

Abstract
Sepsis is a deadly disease characterized by the inability to regulate the inflammatory–coagulation response in which the endothelium plays a key role. The cause of this perturbation remains poorly understood and has hampered the development of effective therapeutics. Matrix metalloproteases (MMPs) are involved in the host response to pathogens, but can also cause uncontrolled tissue damage and contribute to mortality. We found that human sepsis patients had markedly elevated plasma proMMP-1 and active MMP-1 levels, which correlated with death at 7 and 28 days after diagnosis. Likewise, septic mice had increased plasma levels of the MMP-1 ortholog, MMP-1a. We identified mouse MMP-1a as an agonist of protease-activated receptor-1 (PAR1) on endothelial cells. MMP-1a was released from endothelial cells in septic mice. Blockade of MMP-1 activity suppressed endothelial barrier disruption, disseminated intravascular coagulation (DIC), lung vascular permeability as well as the cytokine storm and improved survival, which was lost in PAR1-deficient mice. Infusion of human MMP-1 increased lung vascular permeability in normal wild-type mice but not in PAR1-deficient mice. These findings implicate MMP-1 as an important activator of PAR1 in sepsis and suggest that therapeutics that target MMP1-PAR1 may prove beneficial in the treatment of sepsis.
Keywords: endothelium, MMP-1, MMP-1a, PAR1, sepsis
→See accompanying article http://dx.doi.org/10.1002/emmm.201100146
INTRODUCTION
Severe sepsis is a leading cause of acute hospital admissions and often complicates the clinical course of medical and surgical patients treated for other diseases (Moore et al, 2010; Remick, 2007). Identification of high-risk patients with clinically useful prognostic biomarkers remains a substantial challenge (Shapiro et al, 2009). Sepsis is often characterized by marked derangements of the pro-inflammatory, anti-inflammatory and coagulation responses, a condition known as systemic inflammatory response syndrome (SIRS; Bone et al, 1992; Hotchkiss & Karl, 2003). SIRS develops when the initial, appropriate innate immune response becomes amplified and then dysregulated. High levels of pathogen-associated molecular pattern mediators, such as endotoxin or lipopolysaccharide (LPS) and other bacterial components, can initiate a massive increase in systemic inflammatory mediators—known as a ‘cytokine storm’—that contribute to organ failure and other adverse outcomes (Rittirsch et al, 2008; van der Poll & Opal, 2008). Endothelial barrier integrity is disrupted in sepsis and SIRS causing extravascular fluid leakage and septic shock. Widespread exposure of sub-endothelial tissue factor activates the blood clotting cascade resulting in disseminated intravascular coagulation (DIC), which may further contribute to organ failure and death (Esmon, 2004; Pawlinski et al, 2004; Slofstra et al, 2003).
Currently, there are no single agents that sufficiently treat and sustain septic patients (Dellinger et al, 2008). Therapeutic approaches that block the inflammatory response have largely proven unsuccessful in clinical trials and the incidence of sepsis continues to rise (Dellinger et al, 2008; Dombrovskiy et al, 2007; Remick, 2003). Sepsis and SIRS trigger the coagulation system in which protease-activated receptor-1 (PAR1) plays a pivotal role (Esmon, 2004; Neyrinck et al, 2009; Rittirsch et al, 2008). In this regard, the only approved therapeutic agent for the treatment of severe sepsis is activated protein C (APC), which is a protease agonist of PAR1 (Kerschen et al, 2007; Riewald et al, 2002). However, treatment of septic patients with APC has only shown an absolute reduction in mortality of 6.1% and increases the risk for serious bleeding, therefore, additional therapeutic modalities are critically needed for this disease (Bernard et al, 2001; Dellinger et al, 2008; Kerschen et al, 2007). Thrombin, the prototypical PAR1 agonist, activates PAR1 during sepsis, however, thrombin inhibitors do not completely mitigate against LPS-induced lung vascular permeability and mortality in mouse models (Kaneider et al, 2007) suggesting that other endotoxin-dependent factors may mediate endothelial barrier dysfunction.
Emerging evidence suggests that matrix metalloproteases (MMPs) play critical roles in endothelial function, inflammation and coagulation (Rodriguez et al, 2010; Vanlaere & Libert, 2009). The MMP collagenases include MMP-1, MMP-8, MMP-13 and the mouse ortholog of MMP-1, MMP-1a (Col-A; Balbin et al, 2001). It was recently shown that MMP-1 cleaves and activates PAR1 in the context of cancer and arterial thrombosis (Boire et al, 2005; Goerge et al, 2006; Trivedi et al, 2009). Here, we investigated whether MMP1-PAR1 signalling contributes to the lethal sequelae of sepsis. We found a highly significant correlation between plasma proMMP-1 levels and survival outcomes over 28 days in human patients newly diagnosed with sepsis. In mouse models of sepsis, MMP-1a and collagenase activity rapidly increased in the plasma early after the onset of sepsis. Inhibition of MMP-1a activity had highly beneficial effects on survival, vascular permeability, systemic inflammation and DIC. The effects of inhibition of MMP-1a activity were lost in PAR1-deficient mice but retained in PAR2-deficient mice, indicating that PAR1 is required to mediate the effects of the metalloprotease activity. These findings suggest that MMP1-PAR1 plays an unexpectedly important role in the lethal sequelae of sepsis and that inhibiting MMP1-PAR1 may provide beneficial effects.
RESULTS
Plasma MMP-1 correlates with increased mortality in human sepsis patients
Recent studies (Boire et al, 2005; Trivedi et al, 2009) have identified MMP-1 as a new agonist of PAR1, a pivotal regulator of the inflammatory–coagulation response in sepsis. To investigate the potential role of MMP-1 in human sepsis, we measured proMMP-1 and active MMP-1 in the plasma from 50 patients newly diagnosed with severe sepsis or septic shock. Prior to blood collection or treatment, extensive clinical data was collected including whether the patient had DIC, shock and evidence of organ failure such as hepatobiliary dysfunction (HBD) or acute kidney injury (AKI). Sepsis patients had a significant 18-fold increase in mean levels of proMMP-1 and a significant 8.7-fold increase in mean levels of active MMP-1 in their plasma at time of enrollment relative to healthy controls (Fig 1A and B). Patients with low proMMP-1 levels of ≤1.0 ng/ml had a trend (p = 0.06) towards increased survival over 28 days as compared to patients with proMMP-1 >1.0 ng/ml (Supporting information Fig S1). Active MMP-1 was found to significantly correlate with proMMP-1 levels in plasma (Fig 1C). As a marker of the inflammatory cytokine response, which occurs in septic patients with SIRS, plasma IL-8 was also found to significantly correlate with proMMP-1 (Fig 1D). Forward stepwise regression was performed to model proMMP-1, active MMP-1 and IL-8 on the following predictors: DIC, HBD, gender, AKI and death by day 7 or 28 (Fig 1E). We determined the correlation between the outcome variable (proMMP-1, active MMP-1 or IL-8) and each predictor while controlling for the effect of all the other predictors. ProMMP-1 had a significant positive correlation with both early and late death, HBD, AKI and male gender (Fig 1E). Active MMP-1 had a significant positive correlation with both early and late death, HBD and male gender (Fig 1E). Blood levels of proMMP-1 and active MMP-1 did not significantly differ when compared between the type of microorganism that caused sepsis, site of infection, patient's age or the presence or absence of blood stream infection. There was a non-significant trend of proMMP-1 and active MMP-1 correlating with shock. ProMMP-1 and active MMP-1 levels had a significant negative correlation with DIC suggesting that proMMP-1/active MMP-1 was already depleted following the consumptive coagulopathy and systemic platelet activation (Yaguchi et al, 2004), which had occurred in the DIC patients prior to enrollment in the study. IL-8 was found to be significantly correlated with early death, organ failure (HBD and AKI), but not late death, DIC nor gender (Fig 1E). We also examined whether the other clinical parameters (DIC, HBD, AKI, shock and gender) were independently correlated with survival. Of these, only DIC was significantly correlated (p = 0.03) with worse survival over 28 days.
