

Abstract
Dark exposure initiated in adulthood reactivates robust ocular dominance plasticity in the visual cortex. Here, we show that a critical component of the response to dark exposure is the rejuvenation of inhibitory synaptic transmission, resulting in a decrease in functional inhibitory synaptic density, a decrease in paired-pulse depression, and a reexpression of endocannabinoid-dependent inhibitory long-term depression (iLTD). Importantly, pharmacological acceleration of the maturation of inhibition in dark-exposed adults inhibits the reexpression of iLTD and the reactivation of ocular dominance plasticity. Surprisingly, dark exposure initiated earlier in postnatal development does not rejuvenate inhibitory synaptic transmission or facilitate rapid ocular dominance plasticity, demonstrating the presence of a refractory period for the regulation of synaptic plasticity by visual deprivation.
Introduction
In juveniles, brief monocular deprivation (MD) rapidly shifts the ocular dominance of binocular neurons away from the deprived eye. Over the course of postnatal development, brief monocular deprivation becomes increasingly ineffective. Maturation of perisomatic inhibition mediated by fast-spiking parvalbumin-positive interneurons is believed to constrain Hebbian plasticity at excitatory synapses in the visual cortex, and inhibit rapid ocular dominance plasticity (Kirkwood et al., 1995; Huang et al., 1999; Rozas et al., 2001; Jiang et al., 2005; Di Cristo et al., 2007).
In the rodent primary visual cortex, the number of perisomatic GABAergic synapses onto individual pyramidal neurons increases approximately threefold between eye opening [postnatal day 15 (P15)] and puberty (P35) (Huang et al., 1999; Morales et al., 2002; Chattopadhyaya et al., 2004). During this time, inhibitory synapses transition from an immature state with high release probability but prone to depletion, to a mature state with a low release probability and increased fidelity of transmission (Jiang et al., 2010). The functional maturation of inhibitory synaptic transmission, mediated by an endocannabinoid-dependent long-term synaptic depression [inhibitory long-term depression (iLTD)] is complete by P35.
Acceleration of the maturation of inhibition induces a precocious critical period for ocular dominance plasticity (Huang et al., 1999; Di Cristo et al., 2007). Similarly, dark rearing decelerates the maturation of GABAergic circuits (Morales et al., 2002; Chattopadhyaya et al., 2004; Di Cristo et al., 2007; Jiang et al., 2007, 2010; Kreczko et al., 2009) and the developmental constraint on ocular dominance plasticity (Cynader, 1983; Mower and Christen, 1985; Fagiolini et al., 1994; Guire et al., 1999). However, dark exposure (DE) or suppression of GABA synthesis at P35 does not reduce the number or strength or inhibitory synapses (Morales et al., 2002; Chattopadhyaya et al., 2007; Jiang et al., 2007). This predicts that the developmental constraint on rapid ocular dominance plasticity would be equally irreversible.
Nonetheless, several interventions have been shown to reactivate rapid ocular dominance plasticity in adulthood and implicate the downregulation of intracortical inhibition in this process (Pizzorusso et al., 2002; He et al., 2006; Sale et al., 2007; Maya Vetencourt et al., 2008). Indeed, reactivation of ocular dominance plasticity in adults is induced by a GABAA receptor antagonist (Harauzov et al., 2010) and reversed by enhancing GABAAergic inhibition with a benzodiazepine (Sale et al., 2007; Maya Vetencourt et al., 2008). This work supports the view that inhibitory synaptic transmission imposes limitations on ocular dominance plasticity in adults. However, it is at odds with the view that the developmental maturation of inhibition is irreversible.
We addressed this discrepancy by examining GABAergic synaptic transmission after dark exposure, a potent and noninvasive intervention that reactivates ocular dominance plasticity in adults (He et al., 2006, 2007). Dark exposure initiated in adulthood rejuvenated inhibitory synaptic transmission, resulting in a visual cortex with immature characteristics including robust endocannabinoid-dependent iLTD and rapid ocular dominance plasticity. Surprisingly, dark exposure initiated earlier in development did not stimulate the reexpression of iLTD or facilitate ocular dominance plasticity, demonstrating a refractory period for the rejuvenation of inhibition by visual deprivation.
Materials and Methods
Long–Evans rats were raised on a 12 h light/dark cycle, with food and water available ad libitum. Subjects were moved into a dark room at the indicated age (±2 d), and care was provided under infrared illumination. All procedures conform to the guidelines of the Department of Health and Human Services and the Institutional Animal Care and Use Committees of The Johns Hopkins University and University of Maryland.
Visual cortical slices.
Visual cortical slices (300 μm) were prepared as described previously (Kirkwood and Bear, 1994) in ice-cold dissection buffer containing the following (in mm): 212.7 sucrose, 5 KCl, 1.25 NaH2PO4, 10 MgCl2, 0.5 CaCl2, 26 NaHCO3, 10 dextrose, bubbled with 95% O2/5% CO2, pH 7.4. Slices were transferred to normal artificial CSF (ACSF) for at least 1 h before recording. Normal ACSF was similar to the dissection buffer except that sucrose was replaced by 124 mm NaCl, MgCl2 was lowered to 1 mm, and CaCl2 was raised to 2 mm.
Visualized whole-cell voltage-clamp recordings.
