

Abstract
The majority of human colorectal cancers (CRCs) are initiated by mutations arising in the adenomatous polyposis coli (APC) tumour suppressor gene. However, a new class of non-APC mutated CRCs has been defined that have a serrated histopathology and carry the V600EBRAF oncogene. Here we have investigated the pathogenesis of serrated CRCs by expressing V600EBraf in the proliferative cells of the mouse gastrointestinal tract. We show that the oncogene drives an initial burst of Mek-dependent proliferation, leading to the formation of hyperplastic crypts. This is associated with β-catenin nuclear localization by a mechanism involving Mapk/Erk kinase (Mek)-dependent, Akt-independent phosphorylation of Gsk3β. However, hyperplastic crypts remain dormant for prolonged periods due to the induction of crypt senescence accompanied by upregulation of senescence-associated β-galactosidase and p16Ink4a. We show that tumour progression is associated with down-regulation of p16Ink4a through enhanced CpG methylation of exon 1 and knockout of Cdkn2a confirms this gene is a barrier to tumour progression. Our studies identify V600EBRAF as an early genetic driver mutation in serrated CRCs and indicate that, unlike APC-mutated cancers, this subtype arises by the bypassing of a V600EBraf driven oncogene-induced senescence programme.
Keywords: Braf, intestine, mouse model, senescence, serrated colorectal cancer
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide with ∼655,000 new cases being diagnosed each year. In recent years it has become clear that CRC is not a homogenous disease but can evolve through multiple pathways (Jass, 2007b). The most common pathway is through activation of the Wnt signalling pathway, normally through mutation of the tumour suppressor gene APC (Segditsas & Tomlinson, 2006). The concept of a second ‘serrated neoplasia pathway’ has gained growing acceptance in recent years and refers to a pathway involving the progression from hyperplastic polyps to serrated adenomas and serrated carcinomas (Hawkins et al, 2002; Jass, 2007a; Makinen, 2007). The existence of this pathway was initially suggested on histopathological grounds (Jass et al, 1999), but the discovery of biological and genetic similarities in lesions of this pathway has now provided convincing proof for its existence (Hawkins et al, 2002). Many sporadic tumours of the serrated pathway show a distinct molecular feature of somatic BRAF mutation concordant with a high CpG island methylation phenotype (CIMP-H) and microsatellite instability (MSI+) associated with mutt homologue 1 (MLH1) methylation and for the most part are independent of APC and TP53 mutations (Jass, 2007a). V600EBRAF mutations have been detected in a high proportion of serrated hyperplastic polyps, the proposed earliest lesions of this pathway, suggesting that this oncogene is a founder mutation in this disease (Beach et al, 2005; Chan et al, 2003; Yang et al, 2004).
BRAF is a serine-threonine protein kinase component of the RAS/RAF/MEK/extracellular signal-regulated kinase (ERK) signalling pathway and is activated by somatic mutations, giving rise to gain of function in human cancers (Davies et al, 2002; Garnett & Marais, 2004; Mercer & Pritchard, 2003). Melanoma is the cancer type in which BRAF is mutated most frequently, being present in 50–70% of samples. The V600EBRAF substitution is the most common BRAF mutation in human cancers, including serrated CRCs. In melanomas, this mutation gives rise to constitutive signalling through the MEK/ERK cascade and leads to increased proliferation, transformation and tumourigenicity, characteristics that can all be suppressed by V600EBRAF inhibition (Hingorani et al, 2003; Karasarides et al, 2004; Wellbrock et al, 2004). Despite this evidence for a role in melanoma progression, V600EBRAF is also associated with melanocyte senescence. It is detected in up to 80% of human nevi that remain unchanged for many years (Pollock et al, 2003) and, when over-expressed in human melanocytes in vitro (Michaloglou et al, 2005) and zebrafish melanocytes in vivo (Patton et al, 2005), V600EBraf can induce classical hallmarks of oncogene-induced senescence. Recent studies of a V600EBraf-driven mouse model of melanoma have clarified this dual role by showing, on the one hand, that V600EBRAF is a founder mutation that can drive nevi formation and melanocyte senescence and, on the other, that it can progress melanomas once senescence is surpassed (Dhomen et al, 2009). Although a senescence phenotype has not been previously associated with the development and/or attenuation of traditional CRCs, p16INK4A promoter methylation is a frequent characteristic of serrated carcinomas suggesting that epigenetic inactivation of this senescent marker could be a feature of disease progression (O'Brien et al, 2006; Weisenberger et al, 2006; Yang et al, 2004).
The purpose of this work was to investigate the role of V600EBRAF in the development and progression of CRC. For this reason we used our previously developed Cre-lox-regulated knockin mouse strain in which V600EBraf is expressed from the endogenous Braf gene (Mercer et al, 2005) combined with the AhcreERT mouse strain that allows expression of the Cre recombinase in the proliferative cells of the intestinal crypt (Ireland et al, 2004; Kemp et al, 2004). We show that V600EBraf, through activation of the Mek/Erk pathway, can directly drive the formation of hyperplastic crypts and a serrated epithelium by inducing crosstalk with the Wnt signalling pathway at the level of Gsk3β. Furthermore, we provide evidence that V600EBraf can invoke a senescent programme in the intestine through the upregulation of p16Ink4a and that p16Ink4a down-regulation is necessary for tumour progression. Specifically we show this associated with increased expression of the de novo DNA methyltransferase Dnmt3b and methylation of a CpG island within p16Ink4a exon1. This is the first time senescence has been directly implicated in the progression of intestinal cancer and allows us to identify a distinct difference in the evolution of serrated CRCs compared to APC-mutated CRCs.
RESULTS
Induction of V600EBraf expression in the mouse gastrointestinal (GI) tract
To develop a mouse model of serrated CRC we used the conditional Lox-Stop-Lox-BrafV600E (LSL-BrafV600E) mouse strain that allows induction of V600EBraf expression from its endogenous promoter following Cre expression (Mercer et al, 2005). To induce V600EBraf, the AhcreERT mouse strain was used in which the Cyp1A1 promoter drives expression of the tamoxifen (TM)-regulated Cre recombinase in the stem cells and transit amplifying cells of the mouse GI tract (Ireland et al, 2004; Kemp et al, 2004). Mice heterozygous for the LSL-BrafV600E allele and mice heterozygous for AhcreERT were intercrossed and the untreated double heterozygous mice (referred to as Braf+/LSL-V600E; AhcreERT+/o) were obtained at the expected Mendelian ratio (Table S1 of Supporting Information), were fertile and survived a normal lifespan.
Double heterozygotes and sibling controls (either wild-type (WT) or AhcreERT+/o mice) were injected with β-naphthoflavone (β-NF) and TM and the treated mice were analysed at various time points post-induction (p.i.) (Fig 1A). Induction of the AhcreERT allele in this way is known to induce Cre activity in a range of organs, not just the small intestine (Kemp et al, 2004), and indeed we observed hyperplastic changes in the forestomach and lung of treated Braf+/LSL−V600E; AhcreERT+/o mice (Fig S1 of Supporting Information). Examination of the small intestine by PCR indicated efficient recombination of the LSL-BrafV600E allele within 1 week p.i. and this was sustained throughout the time course (Fig 1B), indicating recombination in the stem cells. Recombination was observed throughout the length of the small intestine and at a lower level in the colon (Fig 1C) as expected from previous studies of the AhcreERT strain (Kemp et al, 2004). By cross-breeding with mice expressing the Cre-reporter allele Rosa26R (Soriano, 1999), Cre-mediated recombination was estimated to be ∼70% in the small intestine (Fig 1D). Similar patterns of β-galactosidase staining were observed in the V600EBraf-expressing epithelium as the control epithelium (Fig 1E). Thus, unlike Apc deletion (Sansom et al, 2004), the V600EBraf mutation does not have a noticeable effect on epithelial migration along the crypt-villus axis.