Figure 1. Plasma MMP-1 levels correlate with poor outcome and mortality in human sepsis patients.

A,B. Human proMMP-1 (A) and active MMP-1 (B) was measured by ELISA in the plasma of healthy volunteers (n = 10) and sepsis patients (n = 50; 29 males, 21 females).
C. Scatterplot with linear regression fit of the natural log (LN) of active MMP-1 levels versus the LN of proMMP-1 levels in healthy individuals and sepsis patients (n = 55).
D. Scatterplot with linear regression fit of the natural log (LN) of IL-8 levels versus the LN of proMMP-1 levels in healthy individuals and sepsis patients (n = 55).
E. Table summarizing forward stepwise regression models where either proMMP-1, active MMP-1 or IL-8 is the dependent variable and disseminated intravascular coagulation (DIC), hepatobiliary dysfunction (HBD), acute kidney injury (AKI), gender and death at day 7 or 28 were used as predictor variables.
Systemic MMP-1a levels rise in the plasma of septic mice
The markedly elevated levels of proMMP-1 observed in the septic patients would lead to the prediction that septic mice may also have a rise in collagenase activity. We induced polymicrobial sepsis in mice using cecal ligation and puncture (CLP; Doi et al, 2009; Kaneider et al, 2005). In the CLP model, the cecum is ligated and punctured resulting in a slow leakage of faecal matter into the peritoneal cavity, which models septic peritonitis in humans. Plasma collected from septic mice had significant increases in collagenase activity at 2 h after CLP surgery (Fig 2A) and within 30 min after sublethal Escherichia coli-derived LPS injection (10 mg/kg; Fig 2B). Peak collagenase activity slightly declined after ∼4–6 h following CLP or LPS injection but still remained elevated versus baseline (Fig 2A and B). Both the CLP and LPS-induced collagenase activity was completely inhibited to baseline by ex vivo addition of the MMP-1 inhibitor, MMP-Inh-1 (FN-439; Odake et al, 1994) (Fig 2A and B).
Figure 2. Systemic levels of mouse MMP-1a and collagenase activity are elevated in septic and endotoxemic mice.

- Time course of DQ-collagenase activity of plasma collected from mice that underwent cecal ligation and puncture (CLP) in the presence or absence of MMP-Inh-1 (3 µM). All samples contained MMP-Inh-8 (12 nM) plus MMP-Inh-9/13 (3 nM). p < 0.05 versus MMP-Inh-1 at corresponding time points (n = 3).
- Collagenase activity of plasma measured as in (A), from mice injected with i.p. LPS (10 mg/kg). (n = 3–8).
- Western blot analysis of 1 µl of plasma collected from mice injected with i.p. LPS and immunoblotted for MMP-1a or MMP-1b. Coomassie blue staining was used as a loading control. Quantification of blot densitometry is shown in the lower panel. p < 0.05 versus vehicle (n = 3–8).
- Conditioned media was collected from Cos-7 cells transiently expressing mouse MMP-1a, MMP-1b or pCMV6Entry vector control and concentrated 10-fold. Media was assayed for collagenase activity assay as in (A) in the presence or absence of 3 µM MMP-Inh-1.
To identify which of the mouse collagenases was responsible for the increases in systemic collagenase activity, we examined the plasma levels of MMP-1a and MMP-1b. MMP-1a (Col-A), the reported MMP-1 ortholog in mice, encodes a collagenase which cleaves Type I collagen, whereas, MMP-1b (Col-B) was reported to have no apparent collagenolytic activity (Balbin et al, 2001). Within 30 min after LPS injection, using a MMP-1a-selective antibody (Ab; Supporting information Fig S2) we detected a ≥3.5-fold increase in MMP-1a in the plasma (Fig 2C). MMP-1a peaked by 2 h and declined at 4–6 h after LPS injection (Fig 2C) which significantly correlated (r = 0.92, p = 0.01) with the collagenase activity curve shown in Fig 2B. Conversely, MMP-1b levels remained at or near baseline during the first 6 h and increased 24–48 h after LPS injection (Fig 2C).
We expressed recombinant mouse MMP-1a and MMP-1b in the conditioned media of Cos7 cells and confirmed that MMP-1a had significant collagenase activity, whereas, MMP-1b lacked detectable activity (Fig 2D). MMP-Inh-1 completely blocked all MMP-1a activity (Fig 2D). Together, these findings suggest that the MMP-1 ortholog, MMP-1a, accounts for the early increase in systemic collagenase activity in septic mice.
Inhibition of MMP-1 activity improves survival in septic mice
To provide evidence that MMP-1 activity plays a (patho)physiological role in an animal model of sepsis, we administered the MMP-1 inhibitor, MMP-Inh-1, immediately after the initiation of the CLP model of septic peritonitis in mice. Following CLP, mice were injected subcutaneously with vehicle or MMP-Inh-1 (5 mg/kg). In untreated (vehicle) wild-type (WT) CF-1 mice, the mortality rate was 90% from CLP-induced sepsis over a 7-days period (Fig 3A). Administration of MMP-Inh-1, but not MMP-Inh-1 that had been inactivated, significantly improved survival of septic CF-1 mice (Fig 3A; Supporting information Fig S3). However, when MMP-Inh-1 was administered at 4 h, survival rates became significantly worse than vehicle-treated animals (Supporting information Fig S3), suggesting that early inhibition of MMP-1 activity is beneficial to the survival of septic mice. This was consistent with previous studies (Kaneider et al, 2007) that showed that PAR1 switches from being a vascular barrier disruptive receptor to a vascular barrier protective receptor during sepsis.
Figure 3. Effect of pharmacologic blockade of MMP-1 activity on the survival of wild-type (WT), PAR1-deficient and PAR2-deficient mice with septic peritonitis.
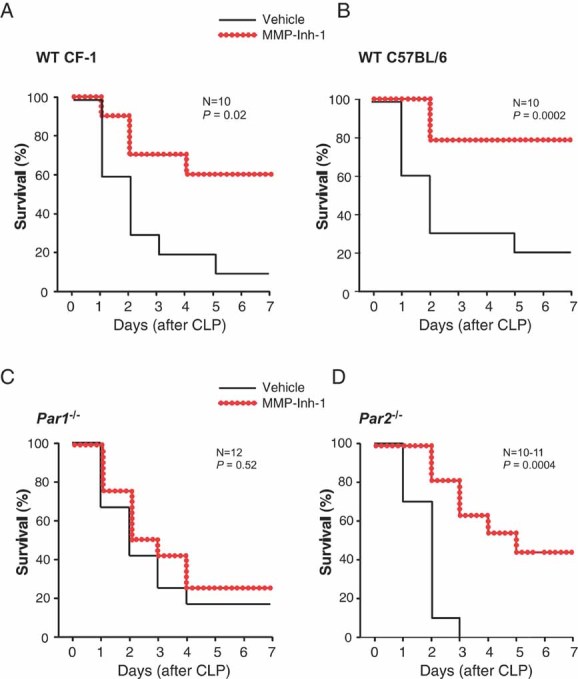
A-D. Survival of (A) WT CF-1 mice, n = 10, (B) WT C57Bl/6 mice, n = 10, (C) Par1−/− C57Bl/6 mice, n = 12 or (D) Par2−/− C57Bl/6 mice, n = 10–11, subjected to CLP then given vehicle or MMP-Inh-1 (5 mg/kg) subcutaneously. Mice received a maintenance dose of 5 mg/kg MMP-Inh-1 or vehicle once every 24 h until day 3.
