

Abstract
Senescence has been implicated as an important mechanism of tumour suppression in a number of human malignancies, including colorectal cancer (CRC). However, we still have a relatively poor understanding of how the underlying mutations that occur in cancer cause senescence and its relevance in vivo. The Apc gene is mutated in approximately 80% of CRC as the initiating event, but rarely elsewhere. In this study we have examined the capacity of Apc loss to induce senescence in the intestinal epithelium compared to the renal epithelium. Within the renal epithelium, loss of Apc function led to an induction of senescence, however, bypassing senescence through combined Apc and p21 or Ink4A gene deletion rapidly initiated renal carcinoma. Within the intestinal epithelium, loss of Apc did not induce senescence. Moreover, combined Apc and p21 or Ink4A loss had no impact upon tumourigenesis. Taken together, these results show that Apc loss in vivo invokes a senescence program in a context-dependent fashion, and implies senescence may play a key barrier to tumourigenesis in the kidney. However, in CRC, escape from senescence is likely to only be a barrier in cancers initiated by other mutations.
Keywords: Apc, colorectal cancer, p21, renal carcinoma, senescence
INTRODUCTION
The adenomatous polyposis coli (APC) tumour suppressor is mutated in approximately 80% of colorectal cancers (CRC) where it is thought to be the key initiating event (Korinek et al, 1997; Morin et al, 1997). The major tumour suppressor function of the APC gene is thought to be as a negative regulator of Wnt signalling. The Apc protein forms part of a destruction complex with glycogen synthase kinase 3 (GSK3), axin and casein kinase 1 (CK1), which binds to β-catenin and allows phosphorylation of β-catenin by GSK3, targeting it for degradation (Bienz & Clevers, 2000). In the absence of Apc this complex no longer forms, β-catenin is not targeted for degradation, and it accumulates and translocates to the nucleus where it interacts with T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors to drive expression of Wnt target genes such as c-myc (He et al, 1998).
Outwith CRC, mutations in the APC gene are rare. However, Wnt pathway activation is observed in cancers such as hepatocellular carcinoma (HCC) where a subset of cancers have activating mutations in β-catenin or loss of negative regulators of the Wnt pathway such as Axin or Axin2 (Giles et al, 2003; Satoh et al, 2000). Similarly, activating mutations in β-catenin have been observed in several cancers including melanoma, ovarian carcinomas, childhood hepatoblastomas and medulloblastomas, desmoid tumours and non-ductal solid pancreatic tumours (Giles et al, 2003). In these cancers, activating mutations of the Wnt pathway are not thought to be an initiating event. Recently, a role for activated Wnt signalling in renal carcinoma has been proposed, as the key renal tumour suppressor protein von Hippel-Lindau (VHL), acts through Jade-1, an E3 ubiquitin ligase, to target β-catenin for degradation (Zhou et al, 2005). Therefore, a mutation in VHL results in the stabilization and activation of the oncogenic β-catenin pathway (Behrens, 2008). Moreover, the promoter of the APC gene is hypermethylated in up to 30% of renal carcinomas (Dulaimi et al, 2004), suggesting APC loss/reduction may play an important role in the progression of renal carcinoma. However, despite this suggested role in progression, familial adenomatous polyposis (FAP) patients (who are germline heterozygous for APC) do not develop renal carcinoma suggesting APC loss is a very poor initiator of tumourigenesis in the kidney. This has been confirmed using proof of principle experiments in the mouse, where both copies of Apc have been removed from the kidney and only a small fraction of mice develop renal carcinoma. Indeed, using the AhCre transgene, which yields constitutive Cre expression within a high proportion of cells of the kidney, less than 1/3 of mice develop renal carcinoma, despite showing the presence of small premalignant lesions at much earlier ages (Sansom et al, 2005). In contrast, deletion of Apc within the intestinal epithelium rapidly leads to a marked crypt-progenitor cell-like phenotype (Sansom et al, 2004) and deletion of Apc within the LGR5+ stem cell zone leads to adenoma formation in as little as 3 weeks (Barker et al, 2009).
Over the past few years there has been great interest in the role of senescence as a bona fide tumour suppression mechanism in vivo. The phenomenon of oncogene induced senescence (OIS) has provoked particular interest, whereby activation of an oncogene leads to senescence, often in association with a deoxyribonucleic acid (DNA) damage response, and the expression of β-galactosidase, hence senescence-associated β-galactosidase (SA β-gal) (Collado et al, 2005; Collado & Serrano, 2006). Evidence to support this has come from both humans and mice (Braig et al, 2005; Chen et al, 2005; Collado et al, 2005; Michaloglou et al, 2005). Staining of pre-malignant lesions (in humans) such as benign nevi, adenomas of the lung and pancreatic intraepithelial neoplasms (PanINs) has shown them all to be characterized by expression of SA β-gal. These lesions also display upregulation of cell cycle arrest proteins such as p16 and p21, both of which have been implicated in senescence in vitro (Serrano, 1997). Similarly, the conditional activation of oncogenes such as v-Raf murine sarcoma viral oncogene homolog B1 (Braf) or Kirsten rat sarcoma viral oncogene homolog (Kras) in mice has led to the formation of premalignant lesions, associated with markers of senescence, that either fail to progress or rarely progress (Dankort et al, 2007; Dhomen et al, 2009). However, controversy remains in the literature over whether these premalignant lesions really exhibit a permanent irreversible growth arrest and indeed other studies have shown proliferation in equivalent premalignant lesions of the lung (Tan et al, 2001). This raises the question of whether these lesions are truly senescent or exhibit a reversible growth arrest.
There is very little evidence indicating that activated Wnt signalling can drive a senescence programme in vivo. This is despite the fact that Apc loss is known to rapidly drive intestinal adenoma formation but tumours from patients constitutively heterozygous for APC are thought to progress over years rather than months. Most tissue culture and in vivo studies have instead shown that Wnt signalling is either required for, or cooperates with, other mutations to overcome senescence (Delmas et al, 2007). However, in lymphoid cells β-catenin activation has been shown to drive senescence ex vivo, suggesting that the Wnt pathway has the capacity to cause senescence, albeit not in a physiological setting (Xu et al, 2008).
In this study, we show that Apc loss drives a context-dependent senescence response. Within the kidney, Apc loss triggers a p21-dependent senescence program, the abrogation of which drives renal carcinoma. Within the intestine, Apc loss does not invoke a senescence programme, rather driving a p21-independent proliferative response leading to rapid tumourigenesis.
RESULTS AND DISCUSSION
Apc loss leads to senescence in the renal epithelium
Our previous studies investigating Apc loss in the renal epithelium using the AhCre transgene to drive constitutive Cre expression within the kidney have suggested that Apc loss is a relatively poor tumour initiating event (Sansom et al, 2005). First to investigate AhCre mediated recombination within the kidney, we intercrossed the AhCre transgene to the Rosa26 Lox stop Lox (LXL) red fluorescent protein (RFP) reporter, Rosa-tdRFP (Luche et al, 2007). The expression of RFP was readily seen in kidneys removed after birth (P3), at weaning and in older mice (6 months) in wholemount kidneys examined using a an Olympus OV100 camera (Fig 1A). To look at the single cell level, we performed immunohistochemical (IHC) staining for RFP and showed the presence of RFP-positive cells throughout the kidneys both immediately after birth and in aged mice. Levels were at approximately 20–30% of the kidney, with the majority of cells being unrecombined (Supporting Information 1A–C).
Figure 1. Apc deletion within the renal epithelium leads to rapid deletion of recombined cells and an upregulation of senescent markers.

