

Abstract
Purpose.
To investigate the pathophysiology of Leber's hereditary optic neuropathy (LHON).
Methods.
Seventy-one subjects from three Chinese families with LHON underwent clinical, genetic, molecular, and biochemical evaluations. Biochemical characterizations included the measurements of the rates of endogenous, substrate-dependent respirations, the adenosine triphosphate (ATP) production and generation of reactive oxygen species using lymphoblastoid cell lines derived from five affected matrilineal relatives of these families and three control subjects.
Results.
Ten of 41 matrilineal relatives exhibited variable severity and age at onset of optic neuropathy. The average age at onset of optic neuropathy in matrilineal relatives of the three families was 5, 11, and 24 years, respectively. Molecular analysis identified the ND1 T3866C (I187T) mutation and distinct sets of polymorphisms belonging to the Eastern Asian haplogroups D4a, M10a, and R, respectively. The I187T mutation is localized at the highly conserved isoleucine at a transmembrane domain of the ND1 polypeptide. The marked reductions in the rate of endogenous, malate/glutamate-promoted and succinate/glycerol-3-phosphate-promoted respiration were observed in mutant cell lines carrying the T3866C mutation. The deficient respiration is responsible for the reduced ATP synthesis and increased generation of reactive oxygen species.
Conclusions.
Our data convincingly show that the ND1 T3866C mutation leads to LHON. This mutation may be insufficient to produce a clinical phenotype. Other modifier factors may contribute to the phenotypic manifestation of the T3866C mutation. The T3866C mutation should be added to the list of inherited factors for molecular diagnosis of LHON. Thus, our findings may provide new insights into the understanding of pathophysiology and valuable information on the management of LHON.
Three Chinese families exhibited a wide range of age and severity of Leber's hereditary optic neuropathy (LHON). Clinical, genetic, molecular, and biochemical evaluations provide the evidence that the mitochondrial ND1 T3866C mutation leads to LHON.
Introduction
Leber's hereditary optic neuropathy (LHON) is a maternally inherited eye disease that generally affects young adults with the rapid, painless, bilateral loss of central vision.1−4 Mutations in mitochondrial DNA (mtDNA) are the molecular bases for this disorder.5−7 Of these, the ND1 G3460A, ND4 G11778A, and ND6 T14484C mutations, which involve genes encoding the subunits of respiratory chain complex I, account for approximately 90% of LHON pedigrees in some countries.8−10 Those LHON-associated mtDNA mutations often occur nearly homoplasmy or homoplasmy. Typical features in LHON pedigrees carrying the mtDNA mutation(s) are incomplete penetrance and male bias among the affected subjects, reflecting the complex etiology of this disease.11,12 The primary defect in these mutations appeared to be a failure in the activity of NADH dehydrogenase,13,14 thereby leading to a deficient function of oxidative phosphorylation, a decrease in adenosine triphosphate (ATP) synthesis, and an increasing generation of reactive oxygen species (ROS). Subsequently, the energy failure and increasing oxidative stress may cause the degeneration of the retinal ganglion cells.3,4
To further elucidate the pathophysiology of LHON, we initiated a systematic and extended mutational screening of mtDNA in a large cohort of Chinese subjects with LHON.15−23 In the previous investigations, we identified the known LHON-associated ND4 G11778A, ND6 T14484C, and ND1 G3460A mutations.15−19 Furthermore, we identified that the ND4 G11696A, ND1 T3394C, ND5 T12338C, and ND6 T14502C mutations are associated with LHON in the Chinese families.20−23 In the present study, we ascertained another three Chinese families with suggestively maternally transmitted LHON. Ten of 41 matrilineal relatives (6 male/4 female) in these families exhibited the variable severity and age at onset in optic neuropathy. Molecular analysis of their mitochondrial genomes identified the T3866C mutation in ND1 gene and distinct sets of polymorphisms belonging to the Eastern Asian haplogroups D4a, M10a, and R.24 Functional significance of the T3866C mutation was evaluated by examining for the rates of endogenous respiration, substrate-dependent respiration, the rate of ATP production, and the generation of ROS using lymphoblastoid cell lines derived from five affected matrilineal relatives carrying the T3866C mutation and from three control individuals lacking the mtDNA mutation.
Materials and Methods
Patients and Subjects
We ascertained three Han Chinese families (Fig. 1) through the School of Ophthalmology and Optometry, Wenzhou Medical College, Wenzhou, China. Informed consent, blood samples, and clinical evaluations were obtained from all participating family members, under protocols approved by the Cincinnati Children's Hospital Medical Center Institute Review Board and the Wenzhou Medical College Ethics Committee. Members of these pedigrees were interviewed at length to identify personal or family medical histories of visual impairments and other clinical abnormalities. All subjects were treated in accordance with the Declaration of Helsinki.
Figure 1. .
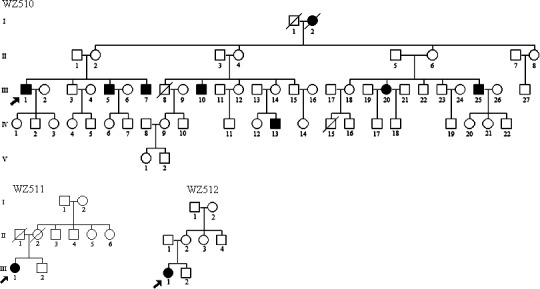
Three Chinese pedigrees with Leber's hereditary optic neuropathy. Vision impaired individuals are indicated by filled symbols. Arrow denotes the probands.
Ophthalmologic Examinations
The following ophthalmologic examinations of proband and other members of this family were conducted: visual acuity, visual field examination (Humphrey Visual Field Analyzer IIi, SITA Standard; Carl Zeiss Meditec, Oberkochen, Germany), visual evoked potentials (VEP; Roland Consult RETI port gamma, flash VEP; Roland Consult, Brandenberg, Germany), and fundus photography (CR6-45NM fundus camera; Canon, Lake Success, NY). The degree of visual impairment was defined according to the visual acuity as follows: normal, >0.3; mild, 0.3 to 0.1; moderate, <0.1 to 0.05; severe, <0.05 to 0.02; and profound <0.02.