Visualized whole-cell voltage-clamp recordings were made from layer II/III pyramidal neurons with glass pipettes filled with intracellular solution (in mm: 140 CsCl, 0.2 CaCl2, 8 NaCl, 2 EGTA, 0.5 NaGTP, 4 MgATP, and 10 HEPES, pH 7.2). Only cells with membrane potentials less than −65 mV, series resistance <20 MΩ, and input resistance >100 MΩ were included. Cells were excluded if input resistance changed >15% over the experiment. Data were filtered at 5 kHz and digitized at 10 kHz using Igor Pro (Wavemetrics). Synaptic currents were recorded at −60 mV in the presence of 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 100 μm 2-amino-5-phosphonovaleric acid (APV), and evoked every 20 s by stimulation of layer IV with 0.2 ms pulses delivered in pairs (interstimulus interval, 100 ms) to compute paired-pulse depression (PPD = 1 − p2/p1, where p1 and p2 are the amplitude of the response to the first and second stimulation, respectively). Stimulation was delivered through concentric bipolar stimulating electrodes (FHC) with intensity adjusted to evoke 100–300 pA responses. Synaptic strength was quantified as the IPSC amplitude. Ten minutes of stable baseline (<10% change) was required before any experimental manipulation. iLTD was induced with theta burst stimulation (TBS), consisting of four theta burst epochs delivered at 0.1 Hz. Each TBS epoch consisted of 10 trains of four pulses (100 Hz) delivered at 5 Hz. Statistical significance was assessed with one-tailed, unpaired t test or two-way ANOVA followed by Tukey's [honestly significant difference (HSD)] post hoc analysis.
Monocular deprivation.
Animals were anesthetized with ketamine/xylazine (50 mg/10 mg/kg, i.p.). The margins of the upper and lower lids of one eye were trimmed and sutured together. The animals were returned to their home cages for 3 d and disqualified in the event of suture opening or infection.
Visually evoked potentials.
Visually evoked potentials (VEPs) were recorded from the surface or layer IV of binocular visual cortex (V1b; ∼7 mm posterior to bregma and 4 mm lateral to the midline) with tungsten microelectrodes (0.5 MΩ) relative to a ground screw in the frontal bone. V1b was exposed through a hole (∼4 mm diameter) in the skull after urethane anesthesia (1.4 mg/kg, i.p.). Electrode placement on the binocular region of V1 was confirmed by capturing a VEP in response to stimulation of the ipsilateral eye. Visual stimuli were full screen horizontal square wave gratings of 0.04 cycles degree−1 reversing at 1 Hz, with 96.28% contrast and 40 cd/m2 luminosity, presented on a computer monitor 25 cm from eyes, in a darkened room. The amplitude of the primary positive (surface) or negative (layer IV) component of the VEP (∼150 ms latency) was used to assess the cortical response to visual stimulation. VEPs were filtered (0.5–60 Hz bandpass digital filter) and averaged (100 repetitions) in synchrony with the stimulus using OpenEX software. Statistical significance was assessed with two-way ANOVA followed by Tukey's HSD post hoc analysis.
Drug solutions.
For systemic injections, diazepam (DZ) and (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo(1,2,3-de)-1,4-benzoxazin-6-yl]-1-napthalenylmethanone (WIN 55212-2) were dissolved in 10% Tween 80, 20% DMSO, and 70% saline to a final concentration of 1 mg/ml. Subjects received diazepam (one time per day; 15 mg/kg diazepam) and WIN (two times per day; 5 mg/kg) for 3 d via intraperitoneal injections. For in vitro experiments, stock solutions of 1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-(1-piperidy)pyrazole-3-carboxamide (AM251) or WIN were dissolved in DMSO and diluted in ACSF to the final concentration immediately before use. AM251, diazepam, and WIN were purchased from Tocris. CNQX, APV, Tween 80, and DMSO were purchased from Sigma-Aldrich.
Results
A refractory period for the rejuvenation of GABAergic transmission
We recently described an endocannabinoid-mediated long-term depression of inhibitory synaptic transmission (iLTD) in layer II/III neurons in the visual cortex that is comparable with iLTD described in other brain regions (Chevaleyre et al., 2006; McBain and Kauer, 2009; Jiang et al., 2010). Cortical iLTD is robust in juveniles, but absent in subjects older than P35. Importantly, dark exposure initiated at P35 does not stimulate reexpression of iLTD (Jiang et al., 2010). However, dark exposure in much older subjects can reactivate ocular dominance plasticity (He et al., 2006, 2007), which prompted us to reexamine the effect of visual deprivation on synaptic plasticity at inhibitory synapses.
Pharmacologically isolated IPSCs were evoked in layer II/III pyramidal neurons by stimulating layer IV in slices of rat primary visual cortex, a configuration shown to recruit inputs mediated by fast-spiking interneurons (Jiang et al., 2010). iLTD was induced with TBS. We confirmed that iLTD was absent at P45 and that 10 d of dark exposure initiated at P35 did not stimulate iLTD reexpression [DE, 96.0 ± 3.7% of baseline 30 min after TBS; normal reared (NR), 94.0 ± 3.4%] (Fig. 1A). However, if dark exposure was initiated in much older subjects (P90), robust iLTD could be subsequently induced by TBS (DE, 72.9 ± 6.4%; NR, 98.3 ± 4.7%) (Fig. 1B). In juveniles, iLTD requires the activation of type 1 endocannabinoid receptors (CB1Rs) and results in a decrease in paired-pulse depression. Similarly, the iLTD that was observed after dark exposure at P90 was blocked by the CB1R antagonist AM251 and was accompanied by a reduction in paired-pulse depression (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

A refractory period for the rejuvenation of inhibition in the visual cortex by 10 d of dark exposure. A, B, IPSC amplitudes recorded in layer II/III pyramidal neurons after TBS (arrow) delivered to layer IV. Absence of iLTD in response to TBS in slices prepared from NR P45 (A, open circles) and P100 visual cortex (B, open circles). Ten days of dark exposure initiated at P90 (B, filled circles) but not P35 (A, filled circles) reactivated significant iLTD. *p < 0.01, one-tail t test at 30 min after TBS. C, The magnitude of iLTD as a function of the age of initiation of dark exposure (filled circles) compared with age-matched normal-reared controls (open circles). Two-way ANOVA, F(7,97) = 3.534, p = 0.002; *p < 0.05 in Tukey's HSD post hoc. D, The magnitude of paired-pulse depression as a function of the age of initiation of dark exposure (filled circles) compared with age-matched normal-reared controls (open circles). Two-way ANOVA, F(7,80) = 2.9193, p = 0.009; *p < 0.05 in Tukey's HSD post hoc. E, F, Dark exposure at P90 (F) but not P35 (E) restored the sensitivity of the IPSC to depression by the CB1R agonist WIN (gray box). The symbols are as in A and B. The numbers in parentheses indicate number of subjects, number of slices.