Figure 1. Recombination of BrafLSL-V600E allele in the gastrointestinal tract.

- Treatment regime. Braf+/LSL-V600E; AhcreERT+/o or control AhcreERT+/o mice were treated with β-NF followed by β-NF + TM 24 h later and harvested at the time points indicated. The time p.i. represents the length of time following the last β-NF/TM injection.
- PCR analysis of BrafLSL-V600E recombination. Genomic DNA was harvested from the small intestine of WT mice, AhcreERT+/o-induced (Cre) or Braf+/LSL-V600E; AhcreERT+/o-induced (VE) mice at 1 or 6 weeks p.i. and subjected to PCR analysis using the primers previously described (Mercer et al, 2005). Recombination of the BrafLSL-V600E allele is indicated by the presence of the BrafLox-V600E PCR product.
- Recombination occurs throughout the length of the gut. Small intestinal samples were cut into 6 sections, with section 1 being the ileum and section 6 the duodenum. Genomic DNA was harvested from each, as well as from the colon, and was subjected to PCR analysis using the recombination primers. Recombination of the BrafLSL-V600E allele was detected in all sections of the small intestine and at a lower level in the colon.
- Monitoring of Cre-mediated recombination using the Rosa26R reporter transgene. An X-gal whole mount stain of the small intestine from β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Rosa26R+/o at 6 weeks p.i. was performed and confirms ∼70% Cre-recombination. Bar = 200 µm.
- X-gal whole mounts from β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Rosa26R+/o (VE) or AhcreERT+/o; Rosa26R+/o (Cre) mice at 6 weeks p.i. were sectioned and photographed. Similar patterns of colonization of recombined cells in the crypts and villi expressing V600EBraf compared to WTBraf are observed. Bars = 50 µm.
V600EBraf induces Mek-dependent crypt hyperplasia
Following induction of V600EBraf expression, lengthening of the crypts was observed within 3 days p.i. (Fig 2A) and the V600EBraf-expressing crypts were found to contain ∼8 more cells in cross-section than control crypts, representing ∼130 more cells in a full 3D crypt (Fig 2B). This increase was observed for all regions of the small intestine (Fig 2B and Figs S2 and S3 of Supporting Information) and for occasional crypts in the large intestine (Fig S4 of Supporting Information). The proportion of crypts with a higher cell number was similar to the level of Cre-induced recombination as assessed by the ROSA26R staining, suggesting that a large fraction of the recombined crypts were hyperplastic (Figs 1D and 2B). The increased cell number was associated with an induction of phospho-Erk and increased staining for proliferation markers, particularly higher up in the crypts (Fig 2C). More mitotic cells were also observed by electron microscopy analysis (Fig 2D). Apoptosis was not significantly altered, as scored histologically (control crypts 23 ± 3.5% SE, VE crypts 20 ± 3% SE; p > 0.05, n = 3 mice of each genotype). Subsequent to the crypt hyperplasia, increased numbers of cells in the villi were observed at 1 week p.i. with the VE-expressing villi containing ∼300 more cells than control villi (Fig 2E and Fig S2 of Supporting Information). This contributed to the induction of a highly serrated epithelium evident at 1 week p.i. (Fig 2F).
Figure 2. 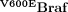 induces crypt hyperplasia.
induces crypt hyperplasia.

- H&E staining of β-NF/TM-treated AhcreERT+/o (Cre) or Braf+/LSL-V600E; AhcreERT+/o (VE) small intestinal sections at 3 days p.i. Bars = 10 µm.
- Increased cell number in VE-expressing crypts at 3 days p.i. H&E-stained ‘swiss’ rolls of section 1 (ileum) were scored blind for number of cells per full crypt. Three animals of each genotype/treatment were analysed and 150 crypts were counted in total. Only intact crypts with a clear cross-section through the lumen were counted. Data are presented as the frequency with which a given number of cells/crypt is represented amongst the crypts counted. The mean frequency for each genotype/treatment is shown.
- Increased expression of proliferation markers. Sections of Cre and VE-expressing crypts at 3 days p.i. were subjected to immunohistochemical analysis with antibodies for phospho-ERK (P-ERK), Ki67, phospho-histone H3 (P-H3) or BrdU (2 h labelling in vivo). Sections were counterstained with haematoxylin. Data presented are representative of three different mice of each genotype. Bars = 10 µm.
- Electron microscopy of WT crypt and VE-expressing crypt at 3 days p.i. showing presence of mitotic cells (black arrows) only in the VE sample. Bars = 10 µm.
- V600EBraf expression induces increased number of cells/villi at 1 week p.i. H&E-stained ‘swiss’ rolls of section 1 (ileum) were scored blind for number of cells per full villus and 150 villi were counted in total from three mice of each genotype. Data are presented as the frequency with which a given number of cells/villus is represented amongst the villi counted. The mean frequency for each genotype is shown.
- V600EBraf induced epithelium serrations at 1 week p.i. H&E stained sections of VE sample at 1 week p.i. and Cre control are shown. Bars = 10 µm
To confirm the involvement of the V600EBraf-Mek-Erk cascade in this proliferative response, Braf+/LSL-V600E; AhcreERT+/o mice were induced to express V600EBraf for 3 days and simultaneously treated every day with the Mek inhibitor PD184352 (Fig 3A). Treatment in this way inhibited the high levels of phosphoErk induced by V600EBraf (Fig 3B) and reversed crypt hyperplasia back to levels similar to that observed in control animals (Fig 3C). This confirms that the hyperplasia induced by short-term V600EBraf expression is mediated by the Mek-Erk pathway.
Figure 3. Crypt hyperplasia is Mek-dependent.
- Treatment regime. Braf+/LSL-V600E; AhcreERT+/o were treated with β-NF followed by β-NF + TM 24 h later. At 1, 2 and 3 days p.i., half of the induced mice were administered with 200 mg/kg PD184352 by intraperitoneal injection. Mice were sacrificed 6 h after the last injection. WT mice were also subjected to the same dose of PD184352 for 3 days.
- Erk phosphorylation is inhibited by PD184352. Protein lysates were prepared from the small intestine of all mice and subjected to Western blot analysis with an antibody for phospho-Erk. Erk2 was used as a loading control.
- PD184352 reverses crypt hyperplasia. H&E-stained ‘swiss’ rolls of section 1 (ileum) were scored blind for number of cells per full crypt. Ninety-four crypts from three VE animals ± PD184352 and three WT animals without PD184352 were counted. Fifty crypts from WT animals + PD184352 were also counted but did not give an altered distribution compared to WT animals without treatment (data not shown). Data are presented as the frequency with which a given number of cells/crypt is represented amongst the crypts counted. The mean frequency for each genotype/treatment is shown.