A and B. Kidneys imaged on OV100 microscope.
A. Mice at P3 and 6 months imaged for RFP. Note kidneys from AhCre mice without the Rosa26 tdRFP reporter (labelled Neg) show little RFP positivity, whilst AhCre Rosa26 tdRFP mice (labelled RFP) on figure show marked RFP positivity.
B. Mice imaged at 2 months (2 mo), top panel and 6 months (6 mo) bottom panels for GFP. Top panel (left kidney), note strong positivity for GFP in AhCre+ Z/EG+ Apc+/+ (labelled GFP). Middle kidney is from AhCre− Z/EG+ Apcfl/fl (labelled Neg) shows only autoflourescence. Right kidney shows no detectable GFP positivity in AhCre+ Z/EG+ Apcfl/fl (labelled Apc) kidney at 2 months. In these mice recombination can only been seen via staining for GFP by IHC (Supporting Information 1D–F) as single cells are below the detection of the OV100. Bottom panel shows at 6 months a small number of positive foci within AhCre+ Z/EG+ Apcfl/fl kidneys, suggesting that Apc deficient cells are being deleted.
C. β-catenin IHC showing that there is only a small number of β-catenin nuclear positive cells within the kidneys of AhCre+ Apcfl/fl mice.
D. Bar graph showing percentage of AhCre+ Apcfl/fl mice with single nuclear β-catenin cells across kidney, premalignant lesions and renal carcinoma at 2, 6 and 12 months of age. At least ten mice were aged to each timepoint.
E. β-catenin IHC showing nuclear localization in small renal lesions.
F–H. IHC performed on AhCre+ Apcfl/fl kidneys, showing scattered cells and premalignant lesions staining for p21 (F and G) and p16 (H and I).
J. Co-immunofluorescence (IF) for β-catenin (red) and p21 (green) showing high expression of both within premalignant lesions from AhCre+ Apcfl/fl mice.
K and L IHC for p21 (K) and p16 (L) in renal carcinoma from AhCre+ Apcfl/fl mice, showing markedly reduced levels compared to premalignant lesions. In all cases scale bars represent 20 µm.
To examine the fate of Apc deficient cells within the kidney, we intercrossed AhCre transgenic mice with mice carrying conditionally inactivatable ‘floxed’ Apc580S alleles (AhCre Apcfl/fl) and the Z/enhanced green fluorescent protein (EGFP) Cre reporter transgene. AhCre Apc+/+ Z/EG+ mice showed green fluorescent protein (GFP) expression throughout the kidney in an equivalent manner to the Rosa26 RFP reporter, both on wholemount examination (Fig 1B) and through IHC staining for GFP (Supporting Information 1E). In contrast at weaning, kidneys from AhCre Apcfl/fl Z/EG+ mice showed very low or no GFP expression via wholemount examination (Fig 1B top panel) and only a small number of GFP positive cells by IHC staining on sections (Supporting Information 1F). These kidneys also showed very low levels of the Apc recombined allele within the kidney (Supporting Information 1G) and there were only a small number of cells that had nuclear β-catenin (which is a marker of Apc loss) (Fig 1C). This data suggests that most of the cells in which Apc was deleted were being lost from the kidney. Histological examination for apoptotic cells and IHC staining for cleaved caspase 3 failed to reveal any evidence for apoptosis, suggesting cells were not being cleared by this mechanism (data not shown). We have a recently shown a similar scenario within the pancreas, where activation of KRASG12D leads to the loss of the majority of recombined cells (approximately 30%) and the pancreata is then reconstituted by wild-type non-recombined cells without any obvious effects on the overall fitness of the mouse (Morton et al, 2010).
When we examined the levels of recombination in older mice, although most of the kidney was negative for GFP (via OV100 analysis), a number of discrete GFP foci were evident suggesting that a number of the Apc deficient cells were being retained and may be the source of renal carcinoma. To investigate this further, we then analyzed the kidneys from Apc deficient mice at 2, 6 and 12 months (Fig 1D). We classified the cells that remained following Apc loss into three types: single cells that were scattered throughout the kidney that had high levels of nuclear β-catenin (Fig 1C), small premalignant lesions (Fig 1E) or renal carcinoma. Although all kidneys from the AhCre Apcfl/fl mice showed a significant number of cells with nuclear β-catenin (Fig 1C) compared to none in wild-type mice (Supporting Information 2A) by 6 months, approximately 40% had developed premalignant lesions (Fig 1E), very few mice developed renal carcinoma (at 1 year of age only 8%, 2 out of 24 mice).
These data suggested that a growth arrest/senescence pathway may be constraining tumour progression within these lesions. Currently, a number of OIS markers have been proposed (Collado & Serrano, 2006). These markers are considered to be tissue specific so a definitive set of markers that work robustly in all tissues is still lacking. Therefore we tested a number of these markers in the AhCre Apcfl/fl mice and found upregulation of p21 and p16 in the occasional, histologically normal cell and in all the premalignant lesions of the AhCre Apcfl/fl kidneys (Fig 1F–I). Within wild-type kidneys, no staining for either p21 or p16 was observed (Supporting Information 2B and C). To test whether p21 and β-catenin staining was coincident in lesions we performed joint immunofluorescence (IF) and found high levels of nuclear p21 in lesions that were characterized by high expression of β-catenin (Fig 1J and see controls in Supporting Information 2 D–G). Moreover, to look at the co-expression of β-catenin and p21 in small foci, we performed staining on serial sections and saw high levels of p21 in these small foci of β-catenin positive cells (Supporting Information 2H–K). Importantly, the rare tumours that formed in the AhCre Apcfl/fl mice lost the expression of p21 (Fig 1K) and p16 (Fig 1L), suggesting that loss of these arrest/senescence markers was contributing to tumour progression.
Given the lack of definite markers of senescence, we also addressed whether there was an accumulation of SA β-gal and a strong reduction or absence of proliferation in the premalignant lesions. Staining for SA β-gal was absent from wild-type kidney and present in premalignant lesions in the AhCre Apcfl/fl mice (Fig 2A and B). Importantly, rare tumours from the AhCre Apcfl/fl mice failed to express SA β-gal (Fig 2C), suggesting that those tumours that grew had escaped from senescence, consistent with the strong reduction in the levels of p21 and p16 within the tumours (Fig 1K and L). We performed IHC staining for two independent markers of proliferation: the cell cycle antigen Ki67 and the replication licensing factor mini-chromosome maintenance 2 (MCM2). The levels of both of these markers remained low within premalignant lesions consistent with an induction of senescence within these lesions (Fig 2D–F). Importantly, a marked induction of proliferation was seen in the rare renal carcinomas that arose in the AhCre Apcfl/fl mice (Fig 2G and H) (Mann–Whitney p = 0.01, n = 5, premalignant lesions vs. tumours). To confirm that the premalignant lesions had low proliferative capacity and high p21 despite nuclear β-catenin, we then performed IHC for these markers on serial sections (Fig 2I–L) and found this to occur. Taken together, this data strongly suggests Wnt pathway activation is causing senescence within the renal epithelium. It should be noted that unlike in vitro senescence where growth arrest can be observed to be permanent and irreversible, in vivo the mutation of other oncogenes or tumour suppressor could potentially overcome this to drive tumourigenesis (Bartkova et al, 2006). Given recent data on the immune clearance/killing of senescent cells, it is tempting to speculate that the reduction/loss of the majority of Apc deficient cells within the kidney is due to the clearance or killing of senescent cells (Xue et al, 2007). Future studies to investigate different immunocompromised strains crossed to the Apc deficient mice will allow us to assess if this is occurring. To this end it is interesting to note that the premalignant lesions (which are pan-keratin positive), are surrounded by a clear stroma which is very high in CD3 positive T cells (Supporting Information 3A and B). Moreover, a number of these stromal cells were Ki67 positive in the Apc deficient kidneys and these were completely absent from the wild-type kidney (Fig 2D and E).
