

Abstract
Activation of EphB receptors by efnB ligands on neuronal cell surface regulates important functions including neurite outgrowth, axonal guidance and synaptic plasticity. Here we show that efnB rescues primary cortical neuronal cultures from necrotic cell death induced by glutamate excitotoxicity and that this function depends on EphB receptors. Importantly, the neuroprotective function of the efnB/EphB system depends on PS1, a protein that plays crucial roles in Alzheimer's disease (AD) neurodegeneration. Furthermore, absence of one PS1 allele results in significantly decreased neuroprotection indicating that both PS1 alleles are necessary for full expression of the neuroprotective activity of the efnB/EphB system. We also show that the ability of brain-derived neurotrophic factor (BDNF) to protect neuronal cultures from glutamate-induced cell death depends on PS1. Neuroprotective functions of both efnB and BDNF however, were independent of γ-secretase activity. Absence of PS1 decreases cell surface expression of neuronal TrkB and EphB2 without affecting total cellular levels of the receptors. Furthermore, PS1 knockout neurons show defective ligand-dependent internalization and decreased ligand-induced degradation of TrkB and Eph receptors. Our data show that PS1 mediates the neuroprotective activities of efnB and BDNF against excitotoxicity and regulates surface expression and ligand-induced metabolism of their cognate receptors. Together, our observations indicate that PS1 promotes neuronal survival by regulating neuroprotective functions of ligand-receptor systems.
Keywords: Presenilin, EphrinB, BDNF, EphB, TrkB, neuroprotection, neurodegeneration, Glutamate excitotoxicity
1. Introduction
Neuronal exposure to toxic insults, including glutamate excitotoxicity and oxidative stress, has been proposed to play crucial roles in neurodegenerative disorders of the CNS including Alzheimer's disease (AD). Although glutamate receptors play key roles in neuronal functions, excessive activation of these receptors results in calcium overload and increased oxidative stress that may result in neuronal cell death (Choi, 1996; Greenamyre and Young, 1989; Mattson, 1997). Neurodegenerative disorders, including AD, are characterized by progressive loss of specific neuronal populations, but the mechanisms underlying this selective vulnerability are not fully understood. Aging, abnormal metabolism of the amyloid precursor protein (APP) and genetic mutations however, may sensitize neuronal populations to excitotoxicity thus promoting neurodegeneration and ultimately dementia (Mattson, 2003). Missense mutations of PS1, a protein important to the production of Aβ peptides and shown to have both γ-secretase-related and -unrelated functions, promote neurodegeneration and familial AD (FAD) in an autosomal dominant inheritance pattern suggesting they may cause gain of neurotoxic functions or dominant loss of essential functions (Robakis, 2011). Additional reports indicate that PS1 regulates cell survival signaling (Baki, et al., 2004), receptor trafficking (Leem, et al., 2002), glutamate release and calcium homeostasis (Tu, et al., 2006; Zhang, et al., 2009) and that PS1 mutations increase neuronal vulnerability to hypoxia and excitotoxicity (Mattson, et al., 2000).
Recent data shows that PS1 functionally interacts with the EphB2 tyrosine kinase receptor (Litterst, et al., 2007), a member of the EphB family of receptors shown to play pivotal roles in development and cell function (Martinez and Soriano, 2005; Pasquale, 2008). Neuronal EphB receptors are activated by binding to their cognate ephrinB (efnB) ligands, including efnB1 and efnB2, at the surface of adjacent neurons a process followed by internalization and degradation of EphB receptors (Litterst, et al., 2007; Pitulescu and Adams, 2010). Ligand-activated EphB2 interacts with extracellular sequence of the NMDA receptor (NMDAR) and recent reports indicate this interaction may be involved in neurodegeneration caused by Aβ oligomers (Cisse, et al., 2011). Furthermore, the efnB-EphB binding regulate neuronal physiology and memory-related functions including neurite outgrowth and synaptic plasticity (Dalva, et al., 2000; Pasquale, 2008). At the molecular level, the efnB-EphB interaction stimulates autophosphorylation of the tyrosine kinase domain of EphB2 (Egea and Klein, 2007; Pasquale, 2008), factor binding to the cytosolic sequence of this receptor as well as endocytosis and degradation of the efnB-EphB complexes (Pitulescu and Adams, 2010). In addition, we showed that the PS1/γ-secretase system participates in the downstream processing of endocytosed EphB2 receptor following ligand stimulation (Litterst, et al., 2007). Here we report that the efnB-EphB system protects cortical neurons from glutamate excitotoxicity and necrotic death and that this function requires the presence of PS1. We also show that PS1 is required for the neuroprotective activity of other factors including brain derived neurotrophic factor (BDNF) and that PS1 regulates the cell surface expression and ligand-induced degradation of EphB2 and TrkB receptors. Surprisingly, however, the neuroprotective function of PS1 is independent of γ-secretase activity. Our data suggest that PS1 promotes neuronal survival by mediating ligand-dependent neuroprotection.