Mitochondrial DNA Analysis
Genomic DNA was isolated from whole blood and cell lines of participants using the Puregene DNA Isolation Kits (Gentra Systems, Minneapolis, MN). The presence of the G3460A, G11778A, and T14484C mutations was examined as detailed elsewhere.8 For the detection of the G3460A mutation, the amplified PCR segments were digested with a restriction enzyme BsaHI,8 while the presence of the T14484C mutation was examined by digesting PCR products with a restriction enzyme MvaI.8 For the examination of the G11778A mutation, the amplified PCR segments were digested with the restriction enzyme Tsp45I.15−17
The entire mitochondrial genome of three probands (WZ510 III-1, WZ511 III-1, and WZ512 III-1) from three families was PCR-amplified in 24 overlapping fragments using sets of the light (L) strand and the heavy (H) strand oligonucleotide primers as described previously.25 Each fragment was purified and subsequently analyzed by direct sequencing in an ABI 3700 automated DNA sequencer using the Big Dye Terminator Cycle Sequencing Reaction Kit (Applied Biosystems Inc. [ABI], Foster City, CA). These sequence results were compared with the updated consensus Cambridge sequence (GenBank accession number: NC_012,920).26
The quantification of mtDNA copy numbers from cell lines was performed by slot blot hybridization.27 In brief, the human DNA slot blots were hybridized with a digoxigenin (DIG)-labeled probe for the mitochondrial 12S rRNA. As an internal control, the blots were stripped and rehybridized with a DIG-labeled human 28S rRNA gene as a control.
Phylogenetic Analysis
A total of 16 vertebrates' mitochondrial DNA sequences were used in the interspecific analysis, including Bos Taurus, Cebus albifrons, Gorilla gorilla, Homo sapiens, Hylobates lar, Lemur catta, Macaca mulatta, Macaca sylvanus, Mus musculus, Nycticebus coucang, Pan paniscus, Pongo pygmaeus, Pongo abelii, Papio hamadryas, Tarsius bancanus, and Xenopus laevis (GenBank) (see Supplementary Material and Supplemental Table 1, http://www.iovs.org/content/53/8/4586/suppl/DC1). The conservation index (CI) was calculated by comparing the human mtDNA variants with other 16 vertebrates.
Table 1. .
Summary of Clinical and Molecular Data for 14 Chinese Families with LHON
|
Pedigree |
Ratio of Affected Males to Affected Females |
Average Age at Onset (y) |
No. of Matrilineal Relatives |
Penetrance (%)* |
G11778A Mutation |
T3866C Mutation |
Mitochondrial DNA Haplogroup |
| WZ4116 | 1:0 | 18 | 7 | 14.3 | + | M8a2 | |
| WZ42 | 2:0 | 14.5 | 25 | 8 | + | D4g2 | |
| WZ43 | 1:0 | 15 | 9 | 11.1 | + | B4a1c | |
| WZ44 | 1:0 | 15 | 17 | 5.9 | + | B5b | |
| WZ45 | 2:0 | 16 | 12 | 16.7 | + | N9a1 | |
| WZ46 | 2:1 | 14 | 19 | 15.8 | + | D4b2b | |
| WZ47 | 2:1 | 27 | 11 | 27.3 | + | C | |
| WZ48 | 1:0 | 16 | 19 | 5.3 | + | M7b1 | |
| WZ141 | 6:0 | 22 | 38 | 15.8 | + | B5 | |
| WZ442 | 3:0 | 17 | 9 | 33.3 | + | F1 | |
| WZ5 | 4:1 | 20 | 14 | 35.7 | + | D4b2b | |
| WZ510 | 3:1 | 24 | 27 | 29.6 | + | D4a | |
| WZ511 | 0:1 | 11 | 8 | 12.5 | + | M10a | |
| WZ512 | 0:1 | 5 | 6 | 16.7 | + | R |
Haplogroup Analyses
The entire mtDNA sequences of three Chinese probands carrying the T3866C mutation were assigned to the Asian mitochondrial haplogroups by using the nomenclature of mitochondrial haplogroups.24
Cell Cultures
Lymphoblastoid cell lines were immortalized by transformation with the Epstein-Barr virus, as described elsewhere.28 Cell lines derived from five matrilineal relatives of three Chinese families (WZ510-III-1, WZ510-III-5, WZ510-III-20, WZ511-III-1, and WZ512-III-1) and from three genetically unrelated control individuals (C1, C2, and A51) were grown in RPMI 1640 medium (Invitrogen, Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS). The bromodeoxyuridine (BrdU)-resistant 143B.TK− cell line was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% FBS.
Oxygen Consumption Measurements
Rates of O2 consumption in intact cells were determined with YSI 5300 Oxygraph (Yellow Springs Instruments, Yellow Springs, OH) on samples of 1 × 107 cells in 1.8 mL special DMEM-glucose lacking glucose medium, supplemented with 10% dialyzed FBS.29 Polarographic analysis of digitonin-permeabilized cells, using different respiratory substrates and inhibitors, to test the activity of the various respiratory complexes, was carried out as detailed elsewhere.30
ATP Measurements
The levels of ATP in cells were measured using the ATP Bioluminescence Assay Kit HS II (Roche Applied Science, Indianapolis, IN) according to the manufacturer's instructions and McKensie et al.31 In brief, samples of 2 × 106 cells were incubated for 2 hours in the record solution (156 mM NaCl, 3 mM KCl, 2 mM MgSO4, 1.25 mM KH2PO4, 2 mM CaCl2, 20 mM HEPES, pH 7.35) with either 10 mM glucose, 10 mM glucose plus 2.5 μg/mL oligomycin (glycolytic ATP generation), or 5 mM 2-deoxy-D-glucose plus 5 mM pyruvate (oxidative ATP production). Cells were lysed and then incubated with the luciferin/luciferase reagents. Samples were measured using a SpectraMAX GEMINI XS microplate luminometer (MDS, Inc., Brandon, FL).