Crypt hyperplasia is not sustained and senescence ensues
In human melanomas, V600EBRAF is associated with the formation of nevi that express senescent markers. It has been proposed that hyperplastic polyps represent equivalent lesions in the serrated CRC pathway (Minoo & Jass, 2006), but this has not yet been substantiated by experimental evidence. To examine the progression of V600EBraf-expressing small intestine further, we examined the small intestine at later time points. We observed the clear induction of senescence as determined by a number of criteria. Firstly, crypt proliferation was suppressed such that there was a marked reduction in the expression of the mitotic marker phospho-histone H3 and a significant decrease in the ability to pulse-label crypt progenitor cells with BrdU (Fig 4A). Quantitation of the phospho-histone H3 staining showed a virtual absence of proliferating cells at late time points indicating stem cell senescence in the majority of crypts (Fig 4A). Secondly, VE-expressing crypts upregulated the expression of senescent markers including senescence-associated (SA)-β-galactosidase (Fig 4B) and p16Ink4A at the protein (Fig 4C) and mRNA level (Fig 4D), although p16Ink4a mRNA levels were slightly lower at the later time point of 10 weeks p.i. (see below). Of note, we consistently failed to observe the induction of other senescent markers including p19Arf, p21Cip1 and p53 (Fig S5 and S6 of Supporting Information). Thirdly, ultrastructural studies using electron microscopy revealed the presence of sessile cells characteristic of senescence. These contained numerous vacuoles in the cytoplasm filled with lipid droplets and autophagic debris as well as disrupted microvilli (Fig 4E).
Figure 4. 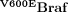 induces crypt senescence.
induces crypt senescence.
- Reduced expression of proliferation markers. Sections of the ileum from VE-expressing crypts at 3 days, 1 week and 6 weeks p.i. were stained with antibodies for BrdU and phospho-histone H3 (P-H3) and counterstained with haematoxylin. The bar chart on the right represents the mean number of phospho-histone H3 positive cells/crypt ± SD from 100 full crypts counted from at least two mice of each genotype/treatment. Bars = 10 µm.
- Expression of SA-β-galactosidase in WT and VE-expressing small intestine at 6 weeks p.i. Bars = 10 µm.
- Increased expression of p16Ink4a protein. Sections of the ileum from WT or VE-expressing crypts at 6 weeks p.i. were stained with an antibody for p16Ink4a and counterstained with haematoxylin. An expanded view showing strong nuclear p16Ink4a staining in the VE sample is shown as an inset. Bars = 10 µm.
- p16Ink4a and Gapdh mRNA expression in WT or VE-expressing small intestine at 3 days, 1 week, 6 weeks and 10 weeks p.i.
- Electron microscopy showing the presence of a sessile, senescent enterocyte in the villi of VE-expressing small intestine at 6 weeks p.i. and a normal enterocyte in the WT control. Bars = 5 µm.
- H&E staining of VE-expressing intestine at 6 weeks p.i. showing crypt loss and epithelial disintegration in parts of the small intestine at this stage. Bar = 20 µm.
- Increased expression of phosphorylated γH2AX and nuclear p53 in areas of epithelial breakdown. Parallel sections of the ileum from VE-expressing sample at 6 weeks p.i. were stained with antibodies for γH2AX phosphorylation (left) or p53 (right) and counterstained with haematoxylin. Bars = 10 µm.
All of the aforementioned effects were widespread throughout the small intestine up to 6 weeks p.i. However, we also found evidence of breakdown of the epithelium and crypt loss in some localized areas of the small intestine at 6 weeks p.i. (Fig 4F). These areas contained a high number of DNA-damaged cells as indicated by the prevalence of phospho-γH2AX positive cells and cells with nuclear p53 (Fig 4G). Control gut samples and VE-expressing gut samples with an intact epithelium were, for the most part, negative for phospho-γH2AX and nuclear p53 staining (Fig S6 of Supporting Information).
p16Ink4a repression is associated with tumour progression
To further investigate the role of p16Ink4a in progression of serrated CRC, we crossed the Braf+/LSL-V600E; AhcreERT+/o mice to Cdkn2a (Ink4a/ArfΔEx2,3) mutated mice (Serrano et al, 1996) and induced V600EBraf expression. Braf+/LSL-V600E; AhcreERT+/o-induced mice survived up to 20 weeks p.i. (Fig 5A) but combined knockout of Cdkn2a reduced this survival such that none of the mice survived past 6 weeks p.i. (Fig 5A). The VE;Cdkn2aΔEx2,3/ΔEx2,3 mice developed aggressive tumours in a range of tissues including the liver, lung and stomach (data not shown) that contributed to the general malaise of these animals (data not shown). In addition, Cdkn2a mutation led to widespread and sustained epithelial hyperplasia (Fig 5B) and a complete abrogation of the ability of V600EBraf to induce crypt senescence as assessed by the lack of SA-β-galactosidase staining (Fig 5C).
Figure 5. Cdkn2a mutation overcomes senescence.
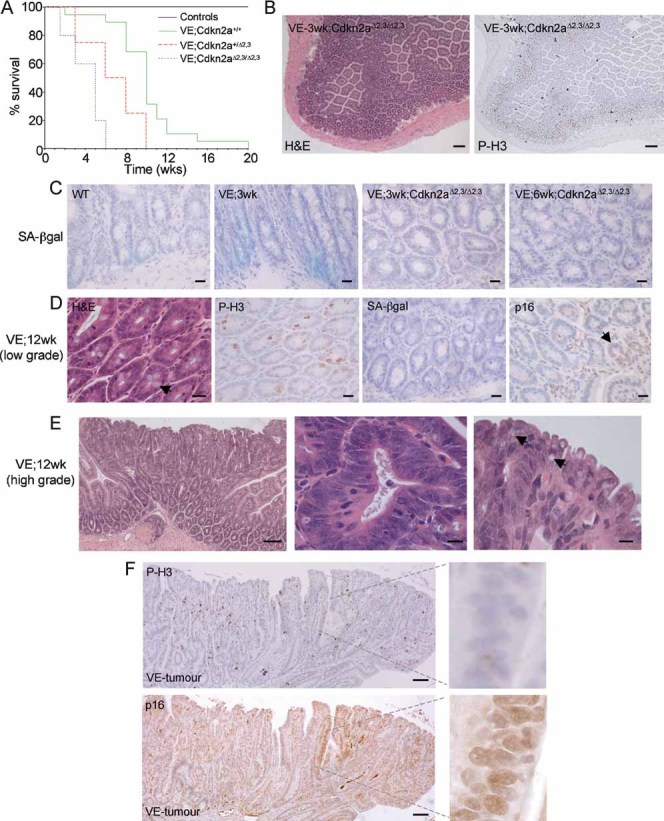
- Kaplan–Meier plots showing survival of mice used in this study. The β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Cdkn2a+/+ (VE;Cdkn2a+/+) group contained 22 animals, the β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Cdkn2a+/Δ2,3 (VE;Cdkn2a+/Δ2,3) group contained 9 animals and the β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Cdkn2aΔ2,3/Δ2,3 (VE;Cdkn2aΔ2,3/Δ2,3) group contained 9 animals. Controls included β-NF/TM-treated Braf+/LSL-V600E; AhcreERTo/o; Cdkn2a+/+ (4 mice), Braf+/+; AhcreERT+/o; Cdkn2a+/+ (5 mice), Braf+/+; AhcreERT+/o; Cdkn2a+/Δ2,3 (3 mice), Braf+/+; AhcreERT+/o; Cdkn2aΔ2,3/Δ2,3 (3 mice) and uninduced Braf+/+; AhcreERTo/o; Cdkn2a+/+ (>100 mice).
- H&E staining (left panel) and immunostaining with phospho-histone H3 (right panel) of small intestine from β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Cdkn2aΔ2,3/Δ2,3 mice at 3 weeks p.i. indicating areas of sustained epithelial proliferation. Bar = 100 µm.
- SA-β-galactosidase staining of intestine from WT, β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o mice at 3 weeks p.i. and β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o; Cdkn2aΔ2,3/Δ2,3 mice at 3 weeks and 6 weeks p.i. Sections were counterstained with haematoxylin. Bars = 10 µm.