The protective effects of inhibiting MMP-1 activity are lost in PAR1-deficient mice
To confirm the potential involvement of PAR1 or PAR2 in the MMP-1-dependent survival in sepsis, we used mouse strains deficient in either Par1 or Par2. Whereas, administration of MMP-Inh-1 improved survival in septic WT C57BL/6 mice (Fig 3B), this protective effect was lost in the PAR1-knockout strain (Par1−/−; Fig 3C). WT C57Bl/6 mice treated with MMP-Inh-1 also had significantly increased survival compared to PAR1-knockout mice treated with MMP-Inh-1. Conversely, administration of MMP-Inh-1 improved survival in septic PAR2-knockout mice, similar to results observed in WT mice demonstrating that the observed protective effects do not require PAR2 (Fig 3D). Together, these findings suggest that PAR1 is essential for the protective effects of MMP-Inh-1.
Endotoxin causes early loss of MMP-1a from the aortic endothelium of mice
During sepsis and SIRS, the endothelium is one of the first tissues to be exposed to invading pathogens, endotoxins and other inflammatory mediators. The resulting loss of barrier function by the endothelial cells leads to vascular leakage, coagulation, hypotension and septic shock. MMP-1 is expressed in human endothelial cells and regulates endothelial function and stability (Game et al, 2003; Naito et al, 2002; Struewing et al, 2009). To gain insight into the source of MMP-1a in the plasma of septic mice, we performed immunohistochemistry on the aortic endothelium en face from mice injected with vehicle or LPS, or from mice that underwent sham or CLP surgery. MMP-1a was localized in a punctate pattern in the aortic endothelium of vehicle or sham treated mice (Fig 4A, Supporting information Fig S4). After 1 h of LPS treatment or 4 h after CLP surgery, MMP-1a was found to be markedly decreased in the aortic endothelium, suggesting that MMP-1a is released from the endothelium after exposure to systemic endotoxin or septic peritonitis. The specificity of the MMP-1a Ab was confirmed by blocking the positive staining of the aorta preparations with the MMP-1a peptide epitope (Supporting information Fig S5).
Figure 4. Bacterial endotoxin causes loss of MMP-1a from the endothelium of mice and endothelial barrier disruption by endotoxin and MMP-1 are mediated by PAR1.
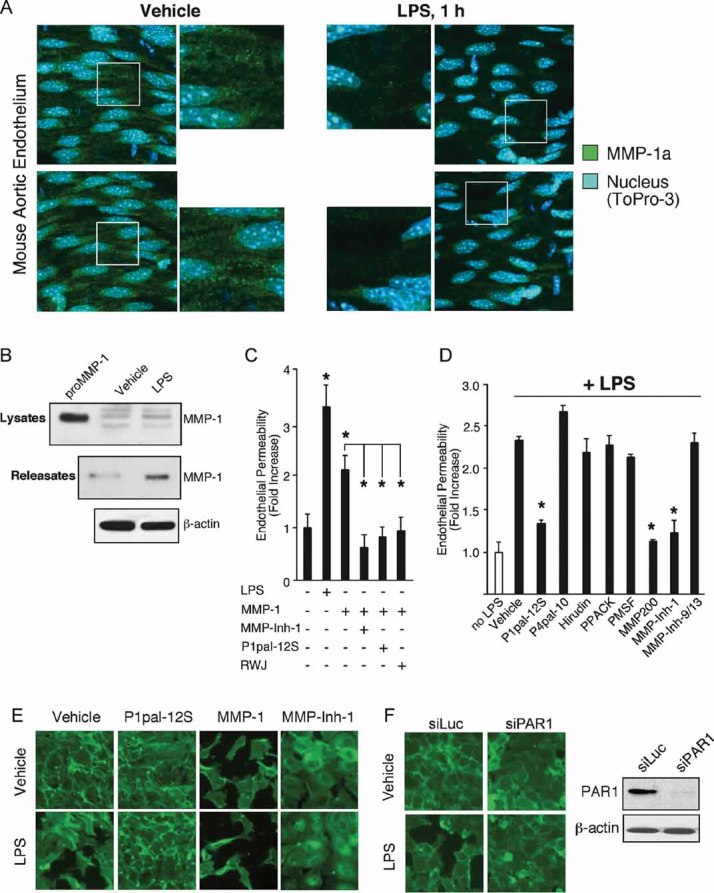
- Whole mount immunohistochemistry of the aortic endothelium en face from mice injected with vehicle or LPS (10 mg/kg) after 1 h and stained for MMP-1a (green) and the nucleus (ToPro-3; blue). Shown are representative projection images taken using a confocal microscope. (n = 3).
- Endothelial cells were treated with vehicle (0.2% DMSO) or LPS (10 µg/ml) for 1 h. Lysates and releasates from the conditioned media were analysed by MMP-1 Western blot (n = 3–4). Recombinant proMMP-1 was used as a positive control (lane 1).
- Permeability of quiescent HUVEC monolayers grown to confluence on Transwell membranes (3 µm pores), and stimulated with LPS endotoxin (1 µg/ml), MMP-1 (0.3 nM), MMP-Inh-1 (3 µM), P1pal-12S (0.3 µM) or RWJ-56110 (RWJ, 1 µM). After 4 h, Evans blue was added to the upper well and leakage over 15 min into the bottom well was measured as absorbance at 620 nm. (n = 3).
- Permeability of EA.hy926 endothelial cells measured as in (B), stimulated with LPS (1 µg/ml) and then treated immediately with vehicle, 0.3 µM P1pal-12S or P4pal-10, or various protease inhibitors at threefold above IC50. Evans blue leakage was measured as in (B). (n = 3).
- Barrier integrity of EA.hy926 monolayers exposed to vehicle or LPS (10 µg/ml) and then treated with vehicle, P1pal-12S, MMP-1 or MMP-Inh-1. After 4 h, cells were stained with FITC-phalloidin. (n = 5–7).
- Barrier integrity of EA.hy926 cells transfected with siRNA for Par1 (siPAR1) or luciferase (siLuc); after 48 h cells were stimulated with vehicle or LPS for 4 h and stained with FITC-phalloidin. (n = 4).
The MMP-1a Ab (2 mg/kg), which was administered only once, 1 h prior to CLP surgery, also significantly improved survival in the mouse CLP sepsis model relative to control, albeit not to as great a degree as MMP-Inh-1 (Supporting information Fig S6). Together, these findings are consistent with the notion that MMP-1a released from the endothelium of endotoxemic mice contributes to the mortality observed in the mouse sepsis models.
Involvement of human MMP1-PAR1 in endotoxin-mediated endothelial barrier disruption
We found that endotoxin treatment of human endothelial cells also caused release of MMP-1 into the conditioned media within 1 h (Fig 4B). Under these same conditions, LPS caused a significant disruption of the endothelial barrier integrity as assessed by leakage of Evans Blue dye through the endothelial monolayers (Fig 4C). We examined the potential contribution of MMP1-PAR1 signalling to the observed LPS-induced endothelial cell permeability. Ectopic addition of 0.3 nM MMP-1 induced a twofold increase in endothelial cell permeability (Fig 4C). Treatment with MMP-Inh-1 completely blocked the MMP-1-induced permeability (Fig 4C). Likewise, inhibition of PAR1 with RWJ-56110 (Leger et al, 2006) or the PAR1 antagonist pepducin, P1pal-12S (Kaneider et al, 2007), completely blocked the MMP-1-induced endothelial cell permeability, consistent with the observed effects being dependent on PAR1 (Fig 4C).
