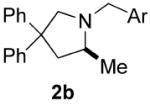
The N-protecting groups on the products in Table 2 were easily removed to reveal the free amines via catalytic transfer hydrogenation25 with ammonium formate as hydrogen source. For example, treatment of the cyclized product 2b (Table 2, entry 5) with palladium on activated carbon and ammonium formate at 68 °C in methanol afforded the 2-methylpyrrolidine 2c in 92% yield.
 |
In summary, we have developed the first enantioselective rhodium-catalyzed intramolecular hydroamination of unactivated terminal alkenes. A variety of enantioenriched pyrrolidines have been synthesized in up to 91% ee using a binaphthyl-based electron-rich phosphine ligand. Further investigations to increase the generality of the process and to expand the scope to include intermolecular reactions are currently underway in our laboratories.
Experimental Section
Typical procedure: The aminoalkene (0.50 mmol) was added to a dry, screw-capped test tube at room temperature and the tube was then transferred into a glovebox. At this point, [Rh(COD)2]BF4 (0.025 mmol, 10.2 mg), phosphine ligand (0.030 mmol) and 0.5 mL of dioxane were added. The vial was sealed with a PTFE cap and removed from the glovebox. The reaction mixture was stirred at the temperature and for the time shown in Table 2. The reaction mixture was allowed to cool to ambient temperature, brine (20 mL) was added and the resulting mixture was extracted with CH2Cl2 (2 × 20 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo. The crude product mixture was purified by flash chromatography on silica gel.
Supplementary Material
Table 1.
Effect of Ligand on the Enantioselectivity for Intramolecular Hydroamination[a]

| |||
|---|---|---|---|
| entry | ligand | yield [%][b] | ee [%][c] |
| 1 | L1 | 95 | 38 (S) |
| 2 | L2 | 83 | 20 (S) |
| 3 | L3 | 8 | n. d. |
| 4 | L4 | 95 | 39 (S) |
| 5 | L5 | 94 | 42 (S) |
| 6 | L6 | 96 | 53 (S) |
| 7 | L7 | 98 | 70 (S) |
| 8 | L8 | 95 | 75 (S) |
| 9 | L9 | 94 | 80 (S) |
| 10 | L10 | 9 | n. d. |
Reaction conditions: aminoolefin (0.15 mmol), 5 mol% Rh and 6 mol% ligand in 0.15 mL of dioxane at 70 °C for 7-15 h.
GC yields.
The ee values were determined by chiral HPLC analysis on a Chiralcel OJ column. The absolute configuration was determined by converting 1b to the known (S)-MTPA amide. See supporting information for details.
Footnotes
Generous financial support from the National Institutes of Health (GM46059) is gratefully acknowledged. We thank Merck and Boehringer-Ingelheim for additional funds. The Varian 300 MHz used in this work was purchased with funding from the National Institutes of Health (GM 1S10RR13886-01). We are grateful to Dr. David S. Surry (MIT) for initial studies on asymmetric hydroamination process and help in the preparation of the manuscript.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For recent reviews on asymmetric hydroamination, see: Chemler SR. Org Biomol Chem. 2009;7:3009–3019. doi: 10.1039/B907743J.; Muller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795–3892. doi: 10.1021/cr0306788.; Aillaud I, Collin J, Hannedouche J, Schulz E. Dalton Trans. 2007:5105–5118. doi: 10.1039/b711126f.; Hii KK. Pure Appl Chem. 2006;78:341–349.; Hultzsch KC. Adv Synth Catal. 2005;347:367–391.; Hultzsch KC. Org Biomol Chem. 2005;3:1819–1824. doi: 10.1039/b418521h.; Hong S, Marks TJ. Acc Chem Res. 2004;37:673–686. doi: 10.1021/ar040051r.; Roesky PW, Muller TE. Angew Chem Int Ed. 2003;42:2708–2710. doi: 10.1002/anie.200301637.