To further explore the unexpected interaction between visual deprivation and age, we examined the effects of 10 d of dark exposure initiated at ages ranging from P21 to P90 on the magnitude of iLTD and paired-pulse depression. Dark exposure enhanced iLTD if initiated at P21 (iLTD, 22.4 ± 5.3% depression from baseline), when cortical inhibitory circuitry is immature (Huang et al., 1999; Morales et al., 2002; Chattopadhyaya et al., 2004), but had little effect if initiated at P35–P49 (4.0 ± 3.7, 1.0 ± 6.6%). Nevertheless, dark exposure enabled robust iLTD if initiated at P70–P90 (18.5 ± 6.2, 28.7 ± 6.5%) (Fig. 1C). The U-shaped relationship between age at initiation of DE and magnitude of iLTD contrasts with the uniformly small iLTD evoked in NR age-matched P31–P100 controls (Fig. 1C). A parallel U-shaped relationship exists between the age at initiation of dark exposure and the magnitude of paired-pulse depression (Fig. 1D). The loss of iLTD observed over development is accompanied by a reduction in CB1R levels and a loss of response to CB1R activation (Jiang et al., 2010). We therefore asked whether adult dark exposure reversed these processes. We found that dark exposure initiated at P90, but not P45, restored the sensitivity of the IPSC to depression by the CB1R agonist WIN (10 μm, 10 min; DE P45: 92.7 ± 7.1%; NR P45: 98.0 ± 6.6%) (Fig. 1E) (DE P90: 57.4 ± 5.1%; NR P90: 88.7 ± 4.7%) (Fig. 1F). Similarly, quantitative PCR confirmed that DE at P90, but not P45 induced in a significant increase in the level of CB1R mRNA in the visual cortex (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Thus, dark exposure rejuvenates many aspects of inhibitory synaptic transmission but is only effective if initiated before or after a refractory period.
The anatomical and functional maturation of inhibition can be decelerated by dark rearing from birth but is unaffected by dark exposure initiated at P35 (Morales et al., 2002; Chattopadhyaya et al., 2007; Jiang et al., 2007). We therefore tracked changes in the maximal evoked IPSC to ask whether the density of functional inhibitory synapses onto individual pyramidal neurons could be regulated by visual experience in adulthood (Choi et al., 2002; Morales et al., 2002; Goldberg et al., 2005). Dark exposure initiated at P35–P38 did not decrease IPSC magnitude (DE: 4.32 ± 0.27 nA; NR: 4.22 ± 0.32 nA) (Fig. 2A) or the input/output relationship across a range of stimulus intensities (5–100 mA; two-way ANOVA, F(1,225) = 0.71, p = 0.79). In contrast, dark exposure initiated at P90–P93 induced a significant decrease in the maximal IPSC amplitude (DE: 2.97 ± 0.37 nA; NR: 4.53 ± 0.36 nA) (Fig. 2B) and a reduction in the input/out relationship (two-way ANOVA, F(1,145) = 12.59, p = 0.0013). Together, this demonstrates a parallel refractory period for the rejuvenation of many aspects of inhibitory synaptic transmission, including endocannabinoid-dependent synaptic depression, regulation of presynaptic neurotransmitter release, and functional connectivity.
Figure 2.