To quantify the relative contribution of LPS-released MMP-1 on PAR1-dependent barrier permeability, we tested the effects of a panel of inhibitors added concomitantly with endotoxin. LPS treatment alone (vehicle) increased endothelial permeability by 2.3-fold (Fig 4D). The PAR1 antagonist pepducin, P1pal-12S, significantly reduced the LPS-induced permeability by 75% (Fig 4D). Conversely, the PAR4 antagonist pepducin, P4pal-10, had no effect on LPS-induced permeability. Serine protease inhibitors, including hirudin (specific to thrombin), PPACK and PMSF had no significant effect on the LPS-induced permeability (Fig 4D). However, the broad-spectrum MMP inhibitors, MMP-200 and MMP-Inh-1 significantly blocked LPS-induced permeability (Fig 4D). MMP-Inhibitor-9/13 (inhibits MMP-9 and -13) had no effect on LPS-induced endothelial cell permeability (Fig 4D). These findings are consistent with the notion that the LPS-induced endothelial barrier disruption is modulated through MMP-1 and PAR1. Similar with previous findings, these results also suggest that activation of PAR1 by MMP-1 is endothelial barrier-disruptive (Kaneider et al, 2007).
To directly demonstrate the effects of MMP-1 on endothelial monolayer integrity, we stained filamentous actin with FITC-phalloidin. Treatment with vehicle, P1pal-12S or MMP-Inh-1 had no effect on endothelial cell morphology, whereas, MMP-1 caused endothelial cell monolayers to contract and lose cell–cell contacts similar to that previously seen with other PAR1 agonists such as thrombin (Kaneider et al, 2007; Fig 4E). When cells were treated with LPS alone, the endothelial monolayer was disrupted (Fig 4E). Blockade of PAR1 with P1pal-12S or MMP-1 with MMP-Inh-1 suppressed the LPS-induced endothelial barrier disruption (Fig 4E). Concomitant addition of MMP-1 with LPS exacerbated the barrier disruption.
To independently confirm the role of PAR1 in the observed LPS-induced endothelial barrier disruption, we used siRNA to knockdown the expression of Par1. Treatment of endothelial cells with Par1 siRNA significantly silenced PAR1 protein expression as compared to control luciferase siRNA (Fig 4F). Silencing of Par1 attenuated LPS-induced endothelial cell contraction and barrier disruption as compared to siRNA control.
Effects of MMP-1a and MMP-1 activity on PAR1-dependent migration and Rho signalling
To provide independent evidence that the mouse MMP-1a is activating PAR1 signalling, we treated PAR1-expressing MCF-7 cells and PAR1-null MCF-7 cells with plasma collected from LPS or vehicle-treated mice, or from mice that were subjected to either CLP or sham surgery. Plasma from LPS-treated mice resulted in a threefold increase in chemotaxis of PAR1-MCF-7 cells relative to plasma from vehicle-treated mice, but had little effect on the PAR1-null MCF-7 cells (Fig 5A). Immunodepletion of MMP-1a or pharmacologic inhibition of either MMP-1a activity with MMP-Inh-1 or PAR1 with RWJ-56110 completely blocked the observed increase in chemotaxis of the PAR1-MCF7 cells towards the LPS-plasma, consistent with MMP-1a activating PAR1 (Fig 5A). Conversely, the inhibitors of MMP-1a and PAR1 had little effect on the migration of the PAR1-null MCF-7 cells towards the plasma from LPS-treated mice. PAR1-MCF7 displayed increased migration towards plasma from mice that underwent CLP surgery as compared to sham control. Inhibition of either MMP-1a activity with MMP-Inh-1 or PAR1 with RWJ-56110 reduced the increase in chemotaxis of the PAR1-MCF7 cells towards CLP-plasma (Fig 5A).
Figure 5. Effects of MMP-1a and MMP-1 on PAR1-dependent migration and Rho-GTP activity.

- Plasma from mice injected with vehicle or LPS was collected after 2 h and was incubated with MMP-1a antibody (Ab) or IgG control Ab. Antigen–Ab complexes were immunodepleted using protein A beads. Alternately, mice underwent CLP or sham surgery and plasma was collected after 2 h. Migration of PAR1-expressing MCF7 (N5M5) cells towards plasma in a transwell apparatus was assessed as in the methods. In some wells, RWJ (5 µM) or MMP-Inh-1 (3 µM) was added to the cells. (*p < 0.05; #p = 0.06–0.10).
- CF-1 mice were treated with vehicle or LPS, or underwent sham or CLP surgery. Plasma was collected after 2 h and immunodepleted as in (A). Immunodepletion of MMP-1a was confirmed by Western blot shown in the upper panel. The immunodepleted plasma was then used to treat EA.hy926 cells for 15 min and activated Rho-GTP was quantified by Western blot analysis (n = 3). Shown are densitometry graphs of the Rho-GTP Western blots.
- EA.hy926 cells were treated with LPS and inhibitors. After 15 min, cells were lysed and activated Rho-GTP was quantified by Western blot analysis (n = 3–6). Shown are densitometry graphs of the Rho-GTP Western blots.
PAR1 is a strong activator of G12/13-driven Rho-GTPase activity, which triggers actin-dependent contraction of endothelial cells and loss of barrier function (van Nieuw Amerongen et al, 1998). MMP-1 in particular is a potent activator of PAR1-G12/13-Rho activity (Trivedi et al, 2009). Plasma from vehicle-treated or sham mice had little or no effect on Rho-GTP activity in endothelial cells, whereas, plasma from LPS-treated or CLP mice induced a major 4.7-fold increase in Rho-GTP activity (Fig 5B). Plasma from LPS-treated or CLP mice that was immunodepleted of MMP-1a showed a marked reduction in Rho-GTP activity (Fig 5B).
Endotoxin treatment of human endothelial cells caused a significant 5.8-fold increase in Rho-GTP activity (Fig 5C). The LPS-induced Rho-GTP activity was inhibited 80–100% by MMP-Inh-1 as well as the PAR1 antagonists RWJ-56110 and P1pal-12S (Fig 5C). Ectopic addition of MMP-1 (0.3 nM) caused a 2.8-fold increase in Rho-GTP activity, which was completely blocked by RWJ-56110 (Fig 5C). Collectively, these findings suggest that the endotoxin-dependent Rho activation and the subsequent endothelial barrier disruption are significantly mediated by autocrine activation of PAR1 by MMP-1 and MMP-1a.
Inhibition of MMP-1 activity protects against lung vascular permeability and DIC in septic mice
Sepsis induces severe cardiovascular changes including an increase in cardiac output, a decrease in peripheral resistance and a loss of intravascular fluids, which can lead to severe shock (Annane et al, 2005). We assessed the effect of exogenous systemic administration of active human MMP-1 on lung vascular permeability, a marker of septic shock. MMP-1 was injected into the internal jugular vein of mice and loss of endothelial barrier function assessed by the accumulation of Evans blue dye into the lung interstitium. Injection of MMP-1 alone induced a significant 3.5-fold increase in lung vascular permeability in normal CF-1 WT mice, however, this effect was lost when MMP-1 was heat inactivated (Fig 6A, left panel). In normal C57Bl/6 WT mice, injection of MMP-1 resulted in a significant 2.3-fold increase in lung vascular permeability, which was lost in Par1-deficient mice (Fig 6A, right panel). The peptide agonist of PAR1, TFLLRN, was used as a control and was found to induce a 2.8-fold increase in C57Bl/6 mice, which was completely abolished in Par1-null mice (Fig 6A, right panel). These findings demonstrate that injection of exogenous MMP-1 induces an increase in lung vascular permeability in a PAR1-dependent manner in mice.
Figure 6. Effect of inhibition of MMP-1 activity on lung vascular permeability and DIC in septic mice.

A. CF-1 mice (n = 4–5), C57Bl/6 mice (n = 3–4) or Par1−/− C57Bl/6 mice (n = 4) were injected with pure active human MMP-1 (0.1 mg/kg), heat-inactivated human MMP-1 (0.1 mg/kg; inact. MMP-1), TFLLRN peptide (2.5 mg/kg) or PBS vehicle. After 5 min (peptide) or 15 min (MMP-1), Evans blue dye was injected into the internal jugular vein and allowed to circulate for 30 min and the amount of Evans Blue dye that leaked into the lung interstitium was quantified.