Figure 2. Apc deficient lesions are not proliferative.
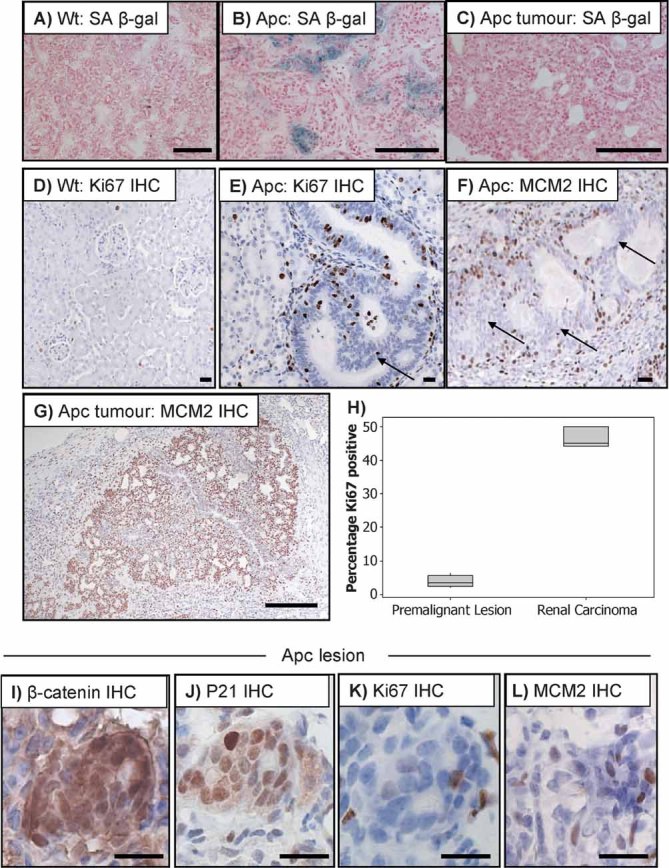
A–C. SA β-galactosidase staining performed on wild-type (A), AhCre+ Apcfl/fl kidneys with premalignant lesions (B) and tumours from AhCre+ Apcfl/fl kidneys (C). Note accumulation of SA β-gal is only observed in the premalignant lesions. Scale bars (A–C) represent 200 µm.
D–E. Ki67 IHC showing only a small subset of cell expressing Ki67 in both wt (D) and small lesions from AhCre+ Apcfl/fl mice (E).
F. MCM2 IHC showing low levels of proliferation in small lesions from AhCre+ Apcfl/fl mice, arrows show epithelial cells with low levels of proliferation. (see Supporting Information 3A for pan keratin staining showing the epithelial cells). Scale bars (D–F) represent 20 µm.
G. IHC for MCM2 showing that renal carcinomas that grow out from AhCre+ Apcfl/fl mice are highly proliferative. Scale bars represent 200 µm.
H. Boxplot showing significantly increased Ki67 staining in renal carcinomas compared to very low levels within the premalignant lesions.
I–L. IHC showing high levels of β-catenin (I) and p21 (J), but low levels of the proliferation markers Ki67 (K), and MCM2 (L) in small Apc deficient renal lesions. Scale bars (I–L) represent 20 µm.
p21 loss robustly cooperates with Apc deletion to cause renal carcinoma
To test the functional significance of the upregulation of OIS markers, we intercrossed AhCre Apcfl/fl mice to p21 knockout mice and aged them until they developed renal carcinoma. Previous studies looking at the effect of p21 gene deletion have shown that p21 knockout mice have a weak predisposition to tumourigenesis, with an average onset of tumour development of 16 months, with the majority of tumours consisting of sarcomas, lymphomas and tumours of vascular and endothelial origins(Martin-Caballero et al, 2001). p21 null mice are also more susceptible to irradiation-induced tumourigenesis (Jackson et al, 2003). Therefore p21 gene deficiency has so far had relatively subtle oncogenic activity in the mouse. However, intercrossing p21−/− mice to our AhCre Apcfl/fl model, where p21 is upregulated in premalignant lesions, had a marked effect. All AhCre Apcfl/fl p21−/− mice rapidly developed signs of renal tumourigenesis (hunching, swelling, anemia due to blood in the urine) and all had to be euthanized by 100 days of age (median lifespan 63 days, Fig 3A). Histological examination of kidneys from these mice shows the development of renal carcinoma in all mice, often with multiple foci (Fig 3B). Renal carcinomas from AhCre Apcfl/fl p21−/− mice did not express SA β-gal (Fig 3C), p16 (Fig 3D) or as expected p21 (Fig 3E), showing that p21 loss is sufficient to overcome the senescence block induced by Apc loss. Importantly, these carcinomas continued to express high levels of β-catenin (Fig 3F), confirming the retention of Apc deficient cells. Immunohistochemistry for Ki67 (Fig 3G) and MCM2 (Fig 3H) showed strong upregulation of both of these proliferation markers, illustrating that p21 deficiency now renders these carcinomas highly proliferative. Given the rampant acceleration of tumourigenesis in a p21 nullizygous background, we also assessed whether heterozygosity for p21 could accelerate tumourigenesis. Remarkably, a rapid acceleration of tumourigenesis in AhCre Apcfl/fl p21+/− mice was observed compared with AhCre Apcfl/fl p21+/+ mice (median lifespan 117 days, Fig 3A). To analyze the kinetics of this, we next examined both lesion area and renal carcinoma formation at 2 months of age (Supporting Information 3C and D). At this stage all AhCre Apcfl/fl p21−/− mice had developed at least one invasive renal carcinoma, as well as having multiple smaller lesions throughout the kidney. Approximately a third of AhCre Apcfl/fl p21+/− mice had developed a renal carcinoma and the majority of mice now had lesions within the kidney. In contrast, AhCre Apcfl/fl mice had very few lesions and no tumours at this time and total lesional area was significantly lower in these mice compared to both AhCre Apcfl/fl p21+/− and AhCre Apcfl/fl p21−/− mice (Mann–Whitney n = 10, p = 0.001, Supporting Information 3D). To confirm that escape from a ‘senescence like’ program was occurring we also aged a small cohort of AhCre Apcfl/fl Ink4a−/− mice. The Ink4A−/− mice lack both p16 and p19. Again all of these developed renal carcinoma by 6 months of age (n = 10, data not shown). Our data provides definitive functional evidence that p21 acts as a key block to tumour progression in the kidney in the absence of Apc. It also highlights the possible reason why FAP patients and ApcMin/+ mice do not develop renal carcinoma, as loss of Apc alone would lead to senescence and a selective disadvantage. Furthermore, our data predicts robust cooperation of the Wnt pathway in renal carcinogenesis once a driver mutation has occurred to block the senescence pathway. To this end we have previously shown that Kras activation in the kidney cooperates with Apc loss to drive renal carcinoma (Sansom et al, 2006). Given the recent data that VHL protein loss may increase Wnt signalling, and that Apc is methylated in renal carcinoma, our findings are relevant to human carcinogenesis. Indeed, recent in vitro and vivo studies have shown that loss of VHL within the kidney results in senescence (Young et al, 2008). Importantly, this study showed the induction of senescence was hypoxia-inducible factor (HIF) independent and therefore may be due to increased levels of Wnt signalling following VHL loss (Behrens, 2008; Chitalia et al, 2008).