2. Materials and Methods
2.1. Materials and antibodies
Polyclonal anti-EphB2 antibody and monoclonal anti-EphB2 antibodies were obtained from Zymed. Anti-TrkB (C-14) was from Santa-Cruz; anti-phospho-TrkB (Tyr516) rabbit mAb was from Cell Signaling, anti-phospho-tyrosine (clone 4G10) was Upstate Biotechnology. Antibodies against Spectrin antibodies were from Millipore and anti-cleaved caspase 3 antibody was from Cell Signaling. Necrostatin-1 was from EMD Chemicals; EfnB1-fc and BDNF were obtained from R&D systems (Minneapolis) and Sigma respectively; Progranulin (PRGN) was prepared as described (Xu, et al., 2011); ZVLL and GSI were from Calbiochem and Z-VAD from R & D Systems. Clustering of recombinant mouse ephrin-B1/Fc ligands and anti-Fc were performed as described and used at 2µg/ml for stimulation (Litterst, et al., 2007).
2.2. Primary neuronal cultures
All animal experiments were carried out in accordance with the rules and regulations at the Mount Sinai School of Medicine. Rat brain cortical neuronal cultures were prepared as described recently (Xu, et al., 2011). Briefly, neurons were plated at a density of 1×105 neurons per cm2 on poly–D-lysine-coated plates in Neurobasal medium supplemented with 2% B27 (Invitrogen, Eugene, OR, USA), L-glutamine (0.5 mM) and penicillin/streptomycin (1% vol/vol) and kept for at least 8 days in vitro (DIV) before use as indicated in Figure legends. Under these conditions post-mitotic neurons represent more than 98% of cultured cells (Xu, et al., 2011). Neurons from mouse brains of embryonic day 15.5 (E15.5) prepared from wild type (WT), transgenic PS1 knock outs (KOs) heterozygous (PS1+/−) or homozygous (PS1−/−) for PS1 were prepared and genotyped for PS1 as described (Baki et al., 2008). Transgenic KOs mice heterozygous for EphB2 (EphB2+/−) and homozygous KOs for EphB1 and EphB3 (EphB2+/−; EphB1−/−; EphB3−/−) as well as triple KOs for EphB1, EphB2 and EphB3 receptors (EphB2−/−; EphB1−/−; EphB3−/−) were obtained from Dr. Henkemeyer (Henkemeyer, et al., 2003). Dissociated brain cells were plated onto poly–D-lysine-coated plates at a density of approximately 60,000/cm2. Cells were maintained in Neurobasal medium supplemented as above and used at 8 days in vitro (DIV). Under these conditions post-mitotic neurons represent more than 98% of cultured cells (Baki, et al., 2008).