B. Lung vascular permeability in CF-1 mice after i.p. injection of LPS (10 mg/kg), and subsequent treatment with subQ MMP Inh-1 (5 mg/kg) or vehicle as measured by Evans Blue dye leakage at the 4 h time point (n = 5).
C. Lung vascular permeability in mice subjected to CLP and injected with vehicle or MMP-Inh-1 immediately after CLP. Permeability was assessed as in (A) at 24 h. p < 0.05 versus vehicle (n = 5).
D-F. CF-1 mice underwent CLP and were injected subQ with vehicle or MMP-Inh-1 (5 mg/kg) immediately after CLP. Blood was drawn by terminal cardiac puncture at 24 or 48 h after the procedure as noted. Platelets were counted in whole blood at 24 h after CLP (D), plasma TAT levels (E) and plasma D-dimer levels (F) were measured by ELISA at 48 h after CLP. (n = 3–9).
Next, mice were challenged with intraperitoneal LPS and then treated with vehicle or MMP-Inh-1. LPS alone (vehicle) caused a sixfold increase in lung vascular permeability (Fig 6B). Treatment with MMP-Inh-1 significantly reduced the LPS-induced vascular leakage (Fig 6B). We confirmed the effects of MMP-inh-1 in the CLP-model of sepsis. CLP induced a threefold increase in lung vascular permeability as compared to sham-operated mice (Fig 6C). Treatment with MMP-Inh-1 significantly blunted the CLP-induced vascular permeability (Fig 6C).
To assess DIC in septic mice, we measured the appearance of thrombin–anti-thrombin III complexes (TAT), fibrin degradation products (D-dimer), and loss of platelets at 24 or 48 h after CLP. The marked thrombocytopenia seen in septic mice was nearly completely prevented by inhibition of MMP-1 activity (Fig 6D). TAT and D-dimer levels increased in mice that underwent CLP surgery as compared to sham operated mice (Fig 6 E and F). Treatment with MMP-Inh-1 significantly reduced TAT levels as compared to vehicle control (Fig 6E). Treatment with MMP-Inh-1 also significantly reduced the CLP-induced D-dimer formation by 60% (Fig 6F). Therefore, inhibition of MMP-1 activity reduces several manifestations of severe sepsis including lung vascular permeability, systemic inflammation and DIC.
Inhibition of MMP-1 activity suppresses the cytokine storm in wild-type mice but not PAR1-deficient mice
During sepsis, the endothelium switches from an anti-thrombotic to a pro-thrombotic state. Endothelial activation of pro-inflammatory mediators and disruption of barrier function leads to tissue factor, collagen/vWF exposure and subsequent activation of the coagulation cascade and platelets (Levi & van der Poll, 2010). Leukocytes are marginated and activated into these exposed subendothelial areas, especially into the lung interstitium in the mouse CLP model (Kaneider et al, 2005), contributing to the observed high levels of pro-inflammatory cytokines such as KC (IL-8 ortholog in humans), TNF-α, MCP-1 and IL-6 (Levi & van der Poll, 2010). We postulated that blockade of MMP-1 activity in septic WT mice but not in Par1−/− mice would interrupt this systemic increase in cytokines. To measure cytokines, mice underwent CLP or sham surgery and were treated with vehicle or MMP-Inh-1. After 24 h, cytokines were measured in the plasma using a multiplexing cytometric bead array and ELISA. In WT mice, CLP caused massive increases in systemic pro-inflammatory cytokines IL-6 (480-fold), the IL-8 ortholog KC (52-fold), TNF-α (5-fold) and IL-10 (3-fold) (Fig 7A). MCP-1 exhibited a 210-fold increase (Fig 7B). By comparison, IL-12p70 and IFNγ were not significantly changed at 24 h (Fig 7B). Quite strikingly, WT mice treated with MMP-Inh-1 showed complete protection from the increases in IL-6, MCP-1, TNF-α and IL-10 and nearly complete protection from increases in KC as compared to vehicle-treated mice (Fig 7A and B). Thus, inhibition of MMP-1 activity resulted in nearly complete protection from the CLP-induced inflammatory cytokine response. In Par1−/− mice, CLP also caused increases in IL-6, KC, TNF-α, IL-10 and a small increase in MCP-1, but did not induce IL12p70 or IFNγ (Fig 7A and B). Notably, MMP-Inh-1 did not significantly reduce the CLP-induced cytokine levels in PAR1-deficient mice demonstrating that PAR1 is required for the observed inhibitory effects of MMP-Inh-1 on systemic cytokines in sepsis.
Figure 7. Inhibition of MMP-1 activity suppresses the cytokine storm in WT mice but not PAR1-deficient mice.

Cytokine levels in WT mice or Par1−/− mice subjected to CLP and injected immediately with either vehicle or MMP-Inh-1 (MMP Inh). After 24 h, plasma was collected and analysed using BD Mouse Inflammation cytometric multiplexing beads and cytokine content was measured using flow cytometry (n = 4–6), except KC was quantified using ELISA (n = 4). p < 0.05 versus vehicle.
DISCUSSION
In this study, we found that proMMP-1 and active MMP-1 levels were markedly increased in the plasma of patients with severe sepsis. Elevated proMMP-1 and active MMP-1 significantly correlated with worsened survival outcomes at days 7 and 28 in septic patients. We also identified mouse MMP-1a as a PAR1 agonist that is released from endothelium in septic mice. Inhibition of MMP-1 activity significantly improved the survival of WT and Par2−/− mice but had no effect in Par1−/− mice, thus demonstrating the dependence of the salutary effects on PAR1 activity. Consistent with this observation, PAR1-deficiency completely ablated the inhibitory effects conferred by the MMP1-inhibitor on the rise in systemic cytokines observed in the WT mice.
MMP-1a levels peaked rapidly in the septic mice indicating that the MMP1-PAR1 system may play an important role as an early responder to bacterial infection and systemic inflammation. In vitro and in vivo experiments pointed to the endothelium as an early source of MMP-1 activity as E. coli endotoxin triggered release of endothelial MMP-1, which in turn activated PAR1-Rho-dependent cell contraction and barrier disruption in an autocrine manner. Other cell types, including platelets and monocytes/macrophages, could also potentially be sources of MMP-1a during the later stages of sepsis (Galt et al, 2002; Geissmann et al, 2010; Libby & Aikawa, 2002; Trivedi et al, 2009). Collectively, these findings are consistent with the notion that MMP-1 plays an important role in regulating PAR1-dependent functions of the endothelium in systemic inflammation and sepsis.
Blockade of MMP-1 activity in septic mice with MMP-Inh-1 was significantly beneficial to survival. Independently, a single injection of MMP-1a Ab also significantly improved survival in septic mice, however, not to as great a degree as MMP-Inh-1. As MMP-Inh-1 has shown inhibitory effects on other collagenases (Odake et al, 1994), one cannot completely rule out that other collagenases are playing a role in the mouse sepsis models. In addition to potential off-target effects on other metalloproteases, the enhanced efficacy of pharmacologic blockade with MMP-Inh-1 could be due to its higher potency and multi-dosing schedule. However, our in vivo data with Par-deficient mice demonstrate that the beneficial effects of MMP-Inh-1 on survival are dependent on the presence of PAR1 but not PAR2.