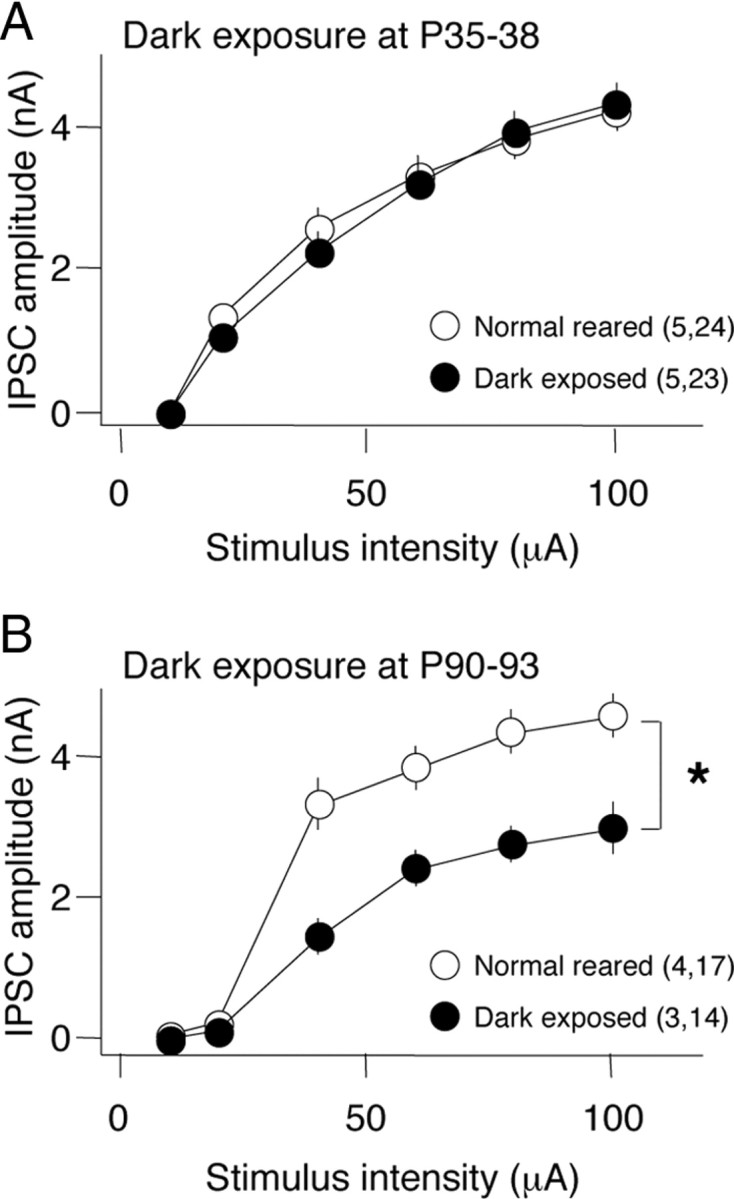
Refractory period for the reduction of maximal IPSC amplitude in the visual cortex by 10 d of dark exposure. IPSC amplitudes evoked by a range of stimulus intensities (5–100 A) recorded in layer II/III pyramidal neurons from slices of visual cortex after 10 d of DE (filled circles) and age-matched NR controls (open circles). Ten days of dark exposure initiated at P90 (B), but not P35 (A), reduced the maximal IPSC evoked in response to layer IV stimulation. *Two-way ANOVA, F(1,145) = 12.59, p = 0.0013. The numbers in parentheses indicate the number of rats, number of slices.
A refractory period for the reactivation of ocular dominance plasticity
The maturation of perisomatic inhibition is widely believed to constrain Hebbian synaptic plasticity at excitatory synapses in the visual cortex and consequently reduce rapid ocular dominance plasticity (Kirkwood et al., 1995; Huang et al., 1999; Rozas et al., 2001; Di Cristo et al., 2007). Dark exposure initiated in adulthood induces a reactivation of juvenile-like ocular dominance plasticity (He et al., 2006, 2007). We therefore used VEPs in response to high contrast gratings (0.04 cycles/degree reversing at 1 Hz) to ask whether there is a refractory period for the reactivation of ocular dominance plasticity by dark exposure. The rodent visual system has a contralateral bias, resulting in twofold larger VEP amplitudes in response to stimulation of the contralateral eye relative to the ipsilateral eye (VEP average ± SEM: 2.33 ± 0.22; n = 4) (Fig. 3A, gray bar). Brief monocular deprivation induces a significant shift in ocular dominance if initiated before P65 (VEP amplitude C/I average ± SEM: P38, 0.96 ± 0.04; P45, 1.03 ± 0.05; P55, 0.93 ± 0.18; P65, 1.94 ± 0.05; P100, 2.33 ± 0.22) (Fig. 3A). The decrease in the VEP contralateral bias was mediated by a decrease in the deprived-eye VEP and an increase in the nondeprived-eye VEP (shown for P45 subjects in supplemental Fig. 3C,D, available at www.jneurosci.org as supplemental material). However, when brief monocular deprivation is preceded by 10 d of DE, a significant shift in ocular dominance was observed in all ages up to P100 (DE P38, 0.74 ± 0.06; DE P45, 1.00 ± 0.04; DE P55, 1.15 ± 0.07; DE P65, 0.89 ± 0.08; DE P100, 0.86 ± 0.08) (Fig. 3A). To separate the effects of dark exposure from baseline ocular dominance plasticity, we normalized the VEP amplitudes from dark-exposed subjects to age-matched normal-reared controls. A U-shape is observed in the relationship between the age at initiation of dark exposure and the deprived-eye depression and the nondeprived-eye potentiation induced by monocular deprivation (Fig. 3B). Thus, dark exposure enhances ocular dominance plasticity but is only effective if initiated before or after a refractory period.
Figure 3.