ROS Measurements
ROS measurements were performed as detailed elsewhere.32,33 Briefly, approximate 2 × 106 cells of each cell line were harvested, resuspended in PBS, supplemented with 100 uM of 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), and 2% FBS. After incubation at 37°C for 20 minutes, cells were washed, resuspended in PBS in the presence of freshly prepared 2 mM H2O2 and 2% FBS, and then incubated at room temperature for another 5 minutes. Finally, cells were resuspended with 1 mL PBS with 0.5% paraformaldehyde. Samples with or without H2O2 stimulation were analyzed by the BD-LSR II flow cytometer system (Beckton Dickinson, Inc., Franklin Lakes, NJ), with an excitation at 488 nm and emission at 529 nm. Ten thousand events were analyzed in each sample.
Statistical Analysis
Statistical analysis was performed by the unpaired, 2-tailed Student's t-test contained in Microsoft Office Excel (Version 2007; Microsoft Inc., Bellevue, WA). Correlation analysis was performed using the curve fitting routine in the Graph Prism package (GraphPad Software, Inc., La Jolla, CA). P indicates the significance, according to the t-test, of the difference between mutant mean and control mean. Differences were considered significant at a P < 0.05.
Results
Clinical and Genetic Evaluation of Three Chinese Pedigrees
In family WZ510, the proband (III-1) suffered from painless, progressive deterioration of bilateral visual impairment at the age of 30 years. He came to the Eye Clinic at the Wenzhou Medical College for further evaluation at the age of 57 years. His visual acuity was counting finger (CF)/30 cm in both eyes, respectively. Visual field testing demonstrated large centrocecal scotomata in both of his eyes. As shown in Figure 2, a fundus examination showed that both of his optic disks were abnormal, including vascular tortuosity of the central retinal vessels, a circumpapillary telangiectatic microangiopathy, and swelling of the retinal nerve fiber layer. Therefore, he exhibited a typical clinical feature of LHON. Seven of the other 27 matrilineal relatives experienced loss of vision with the average of age at onset of 24 years, the age range being from 10 to 44 years. A variable severity of visual impairment was observed among these matrilineal relatives in this pedigree: profound (III-7, III-20), severe (III-5, IV-13), moderate (III-10), mild (III-25) visual loss, and normal vision (other matrilineal relatives).
Figure 2. .
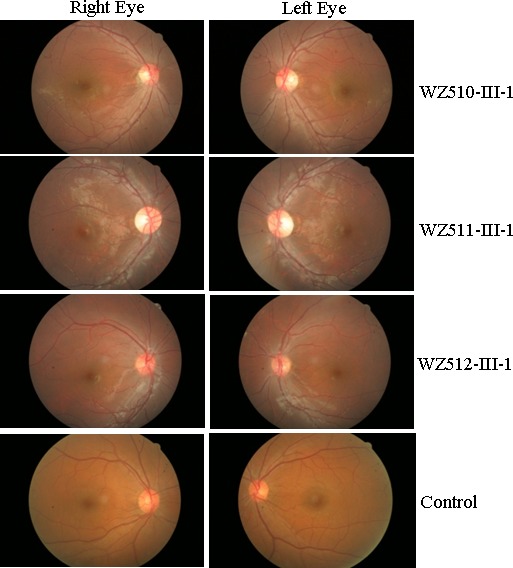
Fundus photograph of three affected subjects (WZ510-III-1, WZ511-III-1, and WZ512-III-1) and one control subject. The figures were taken by fundus photography.
In family WZ511, the proband (III-1) was diagnosed as LHON by the Ophthalmology Clinic at the Wenzhou Medical College at the age of 17 years. She began suffering bilateral visual impairment at the age of 11 years. Her visual acuity was 0.1 in both eyes. IOP was 17 mm Hg and 16 mm Hg in the right and left eyes, respectively. However, none of other members in this family had visual deficit.
In family WZ512, the proband (III-1) suffered from LHON at the age of 5 years. Her visual acuity was hand moving/20 cm in the right and left eyes, respectively. IOP was 22 mm Hg in both her eyes. None of other matrilineal relatives in this pedigree exhibited vision failure.
A comprehensive history and physical examination as well as ophthalmologic examination were performed to identify both personal and family medical histories of visual impairments, and other clinical abnormalities in all available members of three Han Chinese pedigrees. In fact, probands and other matrilineal relatives of these three Chinese families showed no other clinical abnormalities, including diabetes, muscular diseases, hearing dysfunction, and neurologic disorders.
Mitochondrial DNA Analysis
Mutational screening of mtDNA showed the absence of the known LHON-associated G3460A, G11778A, and T14484C mutations in three probands. We then performed the sequence analysis of the entire mtDNA among these probands. As shown in Figure 3, the known T3866C mutation in ND1 gene34 was found in all three subjects. The mutation was present in 6 of 2704 human mitochondrial genomes using the mtDB (Human Mitochondrial Genome Database).35 The T3866C mutation resulted in the replacement of the isoleucine at position 187 with a threonine in the ND1. The isoleucine at this position, 187 in ND1, is localized at the highly conserved residue on the fifth transmembrane domain of this polypeptide (see Supplementary Material and Supplementary Figs. S1, S2, http://www.iovs.org/content/53/8/4586/suppl/DC1). This mutation may alter the tertiary structure of this polypeptide, thereby affecting the function. Further sequence analysis confirmed the presence of the T3866C mutation in all available matrilineal relatives at homoplasmy, but not other members of these families. However, the allele frequency analysis showed that the T3866C mutation was absent in 178 vision-normal Chinese controls.