Figure 3. p21 loss following Apc deletion leads to rapid onset of renal tumourigenesis.

A. Kaplan–Meier survival graph showing a dramatic acceleration of renal tumourigenesis in AhCre+ Apcfl/fl p21−/− mice (Apc p21−/−, n = 31, median lifespan 63 days, blue line) compared with AhCre+ Apc+/+ (Wt n = 20, <400 days) and AhCre+ Apcfl/fl mice (Apc, n = 23, red line, Log rank v Apcfl/fl p21−/−, p < 0.001). Note there was a marked acceleration of tumourigenesis in AhCre+ Apcfl/fl p21+/− mice (Apc p21+/− median lifespan 117 days, n = 17, green line, Log rank v Apcfl/fl p < 0.001).
B. H&E of a renal tumour in AhCre+ Apcfl/fl p21−/− mice.
C–E. Staining shows absence of senescent markers SA β-gal (C), p16 (D) and p21 (E) in AhCre+ Apcfl/fl p21−/− renal tumours. Note there is a small amount non-specific brown staining in the p21 knockout tumours when p21 IHC is performed (black arrow), however all nuclei are blue showing a complete loss of p21 within the tumours (red arrow).
F. β-catenin IHC showing continued Wnt signalling activation in AhCre+ Apcfl/fl p21−/− deficient renal tumours.
G and H Ki67 IHC (G) and MCM2 IHC (H) showing a marked increase in proliferation in AhCre+ Apcfl/fl p21−/− deficient tumours. Scale bars represent 200 µm.
Apc loss does not lead to senescence within the intestine, even within slowly progressing lesions
These data suggest that the reason why Apc loss is not an initiating oncogenic event in the kidney is due to the induction of senescence. Implicit in this hypothesis is that Apc loss in the intestine should not induce senescence. To investigate this we first conditionally deleted Apc within the intestinal epithelium by inducing recombination in the AhCre Apcfl/fl mice using three injections of β-naphthoflavone in a single day. This leads to near 100% recombination within the small intestinal crypt and produces a marked crypt-progenitor cell like phenotype within 4 days of Apc gene deletion (Sansom et al, 2004). Our previous studies have shown that following Apc loss, a small subset of cells upregulate p21 at the leading edge of the phenotype (Fig 4B) (Sansom et al, 2007). To test whether this might be a senescent population of cells, we stained for SA β-gal and found no evidence of positivity (Fig 4C). Likewise no upregulation of p16 was observed (Fig 4D and E). Next, we stained for the proliferation markers MCM2 and Ki67 and found that every Apc deficient cell in the small intestine was MCM2 (Fig 4G) and Ki67 (Fig 4I) positive, highlighting that there is no senescence following Apc loss in the intestine (albeit over the short term). Given that the AhCre transgene yields predominantly small intestinal recombination (Ireland et al, 2004), we also examined proliferation in the colon following Apc loss using the VillinCreER transgenic mice (Andreu et al, 2005). Within the colon crypt, every cell that had nuclear β-catenin following Apc loss was Ki67 positive (Supporting Information 4).
Figure 4. p21 upregulation following Apc deletion does not induce senescence within the intestinal epithelium.
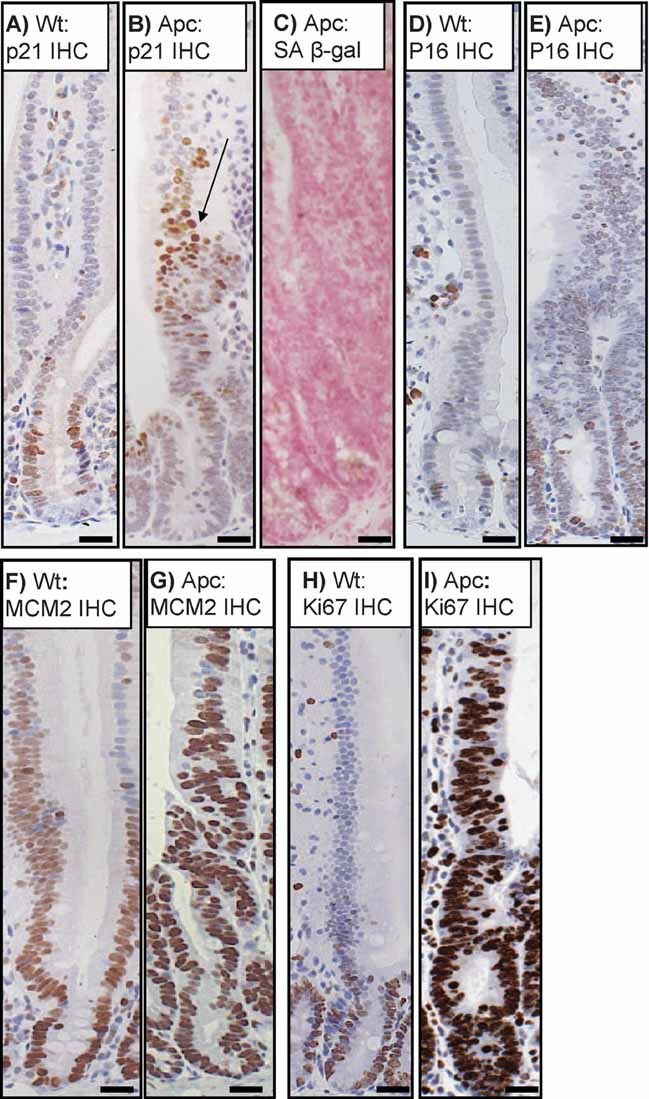
A and B p21 IHC showing a low expression of p21 in a small number of cells in wild-type crypts (labelled Wt) at the crypt villus junction (A), as well as strong upregulation in a subset of cells in the villus following Apc loss in AhCre+ Apcfl/fl mice (labelled Apc) (B).
C. Apc deficient crypts are not senescent as shown by lack of SA β-gal staining.
D and E p16 IHC showing no major upregulation between wt (D) and Apc deficient crypts (E).
F and G IHC showing an upregulation of MCM2 staining in the proliferative crypts of Wt mice (F), MCM2 is upregulated in all Apc deficient cells (G).
H and I IHC showing an upregulation of Ki67 staining in the proliferative crypts of wt mice (H), Ki67 is upregulated in all Apc deficient cells (I). Scale bars represent 20 µm.
To confirm that p21 was not playing a functional role in this process, we induced AhCre Apcfl/fl p21−/− mice at 6 weeks of age with three injections of β-naphthoflavone to yield near constitutive intestinal recombination, and investigated the phenotype of compound mutant Apc p21 knockout intestines. Confirming the lack of any p21-mediated senescence or arrest at the early stages of Apc loss, Apc p21 double knockout intestines were indistinguishable to single Apc knockout intestines (Supporting Information 5).