- 2.Royer J, editor. Asymmetric Synthesis of Nitrogen Heterocycles. Wiley-VCH; Weinheim: 2009. [Google Scholar]
- 3.a) O’Hagan D. Nat Prod Rep. 2000;17:435–446. doi: 10.1039/a707613d. [DOI] [PubMed] [Google Scholar]; b) Mitchinson A, Nadin A. J Chem Soc Perkin Trans 1. 2000;1:2862–2892. [Google Scholar]; c) King AG, Meinwald J. Chem Rev. 1996;96:1105–1122. doi: 10.1021/cr950242v. [DOI] [PubMed] [Google Scholar]
- 4.For examples of hydroamination using lithium complexes, see: Martinez PH, Hultzsch KC, Hampel F. Chem Commun. 2006:2221–2223. doi: 10.1039/b518360j.. Ogata T, Ujihara A, Tsuchida S, Shimizu T, Kaneshige A, Tomioka K. Tetrahedron Lett. 2007;48:6648–6650.
- 5.For recent reports of hydroamination using lanthanide complexes, see: Gribkov DV, Hultzsch KC, Hampel F. J Am Chem Soc. 2006;128:3748–3759. doi: 10.1021/ja058287t.; Riegert D, Collin J, Meddour A, Schulz E, Trifonov A. J Org Chem. 2006;71:2514–2517. doi: 10.1021/jo052322x.; Collin J, Daran J, Jacquet O, Schulz E, Trifonov A. Chem Eur J. 2005;11:3455–3462. doi: 10.1002/chem.200401203.; Kim JY, Livinghouse T. Org Lett. 2005;7:1737–1739. doi: 10.1021/ol050294z.; Hong S, Tian S, Metz MV, Marks TJ. J Am Chem Soc. 2003;125:14768–14783. doi: 10.1021/ja0364672.
- 6.For recent reports of hydroamination using group 4 complexes, see: Zi G, Liu X, Xiang L, Song H. Organometallics. 2009;28:1127–1137.; Gott AL, Clarke AJ, Clarkson GJ, Scott P. Organometallics. 2007;26:1729–1737.; Wood MC, Leitch DC, Yeung CS, Kozak JA, Schafer LL. Angew Chem Int Ed. 2007;46:354–358. doi: 10.1002/anie.200603017.; Watson DA, Chiu M, Bergman RG. Organometallics. 2006;25:4731–4733. doi: 10.1021/om0606791.; Knight PD, Munslow L, O’Shaughnessy PN, Scott P. Chem Commun. 2004:894–895. doi: 10.1039/b401493f.
- 7.For a review on the gem-dialkyl effect, see: Jung ME, Pizzi G. Chem Rev. 2005;105:1735–1766. doi: 10.1021/cr940337h.
- 8.For asymmetric intramolecular hydroamination of alkynes: Luttete LM, Kadota I, Yamamoto Y. J Am Chem Soc. 2004;126:1622–1623. doi: 10.1021/ja039774g.; Patil NT, Luttete LM, Wu H, Pahadi NK, Gridnev ID, Yamamoto Y. J Org Chem. 2006;71:4270–4279. doi: 10.1021/jo0603835.
- 9.For selected examples of asymmetric hydroamination of allenes: LaLonde RL, Sherry BD, Kang EJ, Toste FD, X J Am Chem Soc. 2007;129:2452–2453. doi: 10.1021/ja068819l., Hamilton GL, Kang EJ, Mba M, Toste FD. Science. 2007;317:496–499. doi: 10.1126/science.1145229.; Zhang Z, Liu C, Kinder RE, Han X, Qian H, Widenhoefer RA. J Am Chem Soc. 2006;128:9066–9073. doi: 10.1021/ja062045r.; Nishina N, Yamamoto Y. Angew Chem Int Ed. 2006;45:3314–3317. doi: 10.1002/anie.200600331.
- 10.Lober O, Kawatsura M, Hartwig JF. J Am Chem Soc. 2001;123:4366–4367. doi: 10.1021/ja005881o. [DOI] [PubMed] [Google Scholar]
- 11.For selected examples of asymmetric hydroamination of vinylarenes: Hu A, Ogasawara M, Sakamoto T, Okada A, Nakajima K, Takahashi T, Lin W. Adv Synth Catal. 2006;348:2051–2056.; Utsunomiya M, Hartwig JF. J Am Chem Soc. 2003;125:14286–14287. doi: 10.1021/ja0375535.; Kawatsura M, Hartwig JF. J Am Chem Soc. 2000;122:9546–9547.