- Low-grade adenomatous lesions from β-NF/TM-treated Braf+/LSL-V600E; AhcreERT+/o mice at 12 weeks p.i. H&E staining is indicated on the left panel showing preponderance of goblet cells (arrow). Phospho-histone H3 and SA-β-galactosidase staining show increased proliferation and lack of senescence. p16Ink4a staining (right panel) indicates growth of p16Ink4a negative cells next to an area of high p16Ink4a staining (arrow). Sections were counterstained with haematoxylin. Bars = 12.5 µm.
- H&E staining of tumour from small intestine of Braf+/LSL-V600E; AhcreERT+/o mouse at 12 weeks p.i. showing low- and high-grade areas (left panel), serrated crypts with large, ovoid, fragmented nuclei (middle panel) and tufted tumour borders containing cells with accumulated mucin (arrows in right panel). Bars = 50 µm (left panel), 5 µm (middle and right panels).
- Immunostaining of tumour with phospho-histone H3 (top panel) or p16Ink4a (bottom panel). Circumscribed areas stain strongly for p16Ink4a but weakly for phospho-histone H3. The right panels represent higher magnifications of these areas. Bars = 50 µm.
Although V600EBraf expression in the small intestine yielded widespread senescence, further histological changes were observed in Braf+/LSL-V600E; AhcreERT+/o-induced mice at >6 weeks p.i. Specifically, ∼76% of mice (n = 17) developed numerous areas (15–25/mouse/small intestine) of low-grade adenomatous lesions (Fig 5D) and ∼12% of mice additionally contained more advanced dysplastic tumours (15–25/mouse/small intestine; Fig 5E and F). No tumours were observed in control mice or in the large intestine of mice at >6 weeks p.i. The low-grade areas were well-differentiated, as indicated by the presence of goblet cells, but proliferative as indicated by the high levels of phospho-histone H3 staining and the complete lack of staining for SA-β-galactosidase (Fig 5D). Global p16Ink4a mRNA levels were lower in mice at >6 weeks p.i. than at 6 weeks p.i. (Fig 4D) and, when assessed more directly by immunohistochemical staining, p16Ink4a expression was reduced in these proliferating areas in comparison to neighbouring, senescent areas with high p16INK4a expression (Fig 5D).
The dysplastic tumours appeared to evolve from the lower grade adenomatous lesions (Fig 5E). They contained de-differentiated cells with a loss of apical-basal polarity and large, ovoid, vesicular nuclei (Fig 5E). Dysplastic crypts often formed stellate structures (Fig 5E) and all tumours had tufted, serrated borders with cells containing droplets of accumulated mucin (Fig 5E). The tumours stained strongly for phospho-histone H3 (Fig 5F), indicating high levels of proliferation. Consistent with this observation, p16Ink4a was absent in all tumours (22/22 tumours analysed), except for occasional remnants of high p16Ink4a expression that correlated with low phospho-histone H3 expression (Fig 5F). This result suggests that tumour progression is associated with repression of p16INK4A. The assessment of further progression to invasive carcinomas was not possible because of the death of all mice before 20 weeks p.i.
V600EBraf induces Wnt pathway activation
Intestinal crypt proliferation is dependent on the Wnt signalling pathway. To assess whether this pathway is activated in response to V600EBraf, intestinal sections over the time course were stained with an antibody for β-catenin and compared to the pattern of phospho-Erk staining (Fig 6A). During the hyperplastic phase, at 3 days p.i., increased nuclear β-catenin staining was observed, particularly in cells higher up in the crypts in a similar manner to the phospho-Erk staining (Fig 6A and B). The number of cells with nuclear β-catenin was suppressed to control levels following treatment with PD184352, confirming that crosstalk between the two pathways is Mek-dependent (Fig 6A and B). However, in senescent crypts there was a virtual absence of nuclear β-catenin positive cells (Fig 6A and B) suggesting that suppression of Wnt signalling is an important part of the senescent response. Consistent with this observation, cells within the dysplastic tumours stained for nuclear β-catenin and there was a notable lack of β-catenin at the cell periphery (Fig 6C), confirming that tumour progression is associated with sustained Wnt pathway activation.
Figure 6. Crosstalk with the Wnt pathway.
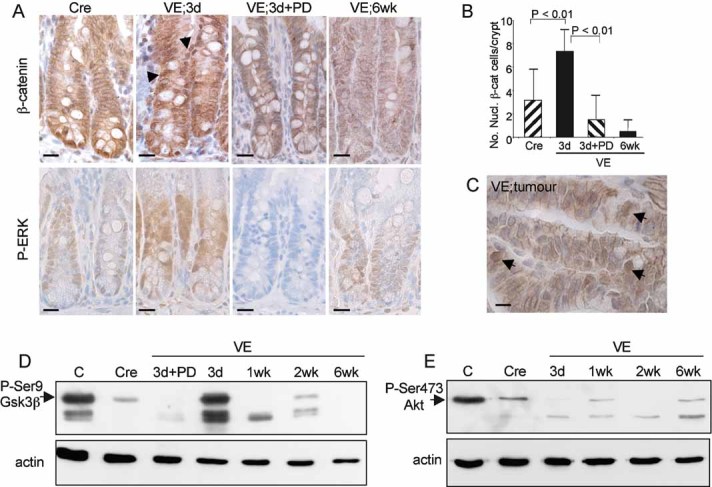
- Immunostaining of Cre control or VE-expressing gut samples at 3 days and 6 weeks p.i. as well as a 3 day sample treated with PD184352 with an antibody for β-catenin (top panels) or phospho-Erk (bottom panels). Sections were counterstained with haematoxylin. Nuclear β-catenin staining is indicated by black arrows. Bars = 10 µm.
- Quantitation of nuclear β-catenin positive cells. The bar chart represents the mean number of nuclear β-catenin positive cells/crypt ± SD from 100 full crypts counted from at least two mice of each genotype/treatment.
- Immunostaining of tumour with antibody for β-catenin. Arrows indicate cells with high levels of nuclear β-catenin. Bar = 5 µm.
- Western blot analysis of phosphorylation of Ser9 of Gsk3β in a control mouse embryonic fibroblast (MEF) sample (C), a Cre control gut sample and VE-expressing gut samples at 3 days–6 weeks p.i. The VE samples at 3 days p.i. treated ± PD184352 are the same as those analysed for phospho-Erk in Fig 3B.
- Western blot analysis of phosphorylation of Ser473 of Akt in a control MEF sample (C), a Cre control gut sample and VE-expressing gut samples at 3 days–6 weeks p.i.
To further assess the mechanism of crosstalk between the two pathways, we assessed phosphorylation of serine 9 of Gsk3β and serine 473 of Akt. We found that there was a significant induction of phospho-Gsk3β phosphorylation at 3 days p.i. and this was significantly suppressed at the later time points as well as following treatment with PD184352 (Fig 6D). By contrast, phospho-serine 473 Akt levels were lower than controls in all VE-expressing samples, and there was no significant alteration during the time course (Fig 6E). Together these data suggest that V600EBraf inactivates Gsk3β in a Mek-dependent, Akt-independent manner.