As Apc mutation occurs as the initiating event in 80% of CRC, and there has been some evidence to suggest that benign adenomas may exhibit features of senescence, we next investigated whether adenomas that are formed following Apc loss show such signs. To do this, we examined OIS in small lesions and adenomas generated from three different models of Apc loss; first, the ApcMin/+ mouse (at 85 days), second, single crypt lesions from the non-stem cell deletion of Apc (at 100 days) and third, adenomas formed through stem cell deletion of Apc (at 35 days). Our previous studies have shown that when Apc is deleted outwith the stem cell zone this leads to protracted tumourigenesis and although adenomas can form, most lesions remain small and do not progress (over a 200–300-day period) (Barker et al, 2009). In all three scenarios, we failed to see upregulation of p16 or SA β-gal (Fig 5D, data not shown), though p21 was expressed within the adenomas of the ApcMin/+ mouse (Fig 5B). To examine the proliferative capacity of these lesions and adenomas we stained for Ki67 and MCM2 (Fig 5C, G, H, K and L). Once again, both lesions and adenomas exhibited high levels of staining showing that Apc loss within the intestinal epithelium does not provoke senescence. Therefore, even in our protracted models we again fail to see senescence in premalignant lesions. The major difference we see between the small lesions and adenomas is in levels of apoptosis; within the small lesions, levels of both proliferation and apoptosis levels are high, whilst in adenomas levels of apoptosis are low, consistent with the notion that apoptotic escape is important for CRC progression (Barker et al, 2009) (apoptotic index, single crypt lesions from non-stem Apc loss 10.4%, adenomas from stem cell deletion 1.5%, Mann–Whitney p = 0.01, n = 5 mice of each genotype).
Figure 5. Apc deletion does not induce OIS within the intestine.

A–D. Small intestinal adenomas arising in ApcMin/+ mice at 85 days. IHC showing high levels of β-catenin (A), p21 (B) and Ki67 (C) within adenomas. (D) Intestinal adenomas from ApcMin/+ mice lack expression of senescence marker SA β-gal.
E. Apc deficient intestinal lesions that grow out from a non-stem cell ‘hit’ (1.0 mg/kg oral gavage), display high levels of β-catenin (E), with some expression of p21 (F) and high levels of proliferative markers Ki67 (G) and MCM2 (H).
I–L. Lgr5-EGFP+-Cre-ER Apcfl/fl mice were treated with a single intraperitoneal injection of tamoxifen to induce recombination in the intestinal stem cell, resulting in intestinal adenoma formation at 24 days. Scale bars (A–H) represent 20 µm. (I) β-catenin IHC showing high levels of expression within intestinal adenomas.
J. p21 IHC showing upregulation in intestinal adenomas.
K and L IHC showing high levels of proliferation as illustrated by Ki67 (K) and MCM2 (L). Scale bars (I–L) represent 200 µm.
To test whether there might be a stage between Apc loss and adenoma formation where senescence may occur, we investigated whether there was any timepoint where we observed senescence from tumour initiation to adenoma formation using stem cell deletion of Apc. Importantly, at all stages up to adenoma formation, there are high levels of Ki67 and 5-bromodeoxyuridine (BrdU) staining (days 10, 15, see Supporting Information 6, adenomas day 20–35, Fig 6). Moreover, all cells within the lesions stain for the proliferative marker MCM2 showing senescence is not occurring. This data is in contrast to when BRAFV600E is activated within the intestine, in this scenario after an initial hyperproliferation there is an induction of senescence. Importantly, the tumours which then form escape from senescence and show methylation of p16 exon 1 (Carragher et al, 2010).
Figure 6. Deletion of p21 or Ink4a does not affect intestinal tumourigenesis.

- Kaplan–Meier survival graph showing no acceleration of intestinal tumourigenesis between AhCre+ Apcfl/+ (Apcfl/+, n = 15, black line) and AhCre+ Apcfl/+ p21−/− (Apcfl/+ p21−/−, n = 24, red line) mice (Log rank, p = 0.476). No significant differences were observed either in total tumour number (Mann–Whitney, p = 0.1893) (B), or average tumour area (Mann–Whitney, p = 0.1346) (C).
- Survival graph showing no acceleration of tumourigenesis between Min (ApcMin/+, n = 20 black line), Min Ink4a+/− (ApcMin/+ Ink4a+/−, n = 23, red line) and Min Ink4a−/− (ApcMin/+ Ink4a−/−, n = 25, green line) mice (Log rank, p = 0.825). No significant differences were observed either in total tumour number (Mann–Whitney p = 0.7405) (E) or average tumour size (Mann–Whitney, p = 0.1601) (F).
Finally, as p21 was expressed in a subset of cells within the adenoma, we tested whether p21 functionally modulated Apc mediated tumourigenesis in the intestine. Thus, 20 AhCre Apcfl/+ p21+/+ and AhCre Apcfl/+ p21−/− mice were induced at weaning with β-naphthoflavone and aged until they developed intestinal tumours as scored by hunching, paling of the feet and weight loss. No significant differences were seen in age at death, tumour burden or average area of tumour in the AhCre Apcfl/+ p21−/− mice compared with the AhCre Apcfl/+ p21+/+ mice (Fig 6A–C, Mann–Whitney, (B) p = 0.3118, (C) p = 0.1346). This data is consistent with a previous study, which showed that p21 deletion did not increase tumourigenesis in Apc1638+/N mice unless they were placed on a western diet of high fat and phosphates and low calcium and vitamin D (Yang et al, 2001). To further test if senescence was playing a role in the intestinal epithelium we also examined whether Ink4a deficiency could accelerate intestinal tumourigenesis. Given the relative longevity of Apcfl/+ mice and the fact that Ink4a−/− mice exhibit spontaneous tumourigenesis from 200 days of age, we approached this question by intercrossing the Ink4a−/− mice to the ApcMin/+ model of tumourigenesis where mice develop intestinal adenomas by 100 days. Once again, no difference in survival, intestinal tumour burden or average area of tumour was observed between ApcMin/+ Ink4a+/+ or ApcMin/+ Ink4a−/− animals (Fig 6D–F, Mann–Whitney, (E) p = 0.7405, (F) = 0.1601). Consistent with this, Apc deficient CRC appears to be one of the few cancers where the INK4A locus is not deleted and in fact high levels of p16INK4a correlate with a bad prognosis and the neighbouring gene MTAP (which is often co-deleted with the INK4A locus) is overexpressed (Bataille et al, 2005; Wassermann et al, 2009).
Taken together, these data show a clear difference in the pattern of cooperating oncogene and tumour suppressor mutations between the intestine and the kidney. This reinforces the notion that deregulation of Wnt signalling following Apc loss is sufficient to drive a proliferative fate in the intestine, and thus other mutations such as those required for escape from senescence are not required within the intestinal epithelium. This is particularly relevant for INK4A loss, as this locus is mutated at very high levels in pancreatic cancer and melanoma, both which have been associated with senescent premalignant lesions (Caldas et al, 1994; Michaloglou et al, 2005).