Recent clinical trials have also shown that the systemic levels of other MMPs including MMP-2, MMP-8, MMP-9, MMP-10 and tissue inhibitor of MMP-1 (TIMP1), correlate well with sepsis severity in humans including coagulation markers, APACHE scores and death (Lorente et al, 2009; Vanlaere & Libert, 2009). Moreover, high levels of MMP-1 in lung fluids have been associated with more ominous disease progression and multi-organ failure in patients with acute lung injury (Fligiel et al, 2006). In mouse models of sepsis and endotoxemia, MMPs have been documented to have both protective effects and damaging effects depending on the MMP, the route and type of infection (Vanlaere & Libert, 2009). For instance, studies using MMP-8-deficient mice have pointed to an anti-inflammatory role for MMP-8 in the lung alveolar space (Owen et al, 2004) and a pro-inflammatory role using an air-pouch model (Tester et al, 2007). It is apparent from both the present work and others that MMPs play varying roles in different stages of sepsis and are important for the normal response to invading pathogens but can also contribute to severe tissue damage and death.
In summary, the present work has identified an autocrine MMP1-PAR1 signalling system on the vascular endothelium that provides a link between inflammation, barrier function, coagulation and sepsis outcomes in models of septic peritonitis and systemic inflammation. Our findings help elucidate the molecular mechanisms involved in the early host responses to sepsis and suggest that MMP-1 may prove to be a useful and independent biomarker for the identification of high-risk sepsis patients and as a predictor of early mortality. Intervention with specific MMP-1 inhibitors might represent a novel treatment strategy in human sepsis if given at the onset of severe sepsis.
MATERIALS AND METHODS
Reagents
MMP-Inh-1 (p-aminobenzoyl-Gly-Pro-D-Leu-D-Ala-NHOH; FN-439) (IC50 = 1 µM for MMP-1; 150 µM for MMP-3; 1 µM for MMP-8; and 30 µM for MMP-9), MMP-Inh-9/13, MMP200 and pure human proMMP-1 were from Calbiochem. Inactive MMP-Inh-1 (p-aminobenzoyl-Gly-Pro-D-Leu-OH) lacking the C-terminal Ala-hydroxamate was synthesized from MMP-Inh-1 using LiOH hydrolysis. Pro-MMP-1 was activated with APMA at 37°C for 35 min and then dialysed overnight to remove the APMA. The pepducins P1pal-12S and P4pal-10 were synthesized with carboxy-terminal amides by standard fluorenylmethoxycarbonyl solid-phase methods (Covic et al, 2002). Pepducins were purified to 95–98% by C4 reverse-phase HPLC and were prepared as stock solutions in dimethyl sulphoxide (DMSO). LPS endotoxin (E. coli O11:B4), Evans blue dye, phenylmethylsulphonylflouride (PMSF) and recombinant hirudin were from Sigma–Aldrich. DQ collagen and oligofectamine were from Invitrogen. Par1-specific siRNA (5′-AAGGCUACUAUGCCUACUACU-3′) and luciferase-specific siRNA (5′-CGTACGCGGAATACTTCGA-3′) were from Dharmacon. RWJ-56110 was a gift from Johnson & Johnson. RhoA antibodies were from Santa Cruz; β-actin Ab was from Sigma–Aldrich; MMP-1 Ab was from Millipore. Polyclonal antibodies to the C-terminus of MMP-1a and MMP-1b were generated by published methods (Kuliopulos et al, 1999) and were purified from rabbit antisera with Sepharose 4B-peptide affinity columns. Enzyme-linked immunoassay (ELISA) kits to assess D-dimer were from Diagnostica Stago, TAT ELISA kits were from Enzyme Research Laboratories. KC, IL-8 and proMMP-1 ELISA kits, and Human Active MMP-1 Fluorokine kits were from R&D systems; the normal range for proMMP-1 in human plasma was 0.179–1.00 ng/ml according to the manufacturer's instructions. Cytometric Bead Array Mouse Inflammation Kits were from BD Biosciences. Endotoxin levels in all cell culture reagents were less than 0.01 EU/ml. Endotoxin levels for reagents injected into mice were less than 20 pg per animal.
Cell culture
Human umbilical vein endothelial cells (HUVEC) were purchased from Lonza. EA.hy926 cells are a transformed HUVEC line (Edgell et al, 1983) and were purchased from ATCC. Cells were grown in EGM-2 medium (Lonza) or Dulbecco's modified Eagle's medium (DMEM) (Cellgro) supplemented with 10% v/v endotoxin-free FBS (Invitrogen) and endothelial cell growth supplement (Fisher). Oligofectamine was used for transfection of siRNA (100 nM per plate) and experiments were conducted 48 h after transfection.
Animal studies
Female CF-1 (6–8 weeks old) were from Charles River Laboratories, C57Bl/6 mice were from Jackson Laboratories. Par1−/− and Par2−/− mice in a C57Bl/6 background were generated as described (Damiano et al, 1999; Darrow et al, 1996). Animal experiments were performed in accordance with guidelines of the NIH and were approved by Tufts Medical Center Institutional Animal Care and Use Committee. CLP was done as described (Ness et al, 2003). Alternately, LPS was injected intraperitoneal at a concentration of 10 mg/kg. Mouse MMP-1a, MMP-1b and MMP-8 levels were measured in plasma using immuno-blot analysis. Platelets were counted in a Hemavet 850 as described (Kaneider et al, 2005). Concentrations of fibrin D-dimer, TAT complexes and KC were measured by ELISA according to the manufacturer's instructions. Cytokine levels in plasma were measured using BD Mouse Inflammatory cytometric muliplexing bead array and a BD FACS Canto II. For quantification of lung vascular permeability, the internal jugular vein was catheterized and 100 µl of Evans blue dye was injected (20 mg/kg). After dye was allowed to circulate for 30 min, mice were euthanized and their lungs flushed with phosphate buffered saline (PBS). Lungs were weighed and Evans blue was extracted overnight at 60°C with formamide and the absorbance at 620 nm was measured as described (Patterson et al, 1992). Collagenase activity in 50 µl of plasma was measured using DQ Collagen type I (Invitrogen) as a fluorescent reporter of collagen cleavage. Collagenase assays contained 10 µg DQ collagen in 50 mM Tris–HCl, pH 7.6, 150 mM NaCl, 5 mM CaCl2 and 0.2 mM NaN3 and cleavage was monitored continuously over time with a fluorescence microplate reader at 538 nm (excitation at 485 nM) at 25°C as described (Boire et al, 2005). For standard curves of MMP-1 activity, proMMP-1 (≥90% purity, from human synovial fibroblasts) was activated using 2 mM APMA at 37°C for 35 min followed by dialysis overnight at 4°C in 50 mM Tris–HCl, pH 7.7, 5 mM CaCl2, 200 mM NaCl. For whole mount immunohistochemistry of the aorta, mice were pressure fixed with 10% formalin. The thoracic aorta was removed and incubated overnight at 4°C with MMP-1a Ab, then incubated with fluorescently tagged secondary Ab for 4 h. The nucleus was stained with Cy5-ToPro-3. The aorta was cut longitudinally and splayed open on a microscope slide with the endothelium facing up. The tissue was mounted with vectashield and imaged using a Leica TCS SP2 confocal microscope.
The paper explained
PROBLEM
Sepsis and associated systemic inflammatory response syndrome (SIRS) is a common and extremely deadly disease that is rising in incidence in both western and developing countries, with an alarming lack of effective treatment options. A major cause of death in sepsis is due to the inability to properly regulate the inflammatory–coagulation response in which the endothelium plays a key role. The molecular causes of this perturbation remain poorly understood and have hampered the development of effective therapeutic interventions for patients with severe sepsis. Emerging evidence suggests that matrix metalloproteases (MMPs) play pivotal roles in the host response to invading pathogens, but in severe sepsis can also cause uncontrolled tissue damage and contribute to mortality.