Figure 3. .
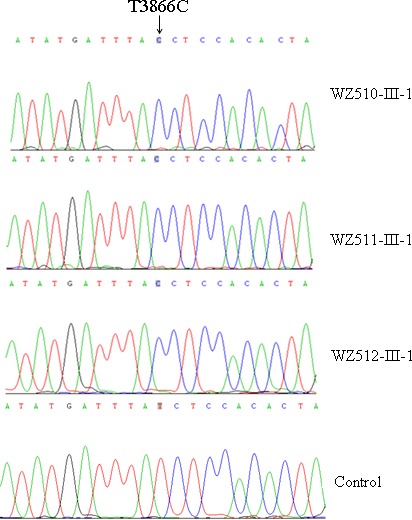
Identification of the ND1 T3866C mutation. Partial sequence chromatograms of ND1 gene from three probands (WZ510-III-1, WZ511-III-1, and WZ512-III-1) and a control subject. An arrow indicates the location of the base changes at position 3866.
In addition to the identical T3866C mutation, as shown in Supplemental Table 2 (see Supplementary Material and Supplementary Table 2, http://www.iovs.org/content/53/8/4586/suppl/DC1), these probands exhibited distinct sets of mtDNA polymorphism belonging to the Eastern Asian haplogroups D4a, M10a, and R, respectively.24 Among 44, 44, and 39 variants of WZ510, WZ511, and WZ512 pedigrees, respectively, their mtDNA shared 22 identical variants. These variants were 13 known variants in the D-loop, 3 known variants in the 12S rRNA gene, 4 known variants in the 16S rRNA gene, 28 (2 novel and 26 known) silent variants, and 19 (2 novel and 17 known) missense mutations in the protein encoding genes.34 These missense mutations are the C5178A (L237M) and A5466G (T333A) in the ND2 gene; the G6915A (V338M) in the CO1 gene; the C8414T (L17F) in the A8 gene; the A8701G (T59A), T8821C (S99A), and A8860G (T112A) in the A6 gene; the G9966A (V254I) in the CO3 gene; the G10197A (A47T) and A10398G (T114A) in the ND3 gene; the G13135A (A267T) and C13561A (L409M) in the ND5 gene; the T14502C (I58V) in the ND6 gene; and the C14766T (T7I), T14979C (I78T), T15071C (Y109H), A15218G (T158A), and A15326G (T194A) in the Cytb gene. These variants in RNAs and polypeptides were further evaluated by phylogenetic analysis of these variants and sequences from 17 vertebrates including mouse,36 bovine,37 and Xenopus laevis.38 The CI of ND1 I187S mutation was 76.5%, which was above the threshold level to be of functional significance in terms of mitochondrial physiology.39 Of other variants, CIs of ATP6 S99A and CO3 V254I variants of WZ511 pedigree, ND3 A47T variant of WZ510 pedigree, ND5 L409M and ND6 I58V variants of WZ512 family, and Cytb T158A variant of WZ511 and WZ512 pedigrees ranged from 76.4% to 100%. Allelic frequency showed that the ATP6 S99A and ND5 L409M variants were absent but CO3 V254I, ND3 A47T, ND6 I58V, and Cytb T158A variants were present among 2704 human mitochondrial genomes using the mtDB.35 Furthermore, other variants were <70% CI, which was below the threshold level to be functional significantly in terms of mitochondrial physiology, proposed by Wallace (2005).39 These data suggest that the ATP6 S99A and ND5 L409M variants but not other variants may be functional significantly.
Lymphoblastoid Cell Lines for Biochemical Assays
Immortalized lymphoblastoid cell lines used for biochemical assays were derived from five affected matrilineal relatives of three Chinese families and three genetically unrelated Chinese controls. The ages of five affected matrilineal relatives (WZ510-III-1, WZ510-III-5, WZ510-III-20, WZ511-III-1, and WZ512-III-1) were 57, 48, 45, 17, and 5 years, respectively; while the ages of three control subjects (C1, C2, and A35) were 25, 29, and 40 years, respectively. The mtDNA haplogroups of three control subjects belonged to D4, M10, and R, respectively.40
The presence and degree of the T3866C mutation in these cell lines were examined. In fact, the T3866C mutation was present in homoplasmy in these cell lines derived from five matrilineal relatives of this Chinese family but absent in the cell lines derived from three Chinese controls. An analysis of the mtDNA content of the individual cell lines was carried out by slot blot hybridization using a DIG-labeled mitochondrial 12S rRNA probe, and by normalizing quantitative differences among DNA samples on the basis of hybridization with a nuclear 28S rRNA probe.27 The average relative levels of mtDNA content of the cell lines derived from controls and affected matrilineal relatives were 125% and 93%, respectively, of those in 143B.TK− cells (9100 molecules per cell).29 There was no significant difference in the mtDNA copy numbers among the mutant and controls cell line.
Oxygen Respiration Defects
The endogenous respiration rates of cell lines derived from five affected individuals carrying the T3866C mutation and from three Chinese control subjects lacking this mutation were measured by determining the O2 consumption rate in intact cells, as described previously.29 As shown in Figure 4A, the rate of total O2 consumption in the lymphoblastoid cell lines derived from two affected individuals ranged between ∼64.1% and 86.4%, with an average consumption of ∼71.6% ± 3.5% relative to the mean value measured in the control cell lines (P = 0.0013).
Figure 4. .