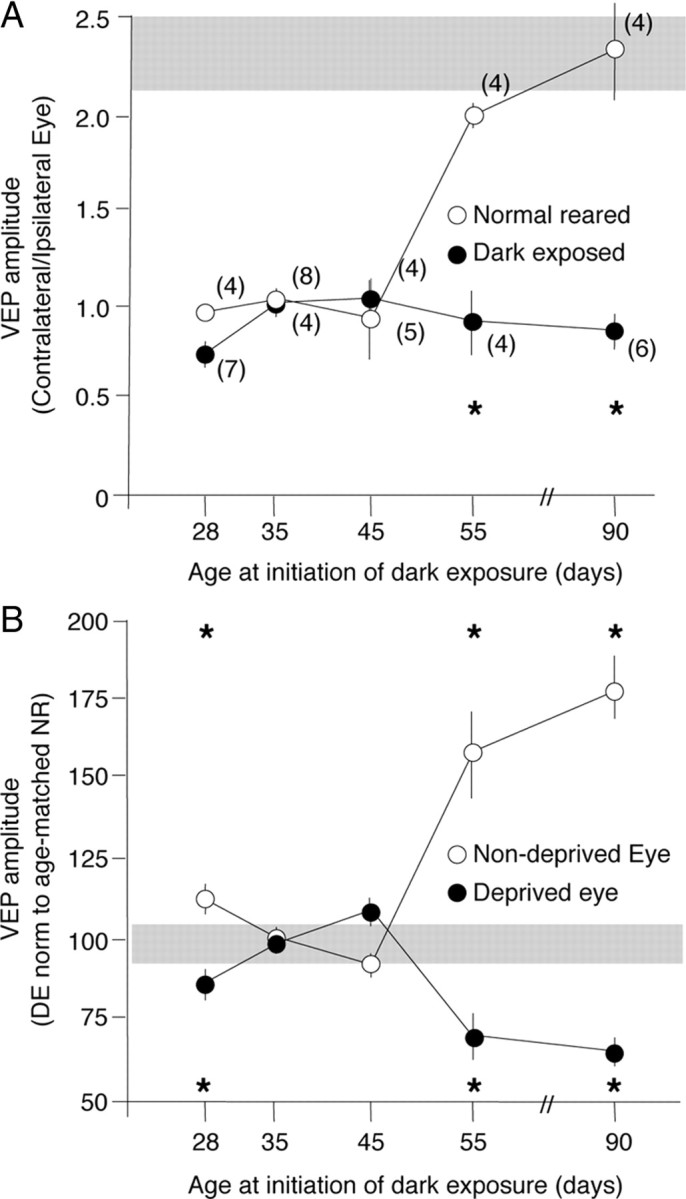
Refractory period for the reactivation of ocular dominance plasticity by 10 d of dark exposure. A, Contralateral bias (VEP amplitude C/I) after monocular deprivation as a function of the age at initiation of 10 d of dark exposure (filled circles) compared with age-matched normal reared controls (open circles). VEPs were recorded from the surface of the binocular region of the right visual cortex after 3 d of left eye deprivation; two-way ANOVA, F(4,40) = 25.22, p < 0.0001; *p < 0.05 versus age-matched NR in Tukey's HSD post hoc. B, Normalization of DE VEP amplitudes to age-matched, normal-reared controls. The refractory period is seen in the response of the deprived eye (filled circles) and nondeprived eye (open circles) to monocular deprivation. Two-way ANOVA, F(4,40) = 55.4, p < 0.0001; *p < 0.05 versus age-matched NR in Tukey's HSD post hoc. The gray boxes indicate normal range of VEP C/I.
Endocannabinoid-mediated regulation of inhibitory synapse maturation and ocular dominance plasticity
We hypothesized that the rejuvenation GABAergic circuits was necessary for the reactivation of ocular dominance plasticity in adults. Therefore, we used two complementary methods to ask whether enhancement of GABAergic inhibition would reduce the reactivation of ocular dominance plasticity by dark exposure at P90. In the juvenile visual system, activation of CB1 receptors with the agonist WIN accelerates the maturation of GABAergic inhibition (Jiang et al., 2010). To ask whether the rejuvenation of GABAergic inhibition by dark exposure at P90 is reversed by CB1R activation, we administered WIN during the last 3 d of a 10 d period of dark exposure (two times per day for 3 d; 5 mg/kg, i.p.). Administration of WIN resulted in a loss of iLTD (WIN: 95.5 ± 7.0% of baseline; vehicle: 75.6 ± 5.11%) (Fig. 4A) and an increase in the maximal IPSC amplitude in P90 DE subjects (WIN: 3.08 ± 0.20 nA; vehicle: 2.40 ± 0.22 nA) (Fig. 4B), suggesting that WIN stimulated the maturation of GABAergic inhibition. Similarly, the reactivation of ocular dominance plasticity by dark exposure at P90 was significantly reduced by systemic injection of WIN (two times per day for 3 d; 5 mg/kg, i.p.). Importantly, enhancement of GABAAR currents with diazepam (one time per day, 15 mg/kg, i.p.) for the last 3 d of dark exposure completely blocked the reactivation of ocular dominance plasticity at P100 (layer IV VEP amplitude C/I: P90 no MD, 2.06 ± 0.26; P90 MD, 2.41 ± 0.20; P90 DE plus Veh, 1.11 ± 0.05; P90 DE plus WIN, 1.67 ± 0.13, p = 0.0072 vs MD plus DE plus Veh; P90 DE plus DZ, 2.04 ± 0.04, p < 0.0001 vs MD plus DE plus Veh in Tukey's HSD post hoc) (Fig. 5A). WIN significantly reduced and diazepam completely eliminated the decrease in deprived-eye VEP and the increase in nondeprived-eye VEP induced by monocular deprivation in P90 DE subjects, and these changes were equally reflected in VEPs recorded from layer IV and the surface of the binocular region of the primary visual cortex (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material).
Figure 4.

Systemic administration of the CB1R agonist WIN (open circles) but not vehicle (filled circles) reverses the effect of adult dark exposure. IPSC amplitudes in layer II/III pyramidal neurons after TBS (arrow) delivered to layer IV. A, WIN reversed the iLTD typically observed in dark-exposed P90 subjects; *p = 0.031, one-tail t test 30 min after TBS. B, WIN reversed the decrease in IPSC amplitude typically observed in dark-exposed, P90 subjects; two-way ANOVA, *p = 0.028.