RESULTS
We discovered that MMP-1 and its mouse homolog, MMP-1a, activate the G protein-coupled protease-activated receptor-1 (PAR1) in an autocrine manner on the surface of endothelial cells to cause endothelial barrier disruption, vascular leakage, systemic inflammation and death. Inhibition of the mouse MMP-1 activity significantly improved the survival of wild-type (WT) and Par2−/− mice but had no effect in Par1−/− mice, thus demonstrating a dependence on PAR1. Sepsis- and endotoxin-induced lung vascular leakage could be attenuated by inhibition of MMP-1 activity. Pharmacologic inhibition of MMP-1a also reduced disseminated intravascular coagulation (DIC) and markedly suppressed the pro-inflammatory cytokine response in WT but not PAR1-deficient mice with sepsis.
IMPACT
These data indicate that the MMP1-PAR1 system plays an important role as an early responder to bacterial infection and systemic inflammation. They may provide the blueprint for a novel prognostic biomarker and completely new treatment modality for high-risk patients.
Endothelial permeability assay
EA.hy926 cells were plated on polycarbonate Transwell membranes with a pore size of 3 µm (Corning) in EGM-2 medium supplemented with 10% v/v fetal bovine serum (FBS). After reaching confluence, cells were treated with vehicle (0.2% DMSO) or LPS (1 µg/ml) and treated immediately with MMP-1 (0.3 nM), MMP-Inh-1 (3 µM), P1pal-12S (0.3 µM), RWJ (1 µM), hirudin (1 U), PPACK (100 µg/ml), PMSF (100 µM), MMP200 (300 nM), MMP-Inh-8 (12 nM) or MMP-Inh-9/13 (3 nM). After 4 h, growth media was removed and DMEM containing Evans blue dye (0.67 mg/ml) was added to the upper chamber and PBS was added to the lower chamber. The leakage of Evans blue dye was quantified in the lower well after 15 min by measurement of absorbance at 620 nm.
Actin staining of endothelial cells
EA.hy926 cells were plated onto tissue culture chamber slides and were grown to confluence. Cells were serum starved for 2 h, then treated with vehicle or LPS (10 µg/ml) and concomitantly treated with either 0.2% v/v DMSO (vehicle), P1pal-12S (0.3 µM) or MMP-1 (0.3 nM). After 4 h, cells were fixed with 1% paraformaldehyde and stained with fluorescein isothiocyanate-conjugated phalloidin.
Rho assay
Confluent EA.hy926 cells were treated with LPS, MMP-1, MMP-Inh-1, RWJ or P1pal-12S for 15 min as described above. The cells were lysed and Rho-GTP was precipitated from cell lysates with glutathione S-transferase (GST)-rhotekin-reduced glutathione-agarose beads as described (Ren & Schwartz, 2000). Rho-GTP was then quantified by immunoblot analysis with monoclonal Ab to RhoA (26C4). A portion of the endothelial cell lysates was reserved and analysed by immunoblot for total Rho as a loading control.
Immunodepletion studies
Mice were injected i.p. with vehicle (PBS) or 10 mg/kg LPS, or underwent sham or CLP surgery. Plasma (100 µl) was collected at 2 h and incubated with 5 µg pure MMP-1a Ab or IgG control. Antigen–Ab complexes were pulled down using protein-A beads. The supernatants (1:500 final dilution from plasma) were then used to treat EA.hy926 cells for 15 min and Rho-GTP was measured as above. Alternately, the supernatants (1:6 final dilution) were used in a migration assay where 50,000 PAR1-expressing MCF-7 cells were plated in the top of a 6 µm transwell filter and migration towards the plasma was measured after 5 h as described (Boire et al, 2005).
Human sepsis samples
The 50 human sepsis samples were randomly selected from the plasma bank from a large multicenter sepsis trial before any experimental therapy was administered. The samples were number coded and de-identified for patient confidentiality. These baseline plasma samples of patients were obtained at the time of enrollment in trial in which all patients met current consensus definitions for severe sepsis or septic shock (Levy et al, 2003). We used the scoring algorithm criteria established by the Japanese Association for Acute Medicine (JAAM) for DIC which gives various scoring points for SIRS, thrombocytopenia, D-dimer (or other fibrin-related markers) and PT. Patients with scores greater than 4 were diagnosed with DIC. The patients all gave informed consent to study participation and all study-related investigations and blood sampling. The plasma samples were drawn in endotoxin-free glassware and separated from blood cells by centrifugation at 4°C for 15 min at 1400 × g and then frozen at −70°C until the samples were analysed. Clinical and laboratory information was collected over 28 days following study entry.
Statistical analysis
Data are expressed as mean ± SE and were analysed using ANOVA followed by the Student-Newman–Keuls post-test. Significance of survival studies were quantified using Kaplan–Meier analysis and log rank (Mantel–Cox) tests. Forward stepwise regression models were chosen by retaining terms whose statistical significance persisted in the presence of other significant effects. Significance was based on an alpha-level of 0.05.
Acknowledgments
We are highly grateful to George Perides, Claudia Derian and Patricia Andrade-Gordon for providing the PAR2-deficient mouse strain and the Tufts Center for Neuroscience Research (P30 NS047243) for use of their confocal microscope. This work was supported in part by an AHA Postdoctoral Fellowship (to SLT), the Austrian Science Fund (J-2342-B05 to NCK) and grants from the National Institutes of Health (HL64701, HL57905 and CA122992 to AK; CA104406 to LC).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
SLT, NCK, SMO and AK conceptualized and designed the experiments; SLT, NCK, SK, CF, GK, KA and AA did the experiments; SLT, NCK, SK, LC, SMO and AK analysed the data and prepared the manuscript.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Annane D, Bellissant E, Cavaillon JM. Septic shock. Lancet. 2005;365:63–78. doi: 10.1016/S0140-6736(04)17667-8. [DOI] [PubMed] [Google Scholar]
- Balbin M, Fueyo A, Knauper V, Lopez JM, Alvarez J, Sanchez LM, Quesada V, Bordallo J, Murphy G, Lopez-Otin C. Identification and enzymatic characterization of two diverging murine counterparts of human interstitial collagenase (MMP-1) expressed at sites of embryo implantation. J Biol Chem. 2001;276:10253–10262. doi: 10.1074/jbc.M009586200. [DOI] [PubMed] [Google Scholar]
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- Boire A, Covic L, Agarwal A, Jacques S, Sharifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- Covic L, Gresser AL, Talavera J, Swift S, Kuliopulos A. Activation and inhibition of G protein-coupled receptors by cell-penetrating membrane-tethered peptides. Proc Natl Acad Sci USA. 2002;99:643–648. doi: 10.1073/pnas.022460899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano BP, Cheung WM, Santulli RJ, Fung-Leung WP, Ngo K, Ye RD, Darrow AL, Derian CK, de Garavilla L, Andrade-Gordon P. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J Pharmacol Exp Ther. 1999;288:671–678. [PubMed] [Google Scholar]
- Darrow AL, Fung-Leung WP, Ye RD, Santulli RJ, Cheung WM, Derian CK, Burns CL, Damiano BP, Zhou L, Keenan CM, et al. Biological consequences of thrombin receptor deficiency in mice. Thromb Haemost. 1996;76:860–866. [PubMed] [Google Scholar]
- Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, Reinhart K, Angus DC, Brun-Buisson C, Beale R, et al. Surviving sepsis campaign: International Guidelines for Management of Severe Sepsis and Septic Shock: 2008. Crit Care Med. 2008;36:296–327. doi: 10.1097/01.CCM.0000298158.12101.41. [DOI] [PubMed] [Google Scholar]