- 12.Zhang Z, Lee SD, Widenhoefer RA. J Am Chem Soc. 2009;131:5372–5373. doi: 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For asymmetric addition of aromatic amines to bicyclic olefins, see: Dorta R, Egli P, Zurcher F, Togni A. J Am Chem Soc. 1997;119:10857–10858.; Zhou J, Hartwig JF. J Am Chem Soc. 2008;130:12220–12221. doi: 10.1021/ja803523z.
- 14.For selected examples of rhodium-catalyzed non enantioselective hydroamination of olefins: Liu Z, Hartwig JF. J Am Chem Soc. 2008;130:1570–1571. doi: 10.1021/ja710126x.; Takemiya A, Hartwig JF. J Am Chem Soc. 2006;128:6042–6043. doi: 10.1021/ja058299e.; Utsunomiya M, Kuwano R, Kawatsura M, Hartwig JF. J Am Chem Soc. 2003;125:5608–5609. doi: 10.1021/ja0293608.
- 15.a) Old DW, Wolfe JP, Buchwald SL. J Am Chem Soc. 1998;120:9722–9723. [Google Scholar]; b) Aranyos A, Old DW, Kiyomori A, Wolfe JP, Sadighi JP, Buchwald SL. J Am Chem Soc. 1999;121:4369–4378. [Google Scholar]; c) Wolfe JP, Buchwald SL. Angew Chem Int Ed. 1999;38:2413–2416. doi: 10.1002/(sici)1521-3773(19990816)38:16<2413::aid-anie2413>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]; d) Martin R, Buchwald SL. Acc Chem Res. 2008;41:1461–1473. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Surry DS, Buchwald SL. Angew Chem Int Ed. 2008;47:6338–6361. doi: 10.1002/anie.200800497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Bender CF, Hudson WB, Widenhoefer RA. Organometallics. 2008;27:2356–2358. [Google Scholar]; b) Bender CF, Widenhoefer RA. J Am Chem Soc. 2005;127:1070–1071. doi: 10.1021/ja043278q. [DOI] [PubMed] [Google Scholar]
- 17.Han X, Widenhoefer RA. Angew Chem Int Ed. 2006;45:1747–1749. doi: 10.1002/anie.200600052. [DOI] [PubMed] [Google Scholar]
- 18.Jin Y, Buchwald SL. J Am Chem Soc. 2000;122:12051–12052. [Google Scholar]
- 19.Chieffi A, Kamikawa K, Ahman J, Fox JM, Buchwald SL. Org Lett. 2001;3:1897–1900. doi: 10.1021/ol0159470. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Fortanet J, Kessler F, Buchwald SL. J Am Chem Soc. 2009;131:6676–6677. doi: 10.1021/ja9025193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For chiral monodentate phosphine ligand MOP in asymmetric synthesis, see: Hayashi T. Acc Chem Res. 2000;33:354–362. doi: 10.1021/ar990080f.
- 22.Hamada T, Chieffi A, Ahman J, Buchwald SL. J Am Chem Soc. 2002;124:1261–1268. doi: 10.1021/ja011122+. [DOI] [PubMed] [Google Scholar]
- 23.Enantioenriched 2-methylpiperidine can be synthesized by this method in moderate yield and ee. For example, cyclization of N-benzyl-5-hexen-1-amine is promoted by 10 mol% [Rh(COD)2]BF4 and 12 mol% L8 in 42% yield and 32% ee.
- 24.Arp FO, Fu GC. J Am Chem Soc. 2006;128:14264–14265. doi: 10.1021/ja0657859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Ram S, Spicer LD. Tetrahedron Lett. 1987;28:515–516. [Google Scholar]; b) Bieg T, Szeja W. Synthesis. 1985:76–77. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.