Figure 5.

Reversal of the effects of dark exposure on ocular dominance plasticity in adults. A, Inset, P90 experimental design. The ocular dominance plasticity reactivated by dark exposure at P90 is attenuated by diazepam and WIN. One-way ANOVA, F(4,24) = 12.212, p < 0.0001; *p < 0.05 versus age-matched NR in Tukey's HSD post hoc. B, Inset, P45 experimental design. The ocular dominance plasticity present in P45 NR subjects is inhibited by diazepam, but unaffected by dark exposure or WIN. One-way ANOVA, F(4,21) = 30.42, p < 0.0001; *p < 0.05 versus age-matched NR in Tukey's HSD post hoc. VEPs were recorded from layer IV of the binocular region of the right visual cortex after 3 d of left eye deprivation. Error bars indicate SEM.
This suggests that an immature state of inhibitory synaptic transmission, induced by dark exposure in adults, permits the reactivation of ocular dominance plasticity. Curiously, however, we observed significant ocular dominance plasticity at P55, despite the evidence that perisomatic inhibition is mature at this age (Kirkwood et al., 1995; Hensch et al., 1998; Huang et al., 1999; Rozas et al., 2001; Di Cristo et al., 2007; Jiang et al., 2010). To ask whether the persistence of ocular dominance plasticity was due to submaximal inhibition, GABAAR currents were enhanced with diazepam. Systemic diazepam (one time per day for 3 d; 15 mg/kg, i.p.; initiated at P53) inhibited ocular dominance plasticity in P55 subjects. As expected, neither dark exposure or WIN administration (two times per day for 3 d; 5 mg/kg, i.p.; during the last 3 d of dark exposure) regulated ocular dominance plasticity at this age (layer IV VEP amplitude C/I P45 no MD, 1.96 ± 0.18; P45 MD, 1.00 ± 0.05; P45 DE plus Veh, 1.12 ± 0.11; P45 DE plus WIN, 0.94 ± 0.14; P45 DZ, 2.03 ± 0.03) (Fig. 5B). Diazepam, but not dark exposure or WIN, inhibited the decrease in deprived-eye VEP and the increase in nondeprived-eye VEP induced by monocular deprivation in P45 subjects, which was equally reflected in VEPs recorded from layer IV and the surface of the binocular region of the primary visual cortex (supplemental Fig. 3C,D, available at www.jneurosci.org as supplemental material). This suggests that, at P55, inhibitory circuitry has developed both functionally and anatomically but is not maximally recruited by visual experience.
Discussion
Dark exposure initiated in adulthood reactivates robust ocular dominance plasticity in the mammalian visual cortex. Here, we show that a critical step in the reactivation of ocular dominance plasticity in adults is the rejuvenation of inhibitory synaptic transmission, resulting in a decrease in functional inhibitory synaptic density, a decrease in paired-pulse depression, and a reexpression of endocannabinoid-dependent iLTD. Pharmacological acceleration of the maturation of inhibition, through activation of CB1Rs, reverses the reexpression of iLTD and the reactivation of ocular dominance plasticity in dark-exposed adults. However, dark exposure initiated earlier in postnatal development does not rejuvenate inhibitory synaptic transmission or facilitate rapid ocular dominance plasticity, demonstrating the presence of a refractory period for the regulation of synaptic plasticity by visual deprivation. The refractory period for the rejuvenation of inhibitory synaptic transmission demonstrates the existence of constraints on the regulation of synaptic function by visual deprivation. In addition, it suggests that the efficacy of therapeutic interventions used to promote ocular dominance plasticity may increase with age.
An important outstanding issue is the location of the synapses that underlie the ocular dominance plasticity observed in dark-exposed adults. Several lines of evidence implicate a role of layer II/III circuits in the reactivation of ocular dominance plasticity. The activation of CB1Rs, which has been implicated in the plasticity of neuronal circuits in cortical layer III, but not layer IV (Crozier et al., 2007; Liu et al., 2008; Li et al., 2009; Yoon et al., 2009), significantly reduces the reactivation of ocular dominance plasticity by dark exposure in adults. Similarly, pyramidal neurons in layer III, which receive substantial excitation from thalamic afferents, exhibit a rejuvenation of inhibitory synaptic transmission after dark exposure in adulthood. Importantly, excitatory synapses onto layer II/III pyramidal neurons retain the capacity to express Hebbian plasticity in adulthood (Jiang et al., 2007), but induction is constrained by GABAergic inhibition (Kirkwood and Bear, 1995; Jang et al., 2009). Together, this suggests that the reduction of inhibition onto layer II/III pyramidal contributes to the reactivation of ocular dominance plasticity by dark exposure.
The rejuvenation of inhibitory synaptic transmission
The experience-dependent maturation of inhibition was previously thought to be irreversible, as visual deprivation during postnatal development retards rather than reverses the developmental progression at inhibitory synapses (Morales et al., 2002; Chattopadhyaya et al., 2004; Di Cristo et al., 2007; Jiang et al., 2007). However, here we show that dark exposure in adulthood rejuvenates inhibitory circuits. Immature inhibition, characterized by sparse synapses with a high density of presynaptic CB1Rs and a high probability of GABA release, is seen in the visual cortex of juveniles and dark-exposed adults. Postnatal visual experience stimulates the maturation of inhibition through an endocannabinoid-dependent long-term depression of GABA release. In contrast, mature inhibition is characterized by dense synapses that no longer express CB1Rs and have a low probability of GABA release (Jiang et al., 2010).