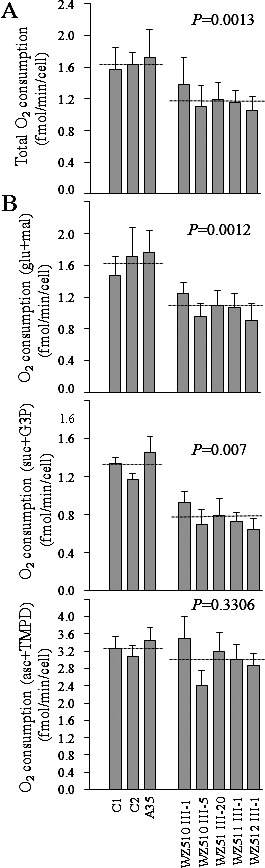
Respiration assays. (A) Average rates of endogenous O2 consumption per cell measured in different cell lines. A total of four determinations were performed on each of the lymphoblastoid cell lines. (B) Polarographic analysis of O2 consumption in digitonin-permeabilized cells of the various cell lines using different substrates and inhibitors. The activities of the various components of the respiratory chain were investigated by measuring, on 1 × 107 digitonin-permeabilized cells, the respiration dependent on malate/glutamate, succinate/G3P, and TMPD/ascorbate. A total of four determinations were performed on each of the lymphoblastoid cell lines: mal/glu, malate/glutamate-dependent respiration; succ/G3P, succinate/G3P-dependent respiration; TMPD/asc, TMPD/ascorbate-dependent respiration. The average of three to six determinations for each cell line is shown. The error bars indicate 2 SE of the mean; P indicates the significance, according to the t-test, of the difference between mutant mean and control mean.
In order to further investigate which enzyme complexes of the respiratory chain were affected in the mutant cell lines, O2 consumption measurements were carried out on digitonin-permeabilized cells, using different substrates and inhibitors.30 As shown in Figure 4B, in the cell lines derived from five affected individuals, the rate of malate/glutamate-driven respiration, which depends on the activities of NADH:ubiquinone oxidoreductase (Complex I), ubiquinol–cytochrome c reductase (Complex III), and cytochrome c oxidase (Complex IV), but usually reflects the rate-limiting activity of Complex I, was very significantly decreased; accounting for 54.6% to 75.9% (∼64% ± 3.7% on the average; P < 0.012) relative to the average rate in the control cell lines. Similarly, the rate of succinate/glycerol-3-phosphate (G3P)-driven respiration, which depends on the activities of Complexes III and IV, but usually reflects the rate-limiting activity of Complex III, were significantly affected in the mutant cell lines, accounting for 49.2% to 70.4% (∼57.5% ± 3.7% on the average; P = 0.0007) relative to the average rate in the control cell lines. Furthermore, the rate of N,N,N',N'-tetramethyl-p-phenylenediamine (TMPD)/ascorbate-driven respiration, which reflects the activity of Complex IV, exhibited a 73.6% to 106.8% oxygen consumption in complex IV activity (∼91.8% ± 5.6% on the average; P < 0.3306) in the mutant cell lines relative to the average rate in the control cell lines. The variations in the rates of malate/glutamate-driven and succinate/G3P-driven respiration among the mutant cell lines derived from affected matrilineal relatives, as compared with the control cell lines, showed a significant correlation with the corresponding variations in the endogenous respiration rates (r = 0.98, P < 0.0001; r = 0.80, P < 0.0027).
Marked Decreases in ATP Generation
The capacity of oxidative phosphorylation in mutant and wild type cells was examined by measuring the levels of cellular ATP using a luciferin/luciferase assay. Populations of cells were incubated in the media in the presence of glucose, glucose with oligomycin, and 2-deoxy-D-glucose with pyruvate.31 The levels of ATP production in mutant cells in the presence of glucose (total cellular levels of ATP) or glucose with oligomycin to inhibit the ATP synthase (glycolysis) were comparable with those measured in the control cell lines (data not shown). By contrast, as shown in Figure 5, the levels of ATP production in mutant cell lines, in the presence of pyruvate and 2-deoxy-D-glucose to inhibit the glycolysis (oxidative phosphorylation), ranged from 42.7% to 48.7%, with an average of 45.2% ± 1% relative to the mean value measured in the control cell lines (P < 0.0001). The variations in the levels of ATP production in mutant cell lines derived from affected matrilineal relatives, as compared with the control cell lines, showed a significant correlation with the corresponding variations in the rates of endogenous respiration, malate/glutamate-driven respiration, and succinate/G3P-driven respiration (r = 0.89, P < 0.0004; r = 0.90, P < 0.0003; r = 0.58, P < 0.0279).
Figure 5. .
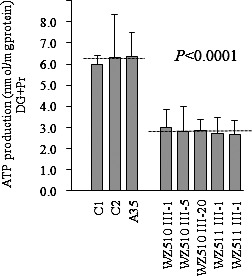
Measurement of cellular ATP levels using bioluminescence assay. Cells were incubated with 5 mm 2-deoxy-d-glucose plus 5 mm pyruvate to determine ATP generation under mitochondrial ATP synthesis. Average rates of ATP level per cell line are shown, with error bars representing doubled standard errors. Average rates of ATP level per cell line are shown. Six to seven determinations were made for each cell line. Graph details and symbols are explained in the legend to Figure 4.
ROS Production Increases
The levels of the ROS generation in the vital cells derived from five affected matrilineal relatives carrying the T3866C mutation and three control individuals lacking the mutation were measured with flow cytometry under normal and H2O2 stimulation.32,33 Geometric mean intensity was recorded to measure the rate of ROS of each sample. The ratio of geometric mean intensity between unstimulated and stimulated cell lines with H2O2 was calculated to delineate the reaction upon increasing level of ROS under oxidative stress. As shown in Figure 6, the levels of ROS generation in the lymphoblastoid cell lines derived from five affected individuals carrying the T3866C mutation ranged from 110.9% to 134.2%, with an average of 122% ± 4.2% (P = 0.0109) of the mean value measured in the control cell lines. The variations in the levels of ROS generation in mutant cell lines derived from affected matrilineal relatives, as compared with the control cell lines, showed a significant correlation with the corresponding variations in the rates of endogenous respiration, malate/glutamate-driven respiration, and succinate/G3P-driven respiration as well as the levels of ATP production (r = 0.76, P < 0.0045; r = 0.67, P < 0.0128; r = 0.69, P < 0.0106; r = 0.70, P < 0.0089).