Tissue specific c-Myc suppression of p21
Within the intestinal epithelium, we have previously shown that loss of c-Myc strongly suppresses the phenotypes of Apc loss (Sansom et al, 2007). One of the key functions of c-Myc is to repress p21 through its interaction with Miz1, and we have previously shown that p21 is de-repressed when Apc and c-myc are co-deleted within the intestinal epithelium (Sansom et al, 2007; Seoane et al, 2002). However, in the renal epithelium we observed a clear induction of p21 coincidentally with the accumulation of β-catenin suggesting that in this context c-Myc expression was not sufficient to repress p21. In other tissues such as the skin, the repression of p21 by c-Myc is crucial for tumourigenesis (Oskarsson et al, 2006). Given that increased c-Myc is not sufficient to repress p21 within the kidney following Apc loss, tumourigenesis in the Apc p21 double knockout may proceed in a c-Myc independent fashion. To test this, we generated triple knockout (TKO) AhCre Apcfl/fl, c-Mycfl/fl p21−/− mice and assessed renal tumour formation in these mice. As can be seen in Fig 7A, renal tumourigenesis still proceeded rapidly in these mice (equivalent to the compound Apc p21 knockout mice), despite the absence of c-Myc. The TKO renal tumours are histologically identical to Apc p21 deficient mice renal tumours (Fig 7B). Similarly, TKO tumours display a continued lack of SA β-gal (Fig 7C), whilst expressing high levels of β-catenin (Fig 7D) and Ki67 (Fig 7G). IHC staining for c-Myc shows complete ablation of protein in TKO tumours (Fig 7F) and an upregulation in Apc p21 tumours (Fig 7E). As with Apc and p21 deficient renal carcinomas, the TKO tumours are highly proliferative as confirmed by MCM2 IHC (Fig 7H). We have also demonstrated that the renal carcinomas that develop in the AhCre Apcfl/fl KrasV12 mice form in the absence of c-Myc protein (see Supporting Information 7). This again contrasts with Apc deficient intestinal epithelium which absolutely requires c-Myc to functionally manifest the phenotypes of Apc loss and tumour formation (Sansom et al, 2007). In these cases, tumours are highly proliferative showing that KrasV12 activation is also able to overcome the senescence following Apc loss. Tumours from these mice again lack p16 (data not shown) and the majority have high levels of p21 suggesting that KrasV12 in this scenario overcomes the ability of p21 to induce growth arrest/senescence (Supporting Information 8). Interestingly there is a small subset of tumours (approximately 10%) that have no p21 staining within them.
Figure 7. c-myc deletion does not affect onset of tumour development.

A. Kaplen–Meier survival graph showing no difference in time to renal tumourigenesis between AhCre+ Apcfl/fl p21−/− (Apc p21, n = 31, black line) and AhCre+ Apcfl/fl c-Mycfl/fl p21−/− mice (Apc c-Myc p21, n = 20, red line) (Log rank p = 0.571), illustrating that c-myc deletion does not affect onset of renal tumourigenesis.
B. H&E showing AhCre+ Apcfl/fl c-Mycf/fl p21−/− triple knockout renal tumours.
C. Absence of SA β-gal staining in AhCre+ Apcfl/fl c-Mycf/fl p21−/− triple knockout renal tumours.
D. β-catenin IHC in AhCre+ Apcfl/fl c-Mycf/fl p21−/− triple knockout renal tumours shows nuclear localization.
E. c-Myc IHC showing upregulation of c-Myc in AhCre+ Apcfl/fl p21−/− renal tumours.
F. c-Myc IHC shows lack of c-Myc expression in AhCre+ Apcfl/fl c-Mycf/fl p21−/− triple knockout renal tumours, showing c-Myc is not required for tumourigenesis. Scale bars (B–F) represent 200 µm.
G and H Strong upregulation of proliferative markers Ki67 (G) and MCM2 (H) showing AhCre+ Apcfl/fl c-Mycf/fl p21−/− triple knockout renal tumours deficient tumours are highly proliferative. Scale bars (G and H) represent 20 µm.
Taken together our data show that Apc loss within the renal epithelium is strongly disadvantageous, with cells upregulating p21 and undergoing senescence. This provides a clear rationale to explain why Apc mutations are not initiating oncogenic events in the kidney. In the intestine, where Apc loss drives carcinogenesis, Apc mutation results in a strong proliferative program. This study therefore yields crucial insights into the context-specific outcome of Wnt signalling and suggests that senescence is not a key barrier to tumourigenesis in CRC that has been initiated with an APC mutation. Several studies have reported SA β-gal positive cells in CRC, coincident with the activation of the DNA damage response (Bartek et al, 2007; Bartkova et al, 2005). More recently it has been shown that the inflammatory mediators required for senescence (such as IL-6) were constraining proliferation of premalignant intestinal lesions and senescent premalignant lesions were observed within CRC (Kuilman et al, 2008). One potential explanation for these findings is that other oncogenic/tumour suppressor mutations may have initiated these lesions and driven senescence, e.g. BRAF or KRAS within the colon. However, so far, KrasG12V and KrasG12D, liver kinase B1 (LKB1) and phosphatase and tensin homolog (PTEN) mutation (Marsh et al, 2008; Sansom et al, 2006; Shorning et al, 2009; Tuveson et al, 2004) have all been investigated in the murine intestinal epithelium, and none induce senescence, indeed most induce proliferation. Of the remaining oncogenic events in CRC, the BRAFV600E mutation is found in approximately 12%, normally in a mutually exclusive manner with Apc gene mutation (Davies et al, 2002). These mutations are associated with a distinct type of CRC: serrated adenocarcinoma. BRAFV600E mutation has been clearly shown to drive senescence in mouse models of lung cancer and melanoma, and human nevi express SA β-gal. It is thus particularly pertinent to this study that BRAFV600E activation in the murine intestine leads to senescence (Carragher et al, 2010) and in this instance p16INK4A loss causes tumour progression. Moreover, although previous studies have shown KrasG12V/D mutation does induce senescence within the small intestine, a recent study has suggested that targeting KRASG12D to the colon leads to OIS (Bennecke et al, 2010). Importantly, combined KRASG12D mutation in the colon with INK4/ARF deletion overcame the senescence and led to rapid serrated adenocarcinoma formation. This was also observed in human serrated adenocarcinoma. Therefore, these studies highlight that the nature of the initiating event produces the selective pressure for specific mutations that drive progression and this may underpin the progression of two distinct forms of CRC (Fig 8 Model). Moreover, it is likely that mutations in tumour suppressors such as APC or oncogenes such as BRAF occur throughout the organism equally, however in most cases these mutations do not lead to a selective advantage and in many cases a disadvantage. Therefore, clear driver mutations are seen in different cancer types due to the impact of that specific mutation in the cell of origin of that particular tumour. It is thus possible that there are many circumstances equivalent to the scenario we observe following Apc loss in the kidney, where a mutation of a bona fide tumour suppressor gene in one context results in tumour development, but drives senescence in another context and blocks tumourigenesis.
Figure 8. Model highlighting that both the initiating mutation and the tissue context determine the biological outcome and downstream tumour progression.

MATERIALS AND METHODS
Mouse experiments
All experiments were performed under the UK Home Office guidelines. Outbred male mice from 6 to 12 weeks of age were used which were segregating for the C57BLJ and S129 genomes. The alleles used were as follows: c-Mycfl, AhCre, Apc580S, p21−/−, Z/EGFP reporter transgene ROSA-tdRFP, Lgr5-EGFP-IRES-creERT2 and VillinCreER (Baena et al, 2005; Barker et al, 2007; Brugarolas et al, 1995; Ireland et al, 2004; Luche et al, 2007; Novak et al, 2000; Sansom et al, 2004). Genotyping was performed by Transnetyx. Semi-quantitative polymerase chain reaction (PCR) for the recombined Apc allele was performed as described in (Sansom et al, 2005).
To address the effect of p21 and Apc deletion within the renal epithelium AhCre, Apcfl/fl mice were bred with p21−/− mice to generate AhCre Apcfl/fl p21−/− mice. As previously described (Sansom et al, 2005), in the absence of the inducer β-naphthoflavone, sporadic Cre mediated recombination occurs in the renal epithelium in the S and comma shaped bodies. Mice were then aged until developing signs of renal failure (blood in the urine, hunching and swollen kidneys).
In order to address the effect of p21 and Apc deletion within the intestinal epithelium, AhCre Apcfl/fl p21−/− mice were given three intraperitoneal (IP) injections of 80 mg/kg β-naphthoflavone in a single day, which yields nearly constitutive recombination in the murine small intestine. Analyses of all intestinal phenotypes were examined at day 4 after induction.