V600EBraf induces CpG methylation of p16INK4a
Human serrated CRCs are associated with the CIMP-H phenotype and so we assessed whether CpG methylation could explain the inactivation of p16INK4a. First, we assessed expression of the de novo DNA methylase Dnmt3b—which has been previously linked to CIMP-H CRCs—and observed its increased expression at the protein (Fig 7A) and mRNA (Fig 7B) levels following induction of V600EBraf expression. High Dnmt3b expression was sustained throughout the time course as well as in tumours (Fig. 7C and D). Using the EMBL-EBI CpG island prediction programme (EMBOSS) (http://www.ebi.ac.uk/Tools/emboss/cpgplot/index.html), we identified three CpG islands within the mouse Cdkn2a locus (Fig 7E) and investigated the methylation status of the island within exon 1 of p16INK4a using bisulphite sequencing (Fig 7F and Fig S7 of Supporting Information). Most CpG residues within this region were unmethylated in the normal gut and there were few changes throughout the time course following induction of V600EBraf. However, of three tumours tested, increased methylation of up to 7 CpG residues was consistently observed.
Figure 7. Methylation of p16INK4a.
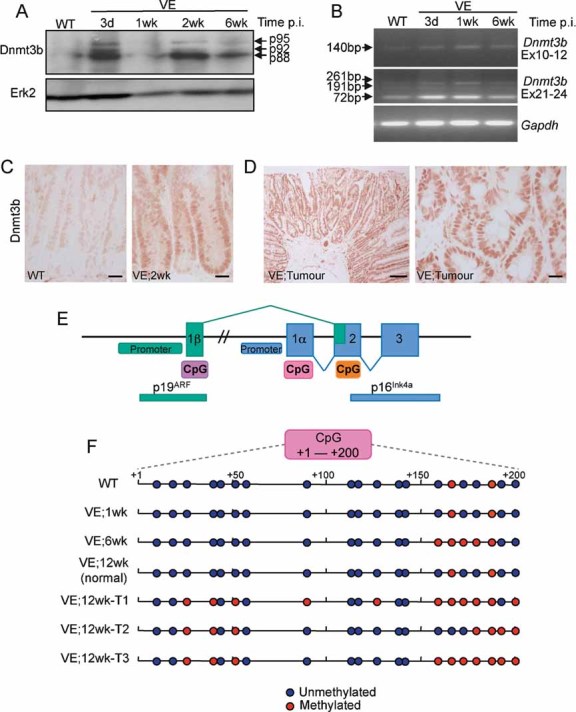
- Dnmt3b protein expression. Protein lysates were subjected to Western blot analysis with an antibody for Dnmt3b. Erk2 was used as a loading control.
- Dnmt3b mRNA expression using reverse transcription polymerase chain reaction (RT-PCR). Amplification across exons 10–12 of Dnmt3b generated a product of 140 bp indicating the lack of exon11 in all mRNA species. Exons 21–24 amplification generated a major species at 72 bp (Δexons 22/23) with minor species at 261 bp (+exons 22/23) and 191 bp (+exon 23/Δexon 22). Thus, the predominant species expressed in the mouse gut lacks exons 11, 22 and 23 and this isoform, along with the minor isoforms, are expressed at higher levels in the VE-expressing gut. Gapdh amplification was used as a control.
- Immunostaining of WT control and VE-expressing gut samples at 2 weeks p.i. with an antibody for Dnmt3b. Bars = 10 µm.
- Immunostaining of tumour from VE-expressing gut at 12 weeks p.i. with an antibody for Dnmt3b. Bars = 50 µm (left) and 10 µm (right).
- Diagram illustrating the organization of the mouse Cdkn2a locus. CpG islands predicted by the EMBOSS programme are shown.
- Bisulphite/sequencing analysis of p16 CpG island at +1 to +200 (with respect to the p16 transcription start site). DNA from WT and VE-expressing gut samples at 1 and 6 weeks p.i. were analysed together with DNA from three tumours (T1–T3) arising from VE-expressing gut at 12 weeks p.i. and a normal region of the gut from the same mouse.
DISCUSSION
The seminal paper by Davies et al (2002) reporting the presence of BRAF mutations in human cancer showed that ∼12% of human CRCs carry missense mutations of this key proto-oncogene. It was subsequently demonstrated that the V600EBRAF mutation has a striking correlation (>90%) with CIMP-H, MMR-deficient CRCs that arise by MLH1 epigenetic inactivation (Deng et al, 2004; Rajagopalan et al, 2002; Wang et al, 2003; Weisenberger et al, 2006). It is now known that these molecular features are concordant with cancers that have a serrated histopathology, thus consolidating the classification of a distinct serrated neoplasia pathway originally pioneered by Jeremy Jass (Jass, 2007a). V600EBRAF mutations have been found in ∼70% of right-sided hyperplastic polyps with serrated morphology and serrated adenomas, the purported precursors in this pathway (O'Brien et al, 2006; Yang et al, 2004). They are not found in histological categories of the traditional adenoma–carcinoma sequence and, furthermore, serrated CRCs are exclusive of APC mutations (Jass, 2007a). Thus, V600EBRAF has been proposed to be a key factor in the development of serrated CRC in an analogous way to APC mutation in traditional CRCs. Here we show that V600EBraf expression from the endogenous Braf promoter in the progenitor cells of the intestine induces a series of molecular and pathological changes highly reminiscent of early stage serrated CRC. These data provide compelling evidence that V600EBraf is an early genetic driver of serrated CRC.
We have found that V600EBraf induces an immediate wave of Mek/Erk-dependent proliferation within intestinal crypts. The consequent increase in cell number can account for the sawtooth-like infolding of the surface epithelium observed following V600EBraf expression (Fig 2F). A serrated epithelium in human CRCs has previously been attributed to inhibition of apoptosis and delayed migration of cells from the crypt to the surface (Parfitt & Driman, 2007; Tateyama et al, 2002). However, our data argue that serrations arise from increased proliferation since we have not observed alterations in apoptosis or migration following V600EBraf expression (Fig 1E).
We have also shown that crypt hyperplasia by V600EBraf is associated with Wnt pathway activation, as indicated by the accumulation of nuclear β-catenin in crypt cells at 3 days p.i. (Fig 6A and B). This activation seems to occur in a cell-intrinsic manner since the number and position of the nuclear β-catenin-positive cells shows a good correlation with the number and position of phospho-Erk-positive cells (Fig 6A). That Wnt pathway activation occurs in this system is not surprising since crypt progenitor cells are known to be dependent on Wnt pathway activation for proliferation (van der Flier & Clevers, 2009). Melanomas with V600EBRAF mutations frequently accumulate nuclear β-catenin and, interestingly, GSK3A and GSK3B were recently shown to be negatively regulated by V600EBraf/Mek/Erk signalling in a phosphoproteomic screen of human melanoma cells following MEK inhibition (Old et al, 2009). Specifically, it was shown that the activating autophosphorylation sites Y279/Y216 of GSK3α/GSK3β were elevated by MEK inhibition and the inhibitory phosphorylation site of Ser9 on GSK3β was repressed by MEK inhibition. We observed a similar response regarding Gsk3β Ser9 phosphorylation in MEK inhibitor-treated V600EBraf-expressing intestine (Fig 6D), thus indicating a possible generic mechanism for crosstalk between the two pathways.
A number of studies have shown that the GSK3 kinases are activated by both the PI3K/PKB and Wnt/β-catenin signalling pathways (MacDonald et al, 2007). However, it has been proposed that different pools of GSK3 are targeted by each pathway (Frame & Cohen, 2001) and, recently, axin binding to GSK3 was shown to prohibit PI3K-activation of the Wnt pathway (Ng et al, 2009). We have found that the activation of Gsk3β in our system does not involve activation of PkB/Akt (Fig 6E), suggesting that the V600EBraf/Mek/Erk pathway influences the axin-bound pool of Gsk3β instead. Such an observation is consistent with the fact that mutations in canonical Wnt pathway components such as APC are mutually exclusive with V600EBRAF mutations (www.sanger.ac.uk/genetics/CGP/cosmic) in CRCs. Little is known regarding the mechanism of control of the Wnt/β-catenin pathway by Erk; such investigations are now essential since therapies that target different components of Wnt/β-catenin pathway are currently in development and could have potential for the treatment of serrated CRCs.