Figure 6. .
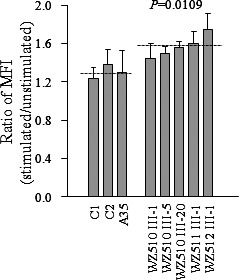
Ratio of geometric mean intensity between levels of the ROS generation in the vital cells with or without H2O2 stimulation. The rates of production in ROS from five affected matrilineal relatives and three control individuals were analyzed by BD-LSR II flow cytometer system with or without H2O2 stimulation. The relative ratio of intensity (stimulated versus unstimulated with H2O2) was calculated. The average of three determinations for each cell line is shown. Graph details and symbols are explained in the legend to Figure 4.
Discussion
In the present study, we investigated the molecular pathogenesis of the LHON-associated ND1 T3866C mutation in three Chinese pedigrees with optic neuropathy. This mutation was only present in the matrilineal relatives of these three families at homoplasmy and not in 178 Chinese controls. The T3866C (I187T) mutation is localized at the highly conserved isoleucine at position 187 on the fifth transmembrane domain of the ND1 polypeptide. The altered structure of the ND1 polypeptide caused by the T3866C mutation appeared to be responsible for significantly mitochondrial dysfunction associated with the LHON in these Chinese families. Furthermore, the occurrence of the T3866C mutation in these genetically unrelated subjects affected by visual impairment further supports that this mutation is involved in the pathogenesis of visual impairment. As shown in Table 1, 10 of 41 matrilineal relatives in these three Chinese families carrying the T3866C mutation exhibited visual impairment, while the average penetrances of visual impairment in the other 11 Chinese pedigrees carrying the G11778A mutation were 19.2%.16,26,40 Furthermore, the average age at onset for visual impairment in these three Chinese families was 5, 11, and 24 years of age, respectively. Conversely, the average age at onset of visual impairment in the other 11 Chinese pedigrees, 66 and 49 Caucasian pedigrees carrying the G11778A mutation was 18, 24, and 28 years, respectively.2,11,16,26,40,41 In addition, the ratio between affected male and female matrilineal relatives is 3:1, 0:1, 0:1 in these Chinese families, whereas this ratio ranged from 2:1 to 6:0 in 11 Chinese families16,21,42 and were 4.5:1 and 3.7:1 in two large cohorts of Caucasian pedigrees carrying the G11778A mutation, respectively.2,11,43 These data strongly indicate that the T3866C mutation is necessary but that by itself is insufficient to induce a clinical expression of LHON, as in the cases of the G11778A, T141484C, and T12338C mutations.13,14,16,18,23 Therefore, the modifier factors should contribute to the phenotypic manifestation of T3866C mutation.
In the present investigation, all lymphoblastoid cell lines carrying the T3866C mutation revealed significant mitochondrial dysfunctions, which could be associated with the T3866C mutation and the contribution of other modifiers. In particular, these cells showed ∼36% decrease in NADH dehydrogenase-dependent respiration after digitonin permeabilization. These data are in good agreement with the observations that there were 30% to 40% reductions in NADH dehydrogenase-dependent respiration in cell lines derived from families carrying the G11778A mutation.13,14,33 These observations strongly indicate that the primary defect in the T3866C mutation was a failure in the activity of NADH dehydrogenase. Remarkably, 42.5% decrease in the rates of succinate/G3P-driven respirations was observed in the mutant cell lines carrying the T3866C mutation. This result was in contrast with the previous investigations indicating that the rate of succinate/G3P-driven respiration in the cell lines carrying the G11778A mutation was comparable with those controls.13,14,44 In fact, the presence of the Cytb T158A variant of WZ511 and WZ512 pedigrees may contribute to the decreased activity of complex III, in the case of one Chinese family carrying the G11778A mutation.33,45 The lack of significant variant in the Cytb gene in the mtDNA of WZ510 pedigree suggests that mutation(s) in nuclear genes may lead to the deficiency of complex-III activity. However, none of the mutations in nuclear genes involved in complex-III activity has been identified as associated with LHON.
There was a very significant correlation between the overall respiratory capacity or the rate of NADH dehydrogenase-dependent respiration or succinate/glycerol-3-phosphate (G3P)-driven respiration and the level of ATP production (P < 0.01) as well as ROS production in the control and mutant cell lines. This correlation is clearly consistent with the importance that the marked decrease in the activities of complexes I and III in the mutant cell lines carrying the T3866C mutation plays a role in decreasing ATP synthesis. In this investigation, ∼45% decrease of ATP synthesis in mutant lymphoblastoid cell lines derived from these Chinese families carrying the T3866C mutation was consistent with those in cybrid cell lines carrying the G3460A, G11778A, A13528G, T14484C, or G14279A mutations.46,47 Moreover, altered activities of complexes I and III can lead to more electron leakage from the electron transport chain, and in turn, increase the generation of ROS.33,48,49 Here, an increase of ROS production was detected in cell lines derived from affected matrilineal relatives carrying the T3866C mutation. The production level of ROS in cells carrying the T3866C mutation appeared to be much lower than those cell lines carrying the G11778A mutation.42,47,50 Remarkably, 2.5-fold more cellular hydroperoxide was detected in neuronal NT2 cells carrying the G11778A mutation.51 This discrepancy is likely attributed to differentiation-specific effects.51 The increasing generation of ROS can cause damage to DNA, lipids, proteins, and membranes. As a result, these biochemical defects can cause dysfunction or apoptosis in retinal ganglion cells, thereby producing the clinical phenotype.52
In summary, this is the first study to investigate the molecular pathogenesis underlying the ND1 T3866C mutation leading to LHON. The T3866C mutation should be added to the list of inherited factors for future molecular diagnosis of LHON. Thus, our findings may provide new insights into the understanding of pathophysiology and valuable information on the management and treatment of LHON.