The paper explained
PROBLEM:
Senescence is thought to play a key tumour suppressor activity during carcinogenesis. However, it is unclear whether there are certain tissues where senescence is particularly potent at blocking tumourigenesis. Moreover, it is uncertain whether there are specific oncogenic/tumour suppressor mutations that drive senescence in a tissue-specific manner.
RESULTS:
We show that deletion of the Apc tumour suppressor gene induces senescence within the kidney and, in contrast, rapid cell proliferation within the intestinal epithelium. Consequently, in the intestinal epithelium, Apc loss alone was sufficient to drive adenoma formation, whilst within the kidney additional loss of p21 was required to initiate renal carcinoma.
IMPACT:
In up to 80% of CRCs, Apc is lost and our data show that senescence does not block tumour initiation in this context. Instead, other oncogenic events such as BrafV600E and KrasG12D mutation drive senescence within the intestinal epithelium, which selects for the loss of tumour suppressor genes that block this process, e.g. INK4. Within the kidney, Apc loss poorly initiates tumourigenesis and only provokes tumourigenesis when combined with a mutation that overcomes senescence. Therefore, initiating mutations drive senescence in a tissue-specific manner and shape the evolutionary landscape for the subsequent mutations that allow tumour progression. These differences could underlie CRC progression and determine whether tumours develop in a serrated or non-serrated pathway.
For tumourigenic studies, AhCre Apcfl/+, AhCre Apcfl/+ p21+/− and AhCre Apcfl/+ p21−/− mice were induced at 6 weeks of age with three injections of 80 mg/kg β-naphthoflavone (IP) and monitored until developing signs of intestinal illness (paling feet, hunching).
Lgr5-EGFP-IRES-creERT2 Apcfl/fl mice were generated by breeding Apcfl/fl and Lgr5-EGFP-IRES-creERT2 mice as previously described (Barker et al, 2009). To induce recombination of Apc within the LGR5 positive stem cells, mice 6 weeks of age were induced with a single IP injection of 3 mg tamoxifen in sunflower oil and monitored until showing signs of intestinal illness.
To generate non-stem cell intestinal adenomas, AhCre+ Apcfl/fl mice were orally gavaged with 1 mg/kg β-naphthoflavone in corn oil and monitored until showing signs of intestinal illness (Barker et al, 2009).
To examine senescence in the colon, VilCreERApcfl/fl mice were given three IP injections of 80 mg/kg tamoxifen daily, to generate near 100% recombination in the colon, and were harvested at day 4 post-induction (El Marjou et al, 2004).
Analysis of survival and tumourigenic cohorts
For each survival cohort, the Kaplan–Meier estimator was used to estimate the survival function of each cohort. p Values were determined from Log rank, and Mann–Whitney tests, where significance was determined as <0.04 (Kaplan & Meier, 1958).
Tissue isolation
At the appropriate time point, mice were killed and both kidneys (including cystic tumours) were removed and fixed in 4% formalin, overnight at 4°C for no more than 24 h before processing and were then paraffin embedded. The small intestine was removed and flushed with water. Intestines were dissected as follows: the proximal 7 cm was mounted ‘en face’ and fixed overnight in methacarn (methanol, chloroform and acetic acid; 4:2:1) and paraffin embedded. The following 5 cm was divided into 1 cm lengths, bundled using surgical tape and then fixed in 4% formaldehyde at 4°C for no more than 24 h before processing. The remainder was fixed in methacarn and then paraffin embedded. For kidney wholemount analysis, kidneys were removed and imaged using the OV100 microscope as described previously (Doyle et al, 2010; Morton et al, 2010).
Assaying apoptosis, mitosis and crypt size in vivo
Apoptosis, crypt size and mitotic index were scored from H&E stained sections as previously described (Sansom et al, 2004). For each analysis, 25 full crypts were scored from at least three mice of each genotype. Apoptosis was independently confirmed by immunohistochemical staining with an antibody against active caspase 3 (1:750, R & D systems).
Assaying proliferation and migration in vivo
In order to examine levels of proliferation, mice were injected with 250 µl of BrdU (GE Healthcare) 2 h prior to being sacrificed. Similarly, in order to score migration, mice were injected with BrdU (GE Healthcare) 24 h prior to being sacrificed. Immunohistochemical staining for BrdU was then performed using an anti-BrdU antibody (BD 1:500). At least three mice were used for each genotype and timepoint.
Immunohistochemistry/immunoflorescence
Primary antibodies used for immunohistochemistry or immunoflorescence are as follows: p21 (1:500 Santa Cruz, sc471), β-catenin (1:50, Transduction labs, 610154), p16 (1:25, Santa Cruz M-156), SA-β-gal (Senescence β-Galactosidase Staining Kit, Cell signalling pH 5.5), MCM2 (1:200, Cell signalling, 4007), Ki67 (1:250, Labvision, VP-K452), c-myc (1:500, Santa Cruz, N-262, sc764), Keratin (1:50, Thermo scientific, MS-343), CD3 (1:100, Dako, A0452), GFP (1:1000, Abcam ab6556), RFP (1:200, Abcam, ab34771).
Acknowledgments
OJS is funded by a Cancer Research UK programme grant. Thanks to Slyvie Robine for providing VillinCreER mice and Manuel Serrano for INK4A mice, Colin Nixon for histology, biological services for animal experiments and Jen Morton for helpful discussions. The Rosa-tdRFP mice were imported from the EMMA repository (strain 02112) and were a kind gift from Hans Jörg Fehling.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
Acquisition of data: A. M. Cole, R. A. Ridgway, L. Parry, S. E. Derkits. Analyses and interpretation of data: A. M. Cole, R. A. Ridgway, S. E. Derkits, O. J. Sansom. Contribution of reagents/materials: N. Barker, H. Clevers, A. R. Clarke. Manuscript preparation: A. M. Cole, R. A. Ridgway, S. E. Derkits, O. J. Sansom. Manuscript editing: R. A. Ridgway, O. J. Sansom.