Oncogenic Kras has been similarly expressed from its own promoter in the intestine using two different transgenic models but these generated drastically different phenotypes from each other as well as from those described here for V600EBraf. Using the analogous AhCre strain, G12VKras was expressed in the mouse small intestine but no proliferative changes were detected (Sansom et al, 2006), intestinal homeostasis was maintained and signalling through the Mek/Erk pathway was not altered, although high levels of feedback regulators of the Erk cascade were induced. Expression of G12DKras in the mouse large intestine using the Fabp-Cre strain promoted extreme hyperplasia through amplification of progenitor cells in a Mek-dependent manner (Haigis et al, 2008). In both cases, oncogenic Kras promoted colon cancer progression in cooperation with Apc deletion. The reason for the differences between V600EBraf and both of the systems expressing oncogenic Kras expression may be attributable to the relative output of the MAPK pathway induced by the two oncogenes and/or the fact that Kras can activate other effector pathways, besides MAPK. KRAS mutation has been viewed as an intermediate step in the classical Vogelstein model of CRC development and the effects observed in the mouse may be reflective of the involvement of KRAS in these CRC types, rather than serrated CRCs.
As in mice manipulated to express V600EBraf in the lung (Dankort et al, 2007) and melanocytes (Dhomen et al, 2009), we show here that V600EBraf induces oncogene-induced senescence (OIS) in the intestine following an initial wave of hyperplasia. Although a number of different OIS markers have been reported (Collado & Serrano, 2006), these are thought to have tissue-specific expression and, for the intestine, we only observed upregulation of SA-βgal and p16Ink4a but not p19Arf, p53 or p21Cip1 (Figs 4, S5 and S6). Although p53 expression was not induced in the initial senescent response to V600EBraf, disintegrated areas of the epithelium were detected at late time points following V600EBraf induction and these demonstrated high levels of DNA damage and high p53 expression (Fig 4G). However, at present it is not clear whether these damaged areas arise as a direct consequence of sustained V600EBraf signalling or as a consequence of the fact that the epithelium is already senescent.
Importantly, this is the first time that OIS has been reported to occur in the intestine. G12VKras was not reported to induce senescence when expressed in the mouse intestine (Sansom et al, 2006), although, when expressed in the lung, premalignant adenomas were formed that express senescent markers (Collado et al, 2005). Similarly, the deletion of Apc in the mouse intestine has not been associated with the induction of senescence, although deletion of Apc in the mouse kidney has been reported to upregulate p21Cip1, p16Ink4a and SA-βgal, and p21CIP1 loss cooperates with Apc deletion in promoting renal carcinomas in the mouse (Owen Sansom, personal communication and accompanying manuscript, DOI 10.1002/emmm.201000101). Taken together, these data indicate that the ability to induce senescence is highly tissue-specific and that cells within the intestine are relatively resistant to senescence, possibly because they need to maintain high levels of proliferation in order to replenish the epithelial lining whereas this is not the case in other tissues. Of note in this regard is the fact that induction of senescence by V600EBraf is associated with suppression of the Wnt/β-catenin pathway, which is required to sustain proliferation in the intestine (Fig 6A and B). The unique ability of V600EBraf to induce OIS in the intestine is undoubtedly related to the downstream signalling pathways it is able to activate or repress and further dissection of these pathways in comparison to those regulated by oncogenic Kras and Apc mutation will be important in fully understanding the mechanisms by which V600EBraf drives intestinal cancer.
Apart from the upregulation of p16Ink4a by V600EBraf, we have also shown that down-regulation of p16Ink4a spontaneously or by knockout of Cdkn2a is associated with resumption of proliferation and tumour progression (Fig 5). Our data are supportive of p16Ink4a being a key player in serrated CRC development rather than p19Arf, since we have not been able to detect p19Arf expression in the intestine at all (Fig S5 of Supporting Information). Our results are consistent with a model whereby V600EBRAF is an early genetic driver mutation that stimulates the formation of hyperplastic crypts but cannot induce further cancer progression as p16Ink4a is induced and senescence ensues. The earliest lesions so far identified to contain V600EBRAF mutations in the human gut are right-sided hyperplastic polyps. These can remain unchanged for many years in a similar way to melanocytic nevi and, in fact, for a long time it was thought that they were non-neoplastic lesions with no malignant potential (Hawkins et al, 2002). Our mouse studies would suggest that hyperplastic polyps also represent senescent lesions that arise through V600EBraf-induced upregulation of p16Ink4a and as such have malignant potential since tumours can arise if p16Ink4a is inactivated. In a recent study, six right-sided human hyperplastic polyps with the V600EBRAF mutation were found not to express p16Ink4a (Sandmeier et al, 2009). However, since this was a small sample set, evaluation of a wider panel of human hyperplastic polyps of proven genetic status needs to be undertaken to clarify this important issue. In the case of melanoma, V600EBRAF mutations are detected in human premalignant nevi that express p16Ink4a (Pollock et al, 2003) and V600EBraf expression in mouse melanocytes drives the formation of senescent nevi that express p16Ink4a (Dhomen et al, 2009). However, deletion of p16Ink4a does not augment V600EBraf-induced melanoma progression in mice (Dhomen et al, 2009) and, in humans, fewer than 2% of sporadic melanomas have mutations that affect p16Ink4a (Aitken et al, 1999; Orlow et al, 2007). Thus, the function of p16Ink4a as a barrier to tumour progression is idiosyncratic to particular tumour types.
The V600EBRAF mutation has a strong correlation with CIMP-H in human CRCs and, interestingly, we have found that expression of V600EBraf in the mouse gut induces high levels of expression of the de novo methylase Dnmt3b (Fig 7A–C). This result is consistent with the observation that high DNMT3b expression is detected in CRCs with the methylator phenotype (Nosho et al, 2009) and with the fact that ectopic expression of V600EBRAF in normal human colon epithelial cells induces methylation of the MLH1 promoter (Minoo et al, 2007). We have shown that tumour progression is associated with the de novo methylation of at least seven CpG residues in p16Ink4a exon1, which strongly suggests that inactivation of p16INK4a by epigenetic mechanisms underpins V600EBraf-driven CRC development. However, it is important to note that these methylation changes are not observed for up to 6 weeks following V600EBraf expression, despite the fact that Dnmt3b levels are high. Thus, an additional, as-yet unidentified factor(s) may be required to suppress p16Ink4a expression. DNA methylation is only part of an epigenetic code that dictates transcriptional activity. Additional key factors are the PcG multiprotein complexes, PRC1 and PRC2 that respectively establish and bind to histone H3 trimethylated on Lys27 (H3K27) and the counteracting histone demethylases UTX and JMJD3. Indeed, recently it was shown that oncogenic RAS and oncogenic BRAF can override PcG-mediated repression of p16INK4a in human fibroblasts by inducing the expression of the H3K27 methylase JMJD3 (Agger et al, 2009; Barradas et al, 2009). We are currently examining these chromatin modifiers as well as the link between increased DNA damage (Fig 4G) and the onset of DNA methylation.