The mechanism by which dark exposure induces the rejuvenation of inhibitory synaptic transmission and the reactivation of ocular dominance plasticity in adults is not yet known. In response to the reduction in synaptic activity induced by dark exposure, preexisting inhibitory synapses in the adult visual cortex may revert to a more immature phenotype, accompanied by an increase in CB1R expression and an increase in presynaptic release probability. Alternatively, dark exposure may increase the turnover of GABAergic terminals by promoting the degradation of factors that restrict axonal outgrowth, such as myelin-derived signaling molecules (McGee et al., 2005) or chondroitin sulfate proteoglycans (Pizzorusso et al., 2002). Interestingly, a significant increase in the condensation of extracellular matrix components into perineuronal nets occurs between P45 and P65 (Pizzorusso et al., 2002), which may impart increased resistance to experience-dependent degradation of a restrictive extracellular environment and contribute to the refractory period.
The role of inhibition in the developmental constraint of ocular dominance plasticity
The late maturation of inhibitory circuitry relative to excitatory circuitry creates a window in postnatal development that is permissive for experience-dependent synaptic plasticity in the visual system. Perisomatic inhibition mediated by fast spiking interneurons exerts a powerful influence on the excitability and plasticity of postsynaptic targets. Although a threshold level of inhibition is necessary to initiate the critical period for ocular dominance plasticity early in postnatal development (Hensch et al., 1998; Huang et al., 1999), a subsequent increase in the strength of perisomatic inhibition has been implicated in the constraint of ocular dominance plasticity later in development (Kirkwood et al., 1995; Rozas et al., 2001; Di Cristo et al., 2007). However, here we show that ocular dominance plasticity is robust after postnatal day 35, the time point at which perisomatic inhibition reaches maturity in the rodent cortex (Huang et al., 1999; Rozas et al., 2001; Morales et al., 2002; Chattopadhyaya et al., 2004). A similar persistence of rapid ocular dominance plasticity beyond the expected age of the maturation of inhibition has been observed in cats and mice (Mower and Guo, 2001; Pham et al., 2004; Lehmann and Löwel, 2008). Interestingly, ocular dominance plasticity at P45 could be inhibited by enhancing GABAA receptor function with diazepam, but not by accelerating the maturation of inhibitory synaptic transmission with the CB1R agonist WIN. This suggests that, at P45, inhibitory synapses are present and mature, but ocular dominance plasticity persists because activity at inhibitory synapses is not maximally recruited by visual experience at this age.
Footnotes
This work was supported by National Institutes of Health Grants R01 EY016431 (E.M.Q.) and R01 EY012124 (A.K.).
References
- Chattopadhyaya B, Di Cristo G, Higashiyama H, Knott GW, Kuhlman SJ, Welker E, Huang ZJ. Experience and activity-dependent maturation of perisomatic GABAergic innervation in primary visual cortex during a postnatal critical period. J Neurosci. 2004;24:9598–9611. doi: 10.1523/JNEUROSCI.1851-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyaya B, Di Cristo G, Wu CZ, Knott G, Kuhlman S, Fu Y, Palmiter RD, Huang ZJ. GAD67-mediated GABA synthesis and signaling regulate inhibitory synaptic innervation in the visual cortex. Neuron. 2007;54:889–903. doi: 10.1016/j.neuron.2007.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Choi SY, Morales B, Lee HK, Kirkwood A. Absence of long-term depression in the visual cortex of glutamic acid decarboxylase-65 knock-out mice. J Neurosci. 2002;22:5271–5276. doi: 10.1523/JNEUROSCI.22-13-05271.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozier RA, Wang Y, Liu CH, Bear MF. Deprivation-induced synaptic depression by distinct mechanisms in different layers of mouse visual cortex. Proc Natl Acad Sci U S A. 2007;104:1383–1388. doi: 10.1073/pnas.0609596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynader M. Prolonged sensitivity to monocular deprivation in dark-reared cats: effects of age and visual exposure. Brain Res. 1983;284:155–164. doi: 10.1016/0165-3806(83)90002-0. [DOI] [PubMed] [Google Scholar]
- Di Cristo G, Chattopadhyaya B, Kuhlman SJ, Fu Y, Bélanger MC, Wu CZ, Rutishauser U, Maffei L, Huang ZJ. Activity-dependent PSA expression regulates inhibitory maturation and onset of critical period plasticity. Nat Neurosci. 2007;10:1569–1577. doi: 10.1038/nn2008. [DOI] [PubMed] [Google Scholar]
- Edagawa Y, Saito H, Abe K. The serotonin 5-HT2 receptor-phospholipase C system inhibits the induction of long-term potentiation in the rat visual cortex. Eur J Neurosci. 2000;12:1391–1396. doi: 10.1046/j.1460-9568.2000.00007.x. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Pizzorusso T, Berardi N, Domenici L, Maffei L. Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 1994;34:709–720. doi: 10.1016/0042-6989(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Goldberg EM, Watanabe S, Chang SY, Joho RH, Huang ZJ, Leonard CS, Rudy B. Specific functions of synaptically localized potassium channels in synaptic transmission at the neocortical GABAergic fast-spiking cell synapse. J Neurosci. 2005;25:5230–5235. doi: 10.1523/JNEUROSCI.0722-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Lickey ME, Gordon B. Critical period for the monocular deprivation effect in rats: assessment with sweep visually evoked potentials. J Neurophysiol. 1999;81:121–128. doi: 10.1152/jn.1999.81.1.121. [DOI] [PubMed] [Google Scholar]