For more information
Cancer Research UK:
http://www.cancerresearchuk.org
Owen Sansom webpage:
Online Mendelian Inheritance in Man – APC:
http://www.ncbi.nlm.nih.gov/omim/611731
Accompanying Research Article:
DOI 10.1002/emmm.201000099
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Andreu P, Colnot S, Godard C, Gad S, Chafey P, Niwa-Kawakita M, Laurent-Puig P, Kahn A, Robine S, Perret C, et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following Apc loss in the mouse intestine. Development. 2005;132:1443–1451. doi: 10.1242/dev.01700. [DOI] [PubMed] [Google Scholar]
- Baena E, Gandarillas A, Vallespinos M, Zanet J, Bachs O, Redondo C, Fabregat I, Martinez C, de Alboran IM. c-Myc regulates cell size and ploidy but is not essential for postnatal proliferation in liver. Proc Natl Acad Sci USA. 2005;102:7286–7291. doi: 10.1073/pnas.0409260102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608–611. doi: 10.1038/nature07602. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J, Bartkova J. DNA damage response as an anti-cancer barrier—damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle. 2007;6:2344–2347. doi: 10.4161/cc.6.19.4754. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LVF, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bataille F, Rogler G, Modes K, Poser I, Schuierer M, Dietmaier W, Ruemmele P, Mühlbauer M, Wallner S, Hellerbrand C, et al. Strong expression of methylthioadenosine phosphorylase (MTAP) in human colon carcinoma cells is regulated by TCF1/[beta]-catenin. Lab Invest. 2005;85:124–136. doi: 10.1038/labinvest.3700192. [DOI] [PubMed] [Google Scholar]
- Behrens J. One hit, two outcomes for VHL-mediated tumorigenesis. Nat Cell Biol. 2008;10:1127–1128. doi: 10.1038/ncb1008-1127. [DOI] [PubMed] [Google Scholar]
- Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, Arkan MC, Kirchner T, Greten FR. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–146. doi: 10.1016/j.ccr.2010.06.013. [DOI] [PubMed] [Google Scholar]
- Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–320. doi: 10.1016/s0092-8674(00)00122-7. [DOI] [PubMed] [Google Scholar]
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters A, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell-cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- Caldas C, Hahn SA, Dacosta LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8:27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
- Carragher LAS, Snell KR, Giblett SM, Aldridge VSS, Patel B, Cook SJ, Winton DJ, Marais R, Pritchard CA. V600EBraf induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med. 2010;2:xx–xx. doi: 10.1002/emmm.201000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZB, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, Bharti A, Seldin DC, Lecker SH, Dominguez I, et al. Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol. 2008;10:1208–1216. doi: 10.1038/ncb1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado M, Serrano M. The power and the promise of oncogene-induced senescence markers. Nat Rev Cancer. 2006;6:472–476. doi: 10.1038/nrc1884. [DOI] [PubMed] [Google Scholar]
- Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M, Benguria A, Zaballos A, Flores JM, Barbacid M, et al. Tumour biology—senescence in premalignant tumours. Nature. 2005;436:642. doi: 10.1038/436642a. [DOI] [PubMed] [Google Scholar]
- Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAF(V600E)-induced lung tumors. Genes Dev. 2007;21:379–384. doi: 10.1101/gad.1516407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Delmas V, Beermann F, Martinozzi S, Carreira S, Ackermann J, Kumasaka M, Denat L, Goodall J, Luciani F, Viros A, et al. Beta-catenin induces immortalization of melanocytes by suppressing p16(INK4a) expression and cooperates with N-Ras in melanoma development. Genes Dev. 2007;21:2923–2935. doi: 10.1101/gad.450107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, Dias SD, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
- Doyle B, Morton JP, Delaney DW, Ridgway RA, Wilkins JA, Sansom OJ. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J Pathol. 2010;222:129–137. doi: 10.1002/path.2748. [DOI] [PubMed] [Google Scholar]
- Dulaimi E, de Caceres II, Uzzo RG, Al-Saleem T, Greenberg RE, Polascik TJ, Babb JS, Grizzle WE, Cairns P. Promoter hypermethylation profile of kidney cancer. Clin Cancer Res. 2004;10:3972–3979. doi: 10.1158/1078-0432.CCR-04-0175. [DOI] [PubMed] [Google Scholar]
- El Marjou F, Janssen KP, Chang BHJ, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Et Biophys Acta-Rev Cancer. 2003;1653:1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Engelman RW, Coppola D, Cantor AB, Wharton W, Pledger WJ. p21(Cip1) nullizygosity increases tumor metastasis in irradiated mice. Cancer Res. 2003;63:3021–3025. [PubMed] [Google Scholar]
- Kaplan EL, Meier P. Nonparametric-estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LCW, Douma S, van Doom R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–1031. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- Luche H, Weber O, Nageswara Rao T, Blum C, Fehling HJ. Faithful activation of an extra-bright red fluorescent protein in “knock-in” Cre-reporter mice ideally suited for lineage tracing studies. Eur J Immunol. 2007;37:43–53. doi: 10.1002/eji.200636745. [DOI] [PubMed] [Google Scholar]
- Marsh V, Winton DJ, Williams GT, Dubois N, Trumpp A, Sansom OJ, Clarke AR. Epithelial Pten is dispensable for intestinal homeostasis but suppresses adenoma development and progression after Apc mutation. Nat Genet. 2008;40:1436–1444. doi: 10.1038/ng.256. [DOI] [PubMed] [Google Scholar]
- Martin-Caballero J, Flores JM, Garcia-Palencia P, Serrano M. Tumor susceptibility of p21(Waf1/Cip1)-deficient mice. Cancer Res. 2001;61:6234–6238. [PubMed] [Google Scholar]
- Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst C, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAF(E600)-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci USA. 2010;107:246–251. doi: 10.1073/pnas.0908428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–155. [PubMed] [Google Scholar]
- Oskarsson T, Essers MAG, Dubois N, Offner S, Dubey C, Roger C, Metzger D, Chambon P, Hummler E, Beard P, et al. Skin epidermis lacking the c-myc gene is resistant to Ras-driven tumorigenesis but can reacquire sensitivity upon additional loss of the p21(Cip1) gene. Genes Dev. 2006;20:2024–2029. doi: 10.1101/gad.381206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Griffiths DFR, Reed KR, Winton DJ, Clarke AR. Apc deficiency predisposes to renal carcinoma in the mouse. Oncogene. 2005;24:8205–8210. doi: 10.1038/sj.onc.1208956. [DOI] [PubMed] [Google Scholar]
- Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci USA. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Meniel VS, Muncan V, Phesse TJ, Wilkins JA, Reed KR, Vass JK, Athineos D, Clevers H, Clarke AR. Myc deletion rescues Apc deficiency in the small intestine. Nature. 2007;446:676–679. doi: 10.1038/nature05674. [DOI] [PubMed] [Google Scholar]
- Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–250. doi: 10.1038/73448. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–734. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- Serrano M. The tumor suppressor protein p16(INK4a) Exp Cell Res. 1997;237:7–13. doi: 10.1006/excr.1997.3824. [DOI] [PubMed] [Google Scholar]
- Shorning BY, Zabkiewicz J, McCarthy A, Pearson HB, Winton DJ, Sansom OJ, Ashworth A, Clarke AR. Lkb1 deficiency alters goblet and paneth cell differentiation in the small intestine. PLoS One. 2009;4:e4264. doi: 10.1371/journal.pone.0004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan DF, Huberman JA, Hyland A, Loewen GM, Brooks JSJ, Beck AF, Todorov IT, Bepler G. MCM2—a promising marker for premalignant lesions of the lung: a cohort study. BMC Cancer. 2001;1:7. doi: 10.1186/1471-2407-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- Wassermann S, Scheel SK, Hiendlmeyer E, Palmovist R, Horst D, Hlubek F, Haynl A, Kriegl L, Reu S, Merkel S, et al. P16(INK4a) is a beta-catenin target gene and indicates low survival in human colorectal tumors. Gastroenterology. 2009;136:196–205. doi: 10.1053/j.gastro.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Xu M, Yu Q, Subrahmanyam R, Difilippantonio MJ, Ried T, Sen JM. Beta-catenin expression results in p53-independent DNA damage and oncogene-induced senescence in prelymphomagenic thymocytes in vivo. Mol Cell Biol. 2008;28:1713–1723. doi: 10.1128/MCB.01360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WC, Mathew J, Velcich A, Edelmann W, Kucherlapati R, Lipkin M, Yang K, Augenlicht LH. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a western-style high-risk diet by altering cell maturation in the intestinal mucosa. Cancer Res. 2001;61:565–569. [PubMed] [Google Scholar]
- Young AP, Schlisio S, Minamishima YA, Zhang Q, Li L, Grisanzio C, Signoretti S, Kaelin WG., Jr VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008;10:361–369. doi: 10.1038/ncb1699. [DOI] [PubMed] [Google Scholar]
- Zhou MI, Foy RL, Chitalia VC, Zhao J, Panchenko MV, Wang HM, Cohen HT. Jade-1, a candidate renal tumor suppressor that promotes apoptosis. Proc Natl Acad Sci USA. 2005;102:11035–11040. doi: 10.1073/pnas.0500757102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.