Supplementary Material
Footnotes
Supported by National Key Technologies R&D Program Grant 2012BAI09B03 from the Ministry of Science and Technology of China (MXG and PJ).
These authors contributed equally to this work and therefore should be regarded as equivalent authors.
Disclosure: X. Zhou, None; Y. Qian, None; J. Zhang, None; Y. Tong, None; P. Jiang, None; M. Liang, None; X. Dai, None; H. Zhou, None; F. Zhao, None; Y. Ji, None; J.Q. Mo, None; J. Qu, None; M.-X. Guan, None
References
- 1. Newman NJ, Wallace DC. Mitochondria and Leber's hereditary optic neuropathy. Am J Ophthalmol. 1990;109:726–730 [DOI] [PubMed] [Google Scholar]
- 2. Brown MD, Wallace DC. Spectrum of mitochondrial DNA mutations in Leber's hereditary optic neuropathy. Clin Neurosci. 1994;2:134–145 [Google Scholar]
- 3. Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. J Med Genet. 2009;46:145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787:518–528 [DOI] [PubMed] [Google Scholar]
- 5. Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science. 1988;242:1427–1430 [DOI] [PubMed] [Google Scholar]
- 6. Howell N. LHON. and other optic nerve atrophies: the mitochondrial connection. Dev Ophthalmol. 2003;37:94–108 [DOI] [PubMed] [Google Scholar]
- 7. Servidei S. Mitochondrial encephalomyopathies: gene mutation. Neuromuscul Disord. 2004;14:107–116 [DOI] [PubMed] [Google Scholar]
- 8. Brown MD, Torroni A, Reckord CL, Wallace DC. Phylogenetic analysis of Leber's hereditary optic neuropathy mitochondrial DNA's indicates multiple independent occurrences of the common mutations. Hum Mutat. 1995;6:311–325 [DOI] [PubMed] [Google Scholar]
- 9. Mackey DA, Oostra RJ, Rosenberg T, et al. Primary pathogenic mtDNA mutations in multigeneration pedigrees with Leber hereditary optic neuropathy. Am J Hum Genet. 1996;59:481–485 [PMC free article] [PubMed] [Google Scholar]
- 10. Mashima Y, Yamada K, Wakakura M, et al. Spectrum of pathogenic mitochondrial DNA mutations and clinical features in Japanese families with Leber's hereditary optic neuropathy. Curr Eye Res. 1998;17:403–408 [DOI] [PubMed] [Google Scholar]
- 11. Riordan-Eva P, Sanders MD, Govan GG, Sweeney MG, Da Costa J, Harding AE. The clinical features of Leber's hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–337 [DOI] [PubMed] [Google Scholar]
- 12. Newman NJ. Leber's hereditary optic neuropathy. Ophthalmol Clin North Am. 1993;4:431–447 [Google Scholar]
- 13. Hofhaus G, Johns DR, Hurkoi O, Attardi G, Chomyn A. Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber's hereditary optic neuropathy. J Biol Chem. 1996;271:13155–13161 [DOI] [PubMed] [Google Scholar]
- 14. Brown MD, Torroni A, Reckord CL, Wallace DC. Functional analysis of lymphoblast and cybrid mitochondria containing the 3460, 11778, or 14484 Leber's hereditary optic neuropathy mitochondrial DNA mutation. J Biol Chem. 2000;275:39831–39836 [DOI] [PubMed] [Google Scholar]
- 15. Qu J, Wang Y, Tong Y, et al. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation in a Chinese family. Invest Ophthalmol Vis Sci. 2006;47:475–483 [DOI] [PubMed] [Google Scholar]
- 16. Qu J, Zhou X, Zhang J, et al. Extremely low penetrance of Leber's hereditary optic neuropathy in eight Han Chinese families carrying the ND4 G11778A mutation. Ophthalmology. 2009;116:558–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qu J, Wang Y, Tong Y, et al. Leber's hereditary optic neuropathy affects only female matrilineal relatives in two Chinese families. Invest Ophthalmol Vis Sci. 2010;51:4906–4912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qu J, Zhou X, Zhao F, et al. Low penetrance of Leber's hereditary optic neuropathy in ten Han Chinese families carrying the ND6 T11484C mutation. Biochim Biophys Acta. 2010;1800:305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tong Y, Sun YH, Zhou X, et al. Very low penetrance of Leber's hereditary optic neuropathy in five Han Chinese families carrying the ND1 G3460A mutation. Mol Genet Metab. 2010;99:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhou X, Wei Q, Yang L, et al. Leber's hereditary optic neuropathy is associated with the mitochondrial ND4 G11696A mutation in five Chinese families. Biochem Biophys Res Commun. 2006;340:69–75 [DOI] [PubMed] [Google Scholar]
- 21. Liang M, Guan M, Zhao F, et al. Leber's hereditary optic neuropathy is associated with mitochondrial ND1 T3394C mutation. Biochem Biophys Res Commun. 2009;383:286–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhao F, Guan M, Zhou X, et al. Leber's hereditary optic neuropathy is associated with mitochondrial ND6 T14502C mutation. Biochem Biophys Res Commun. 2009;389:466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu XL, Zhou X, Zhou J, et al. Leber's hereditary optic neuropathy is associated with the T12338C mutation in mitochondrial ND5 gene in six Han Chinese families. Ophthalmology. 2011;118:978–985 [DOI] [PubMed] [Google Scholar]
- 24. Tanaka M, Cabrera VM, González AM, et al. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004;14:1832–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rieder MJ, Taylor SL, Tobe VO, Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26:967–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23:147 [DOI] [PubMed] [Google Scholar]