- Doi K, Leelahavanichkul A, Yuen PS, Star RA. Animal models of sepsis and sepsis-induced kidney injury. J Clin Invest. 2009;119:2868–2878. doi: 10.1172/JCI39421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid increase in hospitalization and mortality rates for severe sepsis in the United States: a trend analysis from 1993 to 2003. Crit Care Med. 2007;35:1244–1250. doi: 10.1097/01.CCM.0000261890.41311.E9. [DOI] [PubMed] [Google Scholar]
- Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esmon CT. Interactions between the innate immune and blood coagulation systems. Trends Immunol. 2004;25:536–542. doi: 10.1016/j.it.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fligiel SE, Standiford T, Fligiel HM, Tashkin D, Strieter RM, Warner RL, Johnson KJ, Varani J. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol. 2006;37:422–430. doi: 10.1016/j.humpath.2005.11.023. [DOI] [PubMed] [Google Scholar]
- Galt SW, Lindemann S, Allen L, Medd DJ, Falk JM, McIntyre TM, Prescott SM, Kraiss LW, Zimmerman GA, Weyrich AS. Outside-in signals delivered by matrix metalloproteinase-1 regulate platelet function. Circ Res. 2002;90:1093–1099. doi: 10.1161/01.res.0000019241.12929.eb. [DOI] [PubMed] [Google Scholar]
- Game BA, Xu M, Lopes-Virella MF, Huang Y. Regulation of MMP-1 expression in vascular endothelial cells by insulin sensitizing thiazolidinediones. Atherosclerosis. 2003;169:235–243. doi: 10.1016/s0021-9150(03)00165-5. [DOI] [PubMed] [Google Scholar]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerge T, Barg A, Schnaeker EM, Poppelmann B, Shpacovitch V, Rattenholl A, Maaser C, Luger TA, Steinhoff M, Schneider SW. Tumor-derived matrix metalloproteinase-1 targets endothelial proteinase-activated receptor 1 promoting endothelial cell activation. Cancer Res. 2006;66:7766–7774. doi: 10.1158/0008-5472.CAN-05-3897. [DOI] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Agarwal A, Leger AJ, Kuliopulos A. Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat Med. 2005;11:661–665. doi: 10.1038/nm1245. [DOI] [PubMed] [Google Scholar]
- Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschen EJ, Fernandez JA, Cooley BC, Yang XV, Sood R, Mosnier LO, Castellino FJ, Mackman N, Griffin JH, Weiler H. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliopulos A, Covic L, Seeley SK, Sheridan PJ, Helin J, Costello CE. Plasmin desensitization of the PAR1 thrombin receptor: kinetics, sites of truncation, and implications for thrombolytic therapy. Biochemistry. 1999;38:4572–4585. doi: 10.1021/bi9824792. [DOI] [PubMed] [Google Scholar]
- Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1–4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. [DOI] [PubMed] [Google Scholar]
- Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–S34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002;8:1257–1262. doi: 10.1038/nm1102-1257. [DOI] [PubMed] [Google Scholar]
- Lorente L, Martin MM, Labarta L, Diaz C, Sole-Violan J, Blanquer J, Orbe J, Rodriguez JA, Jimenez A, Borreguero-Leon JM, et al. Matrix metalloproteinase-9, -10, and tissue inhibitor of matrix metalloproteinases-1 blood levels as biomarkers of severity and mortality in sepsis. Crit Care. 2009;13:R158. doi: 10.1186/cc8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore LJ, Moore FA, Todd SR, Jones SL, Turner KL, Bass BL. Sepsis in general surgery: the 2005–2007 national surgical quality improvement program perspective. Arch Surg. 2010;145:695–700. doi: 10.1001/archsurg.2010.107. [DOI] [PubMed] [Google Scholar]
- Naito S, Shimizu S, Matsuu M, Nakashima M, Nakayama T, Yamashita S, Sekine I. Ets-1 upregulates matrix metalloproteinase-1 expression through extracellular matrix adhesion in vascular endothelial cells. Biochem Biophys Res Commun. 2002;291:130–138. doi: 10.1006/bbrc.2002.6418. [DOI] [PubMed] [Google Scholar]
- Ness TL, Hogaboam CM, Strieter RM, Kunkel SL. Immunomodulatory role of CXCR2 during experimental septic peritonitis. J Immunol. 2003;171:3775–3784. doi: 10.4049/jimmunol.171.7.3775. [DOI] [PubMed] [Google Scholar]
- Neyrinck AP, Liu KD, Howard JP, Matthay MA. Protective mechanisms of activated protein C in severe inflammatory disorders. Br J Pharmacol. 2009;158:1034–1047. doi: 10.1111/j.1476-5381.2009.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odake S, Morita Y, Morikawa T, Yoshida N, Hori H, Nagai Y. Inhibition of matrix metalloproteinases by peptidyl hydroxamic acids. Biochem Biophys Res Commun. 1994;199:1442–1446. doi: 10.1006/bbrc.1994.1392. [DOI] [PubMed] [Google Scholar]
- Owen CA, Hu Z, Lopez-Otin C, Shapiro SD. Membrane-bound matrix metalloproteinase-8 on activated polymorphonuclear cells is a potent, tissue inhibitor of metalloproteinase-resistant collagenase and serpinase. J Immunol. 2004;172:7791–7803. doi: 10.4049/jimmunol.172.12.7791. [DOI] [PubMed] [Google Scholar]
- Patterson CE, Rhoades RA, Garcia JG. Evans blue dye as a marker of albumin clearance in cultured endothelial monolayer and isolated lung. J Appl Physiol. 1992;72:865–873. doi: 10.1152/jappl.1992.72.3.865. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, Andrade-Gordon P, Frank RD, Mackman N. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood. 2004;103:1342–1347. doi: 10.1182/blood-2003-09-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remick DG. Cytokine therapeutics for the treatment of sepsis: Why has nothing worked. Curr Pharm Des. 2003;9:75–82. doi: 10.2174/1381612033392567. [DOI] [PubMed] [Google Scholar]
- Remick DG. Pathophysiology of sepsis. Am J Pathol. 2007;170:1435–1444. doi: 10.2353/ajpath.2007.060872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8:776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- Shapiro NI, Trzeciak S, Hollander JE, Birkhahn R, Otero R, Osborn TM, Moretti E, Nguyen HB, Gunnerson KJ, Milzman D, et al. A prospective, multicenter derivation of a biomarker panel to assess risk of organ dysfunction, shock, and death in emergency department patients with suspected sepsis. Crit Care Med. 2009;37:96–104. doi: 10.1097/CCM.0b013e318192fd9d. [DOI] [PubMed] [Google Scholar]
- Slofstra SH, Spek CA, ten Cate H. Disseminated intravascular coagulation. Hematol J. 2003;4:295–302. doi: 10.1038/sj.thj.6200263. [DOI] [PubMed] [Google Scholar]
- Struewing IT, Durham SN, Barnett CD, Mao CD. Enhanced endothelial cell senescence by lithium-induced matrix metalloproteinase-1 expression. J Biol Chem. 2009;284:17595–17606. doi: 10.1074/jbc.M109.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester AM, Cox JH, Connor AR, Starr AE, Dean RA, Puente XS, Lopez-Otin C, Overall CM. LPS responsiveness and neutrophil chemotaxis in vivo require PMN MMP-8 activity. PLoS One. 2007;2:e312. doi: 10.1371/journal.pone.0000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi V, Boire A, Tchernychev B, Kaneider NC, Leger AJ, O'Callaghan K, Covic L, Kuliopulos A. Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell. 2009;137:332–343. doi: 10.1016/j.cell.2009.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T, Opal SM. Host–pathogen interactions in sepsis. Lancet Infect Dis. 2008;8:32–43. doi: 10.1016/S1473-3099(07)70265-7. [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, Draijer R, Vermeer MA, van Hinsbergh VW. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ Res. 1998;83:1115–1123. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- Vanlaere I, Libert C. Matrix metalloproteinases as drug targets in infections caused by gram-negative bacteria and in septic shock. Clin Microbiol Rev. 2009;22:224–239. doi: 10.1128/CMR.00047-08. Table of Contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaguchi A, Lobo FL, Vincent JL, Pradier O. Platelet function in sepsis. J Thromb Haemost. 2004;2:2096–2102. doi: 10.1111/j.1538-7836.2004.01009.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.