- Harauzov A, Spolidoro M, DiCristo G, De Pasquale R, Cancedda L, Pizzorusso T, Viegi A, Berardi N, Maffei L. Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J Neurosci. 2010;30:361–371. doi: 10.1523/JNEUROSCI.2233-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HY, Hodos W, Quinlan EM. Visual deprivation reactivates rapid ocular dominance plasticity in adult visual cortex. J Neurosci. 2006;26:2951–2955. doi: 10.1523/JNEUROSCI.5554-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HY, Ray B, Dennis K, Quinlan EM. Experience-dependent recovery of vision following chronic deprivation amblyopia. Nat Neurosci. 2007;10:1134–1136. doi: 10.1038/nn1965. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZJ, Kirkwood A, Pizzorusso T, Porciatti V, Morales B, Bear MF, Maffei L, Tonegawa S. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Jang HJ, Cho KH, Kim HS, Hahn SJ, Kim MS, Rhie DJ. Age-dependent decline in supragranular long-term synaptic plasticity by increased inhibition during the critical period in the rat primary visual cortex. J Neurophysiol. 2009;101:269–275. doi: 10.1152/jn.90900.2008. [DOI] [PubMed] [Google Scholar]
- Jiang B, Huang ZJ, Morales B, Kirkwood A. Maturation of GABAergic transmission and the timing of plasticity in visual cortex. Brain Res Brain Res Rev. 2005;50:126–133. doi: 10.1016/j.brainresrev.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Jiang B, Treviño M, Kirkwood A. Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J Neurosci. 2007;27:9648–9652. doi: 10.1523/JNEUROSCI.2655-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Huang S, de Pasquale R, Millman D, Song L, Lee HK, Tsumoto T, Kirkwood A. The maturation of GABAergic transmission in visual cortex requires endocannabinoid-mediated LTD of inhibitory inputs during a critical period. Neuron. 2010;66:248–259. doi: 10.1016/j.neuron.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Homosynaptic long-term depression in the visual cortex. J Neurosci. 1994;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. (1995) Elementary forms of synaptic plasticity in the visual cortex. Biol Res. 1995;28:73–80. [PubMed] [Google Scholar]
- Kreczko A, Goel A, Song L, Lee HK. Visual deprivation decreases somatic GAD65 puncta number on layer 2/3 pyramidal neurons in mouse visual cortex. Neural Plast. 2009;2009:415135. doi: 10.1155/2009/415135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K, Löwel S. Age-dependent ocular dominance plasticity in adult mice. PLoS One. 2008;3:e3120. doi: 10.1371/journal.pone.0003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bender KJ, Drew PJ, Jadhav SP, Sylwestrak E, Feldman DE. Endocannabinoid signaling is required for development and critical period plasticity of the whisker map in somatosensory cortex. Neuron. 2009;64:537–549. doi: 10.1016/j.neuron.2009.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CH, Heynen AJ, Shuler MG, Bear MF. Cannabinoid receptor blockade reveals parallel plasticity mechanisms in different layers of mouse visual cortex. Neuron. 2008;58:340–345. doi: 10.1016/j.neuron.2008.02.020. [DOI] [PubMed] [Google Scholar]
- Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O'Leary OF, Castrén E, Maffei L. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320:385–388. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Kauer JA. Presynaptic plasticity: targeted control of inhibitory networks. Curr Opin Neurobiol. 2009;19:254–262. doi: 10.1016/j.conb.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales B, Choi SY, Kirkwood A. Dark rearing alters the development of GABAergic transmission in visual cortex. J Neurosci. 2002;22:8084–8090. doi: 10.1523/JNEUROSCI.22-18-08084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower GD, Christen WG. Role of visual experience in activating critical period in cat visual cortex. J Neurophysiol. 1985;53:572–589. doi: 10.1152/jn.1985.53.2.572. [DOI] [PubMed] [Google Scholar]
- Mower GD, Guo Y. Comparison of the expression of two forms of glutamic acid decarboxylase (GAD67 and GAD65) in the visual cortex of normal and dark-reared cats. Brain Res Dev Brain Res. 2001;126:65–74. doi: 10.1016/s0165-3806(00)00139-5. [DOI] [PubMed] [Google Scholar]
- Pham TA, Graham SJ, Suzuki S, Barco A, Kandel ER, Gordon B, Lickey ME. A semi-persistent adult ocular dominance plasticity in visual cortex is stabilized by activated CREB. Learn Mem. 2004;11:738–747. doi: 10.1101/lm.75304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- Rozas C, Frank H, Heynen AJ, Morales B, Bear MF, Kirkwood A. Developmental inhibitory gate controls the relay of activity to the superficial layers of the visual cortex. J Neurosci. 2001;21:6791–6801. doi: 10.1523/JNEUROSCI.21-17-06791.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale A, Maya Vetencourt JF, Medini P, Cenni MC, Baroncelli L, De Pasquale R, Maffei L. Environmental enrichment in adulthood promotes amblyopia recovery through a reduction of intracortical inhibition. Nat Neurosci. 2007;10:679–681. doi: 10.1038/nn1899. [DOI] [PubMed] [Google Scholar]
- Yoon BJ, Smith GB, Heynen AJ, Neve RL, Bear MF. Essential role for a long-term depression mechanism in ocular dominance plasticity. Proc Natl Acad Sci U S A. 2009;106:9860–9865. doi: 10.1073/pnas.0901305106. [DOI] [PMC free article] [PubMed] [Google Scholar]