- 27. Guan MX, Fischel-Ghodsian N, Attardi G. Biochemical evidence for nuclear gene involvement in phenotype of non-syndromic deafness associated with mitochondrial 12S rRNA mutation. Hum Mol Genet. 1996;5:963–972 [DOI] [PubMed] [Google Scholar]
- 28. Miller G, Lipman M. Release of infections Epstein-Barr virus by transformed marmoser leukocytes. Proc Natl Acad Sci U S A. 1973;70:190–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503 [DOI] [PubMed] [Google Scholar]
- 30. Hofhaus G, Shakeley RM, Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483 [DOI] [PubMed] [Google Scholar]
- 31. McKenzie M, Liolitsa D, Akinshina N, et al. Mitochondrial ND5 gene variation associated with encephalomyopathy and mitochondrial ATP consumption. J Bio Chem. 2007;282:36845–36852 [DOI] [PubMed] [Google Scholar]
- 32. Mahfouz R, Sharma R, Lachner J, Aziz N, Agarwal A. Evaluation of chemiluminescence and flow cytometry as tools in assessing production of hydrogen peroxide and superoxide anion in human spermatozoa. Fertil Steril. 2008;92:819–827 [DOI] [PubMed] [Google Scholar]
- 33. Qian Y, Zhou X, Liang M, Qu J, Guan MX. The altered activity of complex III may contribute to the high penetrance of Leber's hereditary optic neuropathy in a Chinese family carrying the ND4 G11778A mutation. Mitochondrion. 2011;11:871–877 [DOI] [PubMed] [Google Scholar]
- 34. Ruiz-Pesini E, Lott MT, Procaccio V, et al. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35:D823–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ingman M, Gyllensten U. mtDB: Human Mitochondrial Genome Database, a resource for population genetics and medical sciences. Nucleic Acids Res. 2006;34:D749–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981;26:167–180 [DOI] [PubMed] [Google Scholar]
- 37. Gadaleta G, Pepe G, De Candia G, Quagliariello C, Sbisa E, Saccone C. The complete nucleotide sequence of the Rattus norvegicus mitochondrial genome: cryptic signals revealed by comparative analysis between vertebrates. J Mol Evol. 1989;28:497–516 [DOI] [PubMed] [Google Scholar]
- 38. Roe A, Ma DP, Wilson RK, Wong JF. The complete nucleotide sequence of the Xenopus laevis mitochondrial genome. J Biol Chem. 1985;260:9759–9774 [PubMed] [Google Scholar]
- 39. Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu J, Qian Y, Li Z, et al. Mitochondrial haplotypes may modulate the phenotypic manifestation of the deafness-associated 12S rRNA 1555A>G mutation. Mitochondrion. 2010;10:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Qu J, Li R, Tong Y, et al. Only male matrilineal relatives with Leber's hereditary optic neuropathy in a large Chinese family carrying the mitochondrial DNA G11778A mutation. Biochem Biophys Res Commun. 2005;328:1139–1145 [DOI] [PubMed] [Google Scholar]
- 42. Qian Y, Zhou X, Hu Y, et al. Clinical evaluation and mitochondrial DNA sequence analysis in three Chinese families with Leber's hereditary optic neuropathy. Biochem Biophys Res Commun. 2005;332:614–621 [DOI] [PubMed] [Google Scholar]
- 43. Nikoskelainen EK. Clinical pictures of LHON. Clin Neurosci. 1994;2:115–120 [Google Scholar]
- 44. Pello R, Martín MA, Carelli V, et al. Mitochondrial DNA background modulates the assembly kinetics of OXPHOS complexes in a cellular model of mitochondrial disease. Hum Mol Genet. 2008;17:4001–4011 [DOI] [PubMed] [Google Scholar]
- 45. Zhou X, Zhang H, Zhao F, et al. Very high penetrance and occurrence of Leber's hereditary optic neuropathy in a large Han Chinese pedigree carrying the ND4 G11778A mutation. Mol Genet Metab. 2010;100:379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Baracca A, Solaini G, Sgarbi G, et al. Severe impairment of complex I–driven adenosine triphosphate synthesis in Leber hereditary optic neuropathy cybrids. Arch Neurol. 2005;62:730–736 [DOI] [PubMed] [Google Scholar]
- 47. Beretta S, Mattavelli L, Sala G, et al. Leber hereditary optic neuropathy mtDNA mutations disrupt glutamate transport in cybrid cell lines. Brain. 2004;127:2183–2192 [DOI] [PubMed] [Google Scholar]
- 48. Lenaz G, Baracca A, Carelli V, D'Aurelio M, Sgarbi G, Solaini G. Bioenergetics of mitochondrial diseases associated with mtDNA mutations. Biochim Biophys Acta. 2004;1658:89–94 [DOI] [PubMed] [Google Scholar]
- 49. Yen MY, Wang AG, Wei YH. Leber's hereditary optic neuropathy: a multifactorial disease. Prog Retin Eye Res. 2006;25:381–396 [DOI] [PubMed] [Google Scholar]
- 50. Porcelli AM, Angelin A, Ghelli A, et al. Respiratory complex I dysfunction due to mitochondrial DNA mutations shifts the voltage threshold for opening of the permeability transition pore toward resting levels. J Biol Chem. 2009;284:2045–2052 [DOI] [PubMed] [Google Scholar]
- 51. Wong A, Cavelier L, Collins-Schramm HE, et al. Differentiation-specific effects of LHON mutations introduced into neuronal NT2 cells. Hum Mol Genet. 2002;11:431–438 [DOI] [PubMed] [Google Scholar]
- 52. Carelli V, La Morgia C, Valentino ML, Barboni P, Ross-Cisneros FN, Sadun AA. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim Biophys Acta. 2009;1787:518–528 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.