In summary, we have established a mouse model that has allowed us to demonstrate that serrated CRC initiates and develops by mechanisms different to traditional CRCs involving APC mutation. Our data show that V600EBraf can drive Erk and Wnt pathway activation and the formation of hyperplastic crypts that subsequently remain dormant for prolonged periods due to the upregulation of p16INK4a. The switch from p16Ink4a expression to repression by epigenetic mechanisms is a key factor in V600EBraf-induced tumour progression. This mouse model provides an essential reagent for further exploring the mechanisms underpinning serrated CRC including the crosstalk between the Wnt and ERK signalling pathways, the role of these pathways in senescence and the mechanisms leading to p16Ink4a inactivation.
The paper explained
PROBLEM:
In the Vogelstein model, human colorectal cancer (CRC) is initiated by mutations arising in the APC tumour suppressor gene. However, ∼12% of CRC samples are non-APC mutated and represent a distinct subclass characterized by a serrated histopathology, the V600EBRAF mutation and the CpG island methylator-high phenotype (CIMP-H). The mechanisms underpinning the evolution of serrated CRCs are not understood at present and are investigated in this study.
RESULTS:
Using a mouse model, this report shows that V600EBraf is an early genetic driver of serrated CRC since it can induce the formation of hyperplastic crypts, the purported early lesions of this disease type. Hyperplastic crypts enter senescence through upregulation of p16INK4a and we show that inactivation of this tumour suppressor gene through DNA methylation is necessary for tumour progression.
IMPACT:
This preclinical study shows that serrated CRCs evolve through a series of genetic changes and biochemical changes that are distinct from those involved in traditional CRCs. These results are important for informing the utility and application of novel biological therapies to the treatment of patients with serrated CRCs.
MATERIALS AND METHODS
Experimental animals and genotyping
Animal experiments were performed under Home Office project license authority. Generation and genotyping of transgenic animals has been described previously for the LSL-BrafV600E (Mercer et al, 2005), AhcreERT (Kemp et al, 2004), Cdkn2a+/Δ2,3 (Serrano et al, 1996) and Rosa26R (Soriano, 1999) mice. β-NF (8 mg/ml) and TM (10 mg/ml) were prepared in corn and sunflower oil respectively and injected i.p. into mice at 8–12 weeks of age. Experimental mice received β-NF (80 mg/kg) and TM (100 mg/kg). Mice were pulse labelled with BrdU (Roche; 10 mg/kg i.p.) for 2 h prior to harvesting. PD184352 (prepared at 10 mg/ml in 10% dimethyl sulphoxide (DMSO), 10% cremophor, 80% water) was injected i.p. at a dose of 200 mg/kg/day.
Histology
Small intestines were fixed, paraffin-embedded, sectioned and stained with haematoxylin and eosin (H&E) by standard procedures. Antibodies used for immunostaining were: PP-Erk (Cell signalling 9101; 1:200), Ki67 (Neomarkers RM-9106-S1; 1:200) phospho-histone H3 (Cell signaling 9701; 1:100), BrdU (Roche; 1:100), p16INK4a (Santa Cruz SC-1207; 1:100), β-catenin (Sigma C2206; 1:2000), phospho-serine 139-γH2AX (Cell Signaling 9718; 1:120), p53 (Cell Signaling 2524; 1:200) and Dnmt3b (Imgenex IMG-184A; 1:160). All staining was performed as described (Mercer et al, 2005) except the β-catenin staining was performed as described (Amini Nik et al, 2005). For X-gal staining, intestines were fixed (2% formaldehyde/0.1% glutaraldehyde/phosphate-buffered saline (PBS)) and demucified (22 mM dithiothreitol (DTT), 10% glycerol, 10 mM Tris pH 8.2, 20% ethanol in saline) prior to staining. For SA-β-galactosidase staining fresh frozen tissues were sectioned and stained using the Cell Signaling kit (9860).
Electron microscopy
Intestines were fixed in 4% formaldehyde/2.5% glutaraldehyde in 0.1 M Sörensens phosphate buffer (pH 7.2) followed by buffered 1% osmium tetroxide. Tissue was dehydrated then infiltrated with a mixture of propylene oxide/agar low viscosity resin before embedding and polymerization. Thin sections (80 nm) were cut using a Reichert Ultracut S ultramicrotome, collected on copper mesh grids and counterstained with 2% aqueous uranyl acetate followed by Reynolds’ lead citrate. Images were captured using a JEOL 1220 electron microscope, with an accelerating voltage of 80 kV, and an SIS Megaview III digital camera with Analysis software.
Western blot analysis
Intestinal tissue was lysed in SDS lysis buffer (50 mM Tris, 2% v/v SDS, 10% v/v glycerol) and Western blots performed as described (Mercer et al, 2005). Antibodies used were: PP-Erk (Cell signalling 9101; 1:500), Erk2 (Santa Cruz 73369; 1:2000), phosphoser9-Gsk3β (Cell Signalling 9323; 1: 1000) phosphoser473-Akt (Cell Signaling 4060; 1:2000), Dnmt3b (Imgenex IMG-184A; 1:160) and actin (Sigma A2103; 1:1000).
RT-PCR
RNA was extracted using an RNeasy kit (Qiagen) and reverse-transcribed using Superscript III (Invitrogen). cDNA was amplified by PCR using Reddymix (Thermo Scientific). p16INK4a primers were: Fwd, 5′-TCTGGAGCAGCATGGAGTCC-3′, Rev, 5′-TCGCAGTTCGAATCTGCACC-3′ using 94°C (15 s), 57°C (30 s), 68°C (1 min) for 35 cycles. Gapdh primers were: Fwd, 5′-AGGTCGGTGTGAACGGATTTG-3′, Rev, 5′-TGTAGACCATGTAGTTGAGGTCA-3′. Dnmt3b primers were: Fwd (exons 10–12), 5′-CTTCAAGCCTACTGGGATCG-3′, Rev (exons 10–12), 5′-TCCCGCCATAGCTATTTGTC-3′, Fwd (exons 21–24), 5′-CCCGGTACTTCTGGGGTAAC-3′, Rev (exons 21–24), 5′-ACACGTCCGTGTAGTGAGCA-3′. Conditions used for Dnmt3b and Gapdh were 94°C (30 s), 55°C (30 s), 72°C (30 s) for 35 cycles. All genes were amplified under conditions in which amplification was still linear.
Bisulphite sequencing
DNA from the time course was collected from the separated epithelial layer and extracted using the Promega Wizard® SV Genomic DNA Purification System. Tumours were manually microdissected from 10 µm thick paraffin-embedded sections and DNA was extracted by incubation in 0.05 M NaOH at 95°C for 15 min after which Tris–HCl pH 8 was added at 10 mM. DNA was bisulphite converted using the DNA Methylation-Gold™ Kit (Zymo Research) and amplified using Platinum Taq (Invitrogen) with 5% DMSO. Primers used were: Fwd, 5′-AGG AGG GAT TTA TTG GTT ATA-3′, Rev, 5′-AAA TAA AAA AAA CAA AAC TTC TCA AAA ATA-3′. PCR products were purified and sequenced using the same primers.
Statistics
Statistical analysis was performed using the Dunnett multiple comparisons test.
Acknowledgments
This work was funded by a Cancer Research UK programme grant (C1362/A6969) and by a Hope Against Cancer studentship awarded to KRS. We give particular thanks to Biomedical Services for their technical expertise. We also thank Suzanne Turner and Manuel Serrano for providing Cdkn2a knockout mice, Jennifer Edwards for H&E staining, Natalie Allcock for facilitating the electron microscopy studies, Nigel Belshaw for methylation advice and Felix Beck, Peter Greaves and Kevin West for pathology advice.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
CAP and RM conceived the project; CAP and LASC wrote the paper; LASC, KRS, VSSA, BP and SMG performed the experiments; SJC provided PD148352 and DJW provided the AhCreER mice.
For more information
COSMIC: Wellcome Trust Sanger Institute database of somatic mutation information in human cancers:
www.sanger.ac.uk/genetics/CGP/cosmic
EMBOSS: EMBL-EBI CpG island prediction programme (EMBOSS):
http://www.ebi.ac.uk/Tools/emboss/cpgplot/index.html
Accompanying Research Article:
DOI 10.1002/emmm.201000101
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Agger K, Cloos PA, Rudkjaer L, Williams K, Andersen G, Christensen J, Helin K. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene- and stress-induced senescence. Genes Dev. 2009;23:1171–1176. doi: 10.1101/gad.510809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aitken J, Welch J, Duffy D, Milligan A, Green A, Martin N, Hayward N. CDKN2A variants in a population-based sample of Queensland families with melanoma. J Natl Cancer Inst. 1999;91:446–452. doi: 10.1093/jnci/91.5.446. [DOI] [PubMed] [Google Scholar]
- Amini Nik S, Hohenstein P, Jadidizadeh A, Van Dam K, Bastidas A, Berry RL, Patek CE, Van der Schueren B, Cassiman JJ, Tejpar S. Upregulation of Wilms’ tumor gene 1 (WT1) in desmoid tumors. Int J Cancer. 2005;114:202–208. doi: 10.1002/ijc.20717. [DOI] [PubMed] [Google Scholar]
- Barradas M, Anderton E, Acosta JC, Li S, Banito A, Rodriguez-Niedenfuhr M, Maertens G, Banck M, Zhou MM, Walsh MJ, et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009;23:1177–1182. doi: 10.1101/gad.511109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach R, Chan AO, Wu TT, White JA, Morris JS, Lunagomez S, Broaddus RR, Issa JP, Hamilton SR, Rashid A. BRAF mutations in aberrant crypt foci and hyperplastic polyposis. Am J Pathol. 2005;166:1069–1075. doi: 10.1016/S0002-9440(10)62327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan TL, Zhao W, Leung SY, Yuen ST. BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res. 2003;63:4878–4881. [PubMed] [Google Scholar]
- Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6:472–476. doi: 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, Truta B, Sleisenger MH, Kim YS. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:191–195. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NJ, Bariol C, Ward RL. The serrated neoplasia pathway. Pathology. 2002;34:548–555. [PubMed] [Google Scholar]
- Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–5202. [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007a;50:113–130. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- Jass JR. Molecular heterogeneity of colorectal cancer: implications for cancer control. Surg Oncol. 2007b;(16):S7–S9. doi: 10.1016/j.suronc.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Jass JR, Biden KG, Cummings MC, Simms LA, Walsh M, Schoch E, Meltzer SJ, Wright C, Searle J, Young J, et al. Characterisation of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J Clin Pathol. 1999;52:455–460. doi: 10.1136/jcp.52.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasarides M, Chiloeches A, Hayward R, Niculescu-Duvaz D, Scanlon I, Friedlos F, Ogilvie L, Hedley D, Martin J, Marshall CJ, et al. B-RAF is a therapeutic target in melanoma. Oncogene. 2004;23:6292–6298. doi: 10.1038/sj.onc.1207785. [DOI] [PubMed] [Google Scholar]
- Kemp R, Ireland H, Clayton E, Houghton C, Howard L, Winton DJ. Elimination of background recombination: somatic induction of Cre by combined transcriptional regulation and hormone binding affinity. Nucleic Acids Res. 2004;32:e92. doi: 10.1093/nar/gnh090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Semenov MV, He X. SnapShot: Wnt/beta-catenin signaling. Cell. 2007;131:1204. doi: 10.1016/j.cell.2007.11.036. [DOI] [PubMed] [Google Scholar]
- Makinen MJ. Colorectal serrated adenocarcinoma. Histopathology. 2007;50:131–150. doi: 10.1111/j.1365-2559.2006.02548.x. [DOI] [PubMed] [Google Scholar]
- Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Mercer K, Giblett S, Green S, Lloyd D, DaRocha Dias S, Plumb M, Marais R, Pritchard C. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. 2005;65:11493–11500. doi: 10.1158/0008-5472.CAN-05-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Minoo P, Jass JR. Senescence and serration: a new twist to an old tale. J Pathol. 2006;210:137–140. doi: 10.1002/path.2047. [DOI] [PubMed] [Google Scholar]
- Minoo P, Moyer MP, Jass JR. Role of BRAF-V600E in the serrated pathway of colorectal tumourigenesis. J Pathol. 2007;212:124–133. doi: 10.1002/path.2160. [DOI] [PubMed] [Google Scholar]
- Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC, Schutte M, Clevers H. Phosphatidylinositol 3-Kinase signaling does not activate the Wnt cascade. J Biol Chem. 2009;284:35308–35313. doi: 10.1074/jbc.M109.078261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosho K, Shima K, Irahara N, Kure S, Baba Y, Kirkner GJ, Chen L, Gokhale S, Hazra A, Spiegelman D, et al. DNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancer. Clin Cancer Res. 2009;15:3663–3671. doi: 10.1158/1078-0432.CCR-08-2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien MJ, Yang S, Mack C, Xu H, Huang CS, Mulcahy E, Amorosino M, Farraye FA. Comparison of microsatellite instability, CpG island methylation phenotype, BRAF and KRAS status in serrated polyps and traditional adenomas indicates separate pathways to distinct colorectal carcinoma end points. Am J Surg Pathol. 2006;30:1491–1501. doi: 10.1097/01.pas.0000213313.36306.85. [DOI] [PubMed] [Google Scholar]
- Old WM, Shabb JB, Houel S, Wang H, Couts KL, Yen CY, Litman ES, Croy CH, Meyer-Arendt K, Miranda JG, et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol Cell. 2009;34:115–131. doi: 10.1016/j.molcel.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlow I, Begg CB, Cotignola J, Roy P, Hummer AJ, Clas BA, Mujumdar U, Canchola R, Armstrong BK, Kricker A, et al. CDKN2A germline mutations in individuals with cutaneous malignant melanoma. J Invest Dermatol. 2007;127:1234–1243. doi: 10.1038/sj.jid.5700689. [DOI] [PubMed] [Google Scholar]
- Parfitt JR, Driman DK. Survivin and hedgehog protein expression in serrated colorectal polyps: an immunohistochemical study. Hum Pathol. 2007;38:710–717. doi: 10.1016/j.humpath.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD, Berghmans S, Mayhall EA, Traver D, Fletcher CD, et al. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005;15:249–254. doi: 10.1016/j.cub.2005.01.031. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, Moses TY, Hostetter G, Wagner U, Kakareka J, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- Sandmeier D, Benhattar J, Martin P, Bouzourene H. Serrated polyps of the large intestine: a molecular study comparing sessile serrated adenomas and hyperplastic polyps. Histopathology. 2009;55:206–213. doi: 10.1111/j.1365-2559.2009.03356.x. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci USA. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Tateyama H, Li W, Takahashi E, Miura Y, Sugiura H, Eimoto T. Apoptosis index and apoptosis-related antigen expression in serrated adenoma of the colorectum: the saw-toothed structure may be related to inhibition of apoptosis. Am J Surg Pathol. 2002;26:249–256. doi: 10.1097/00000478-200202000-00013. [DOI] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA, Burgart LJ, McDonnell SK, Schaid DJ, Thibodeau SN. BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res. 2003;63:5209–5212. [PubMed] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- Yang S, Farraye FA, Mack C, Posnik O, O'Brien MJ. BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol. 2004;28:1452–1459. doi: 10.1097/01.pas.0000141404.56839.6a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.