2.3. Cell survival assays
Three independent assays were used to measure neuroprotection against glutamate toxicity as described (Xu, et al., 2011). Briefly, mature cortical neuronal cultures prepared and kept in Neurobasal medium supplemented as above, were changed to HBSS medium (Invitrogen) for 30 min and then clustered efnB1-Fc or BDNF was added for another 30 min. Glutamate was then added for 3hrs before measurements. For nuclei staining, cells were fixed in 4% paraformaldehyde for 20 min at room temperature and stained with Hoechst 33342 for 10 min. Stained nuclei were then observed under a fluorescence microscope on ultraviolet illumination and counted according to manufacturer’s instructions (Sigma). Numbers of viable neurons were determined in ten fields per well and in each experiment four identical wells were evaluated per condition and per plate. The average number of four wells was used as one experiment per condition in statistical analysis. Mouse cortical neuronal cultures 8 DIV were used as described in figures. Results are expressed as percent of control value. MTT cell viability assay (Denizot, Lang, 1986) was performed according to manufacturer’s instructions (Sigma). In summary, neurons as above cultured on poly-D-lysine-coated 96-well plates were treated with glutamate for 3 hrs and then MTT solution (1 mg/ml) was added to each well. Plates were incubated at 37 °C for 2 hrs and the reaction was terminated by 0.1N HCl in isopropanol for 1 hr. Absorbance was then measured at 560 nm with background subtraction at 620 nm. Data are expressed as a percent of control value. Lactate dehydrogenase (LDH) release assays (Koh and Choi, 1987), were performed using the Cytotoxicity Detection Kit Plus (Roche) according to manufacturer’s instruction. Results are expressed as the percentage of LDH release by non-treated cells.
2.4. Biotinylation of cell surface protein
Neuronal culture were incubated with 1mg/ml Sulfo-NHS-SS-biotin in PBS at 4°C for 1h, washed with 0.1M glycine, and lysed in a RIPA lysis-buffer. The cell extracts were incubated with streptavidin-agarose beads overnight (O/N). The bound proteins were washed three times with PBS and eluted with Laemmli buffer.
2.5. Statistical analysis
At least four independent experiments were performed for statistical analysis of the data shown in each figure. Each independent experiment used neurons from a single embryonic brain and each condition was represented by four wells (see above). All data are expressed as mean ± SEM and were normalized to controls (100%). To evaluate statistical significance of treatments, paired t-tests were performed against the value of untreated basal condition (* p < 0.05; ** p < 0.01; ***p < 0.005).
3. Results
3.1. EfnB1 protects neurons from excitotoxic cell death
EphB receptors are activated by efnB ligands and interact with PS1, a protein involved in neurodegeneration (Litterst, et al., 2007). To examine whether the efnB/EphB system affects neuronal survival, we used glutamate-treated mature rat cortical neuronal cultures of 8–13 DIV and efnB1-Fc recombinant constructs. These contain the extracellular domain of mouse efnB1 fused to Fc and are used as ligands able to bind and activate EphB receptors in vitro mimicking the in vivo ligand effects (Davis, et al., 1994; Litterst, et al., 2007). Figure 1A shows that treatment of our cultures with efnB1-Fc rescues neurons from glutamate-induced cell death measured by counting healthy nuclei stained with Hoechst stain (see Methods). Fc alone or N-cadherin-Fc containing the extracellular domain of N-cadherin fused to Fc had no effect on neuronal survival (data not shown). The neuroprotective function of efnB1 against excitotoxicity was manifested in all mature cortical neuronal cultures 8 to 13 DIV. EfnB1-Fc rescues neurons from glutamate toxicity when it is added to cultures before or at the same time as glutamate but it is inactive if added later than 30 minutes following glutamate treatment (Fig. 1B). Dose-response experiments showed that neuroprotection depends on the concentration of efnB1-Fc with a half maximum effect of about 0.5µg/ml (Fig. 1C). This neuroprotective effect is also detected by the ability of cells to reduce 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to formazan (Morgan, 1998; Xu, et al., 2011), an assay of mitochondrial activity (Figure 1D), and by measuring released lactate dehydrogenase, a widely used assay of cytotoxicity (Figure 1E). The efnB1 neuroprotection against glutamate was evident by measuring neuronal processes using microtubule associated protein (MAP2) immunostaining. Glutamate reduces the number of neuritic extensions per cell, an outcome partially reversed by efnB1-Fc as well as by neurotrophin BDNF (Figs. 2A and 2B). This effect of efnB1 in post-mitotic cortical neurons is noteworthy as it suggests that efnB preserves the integrity of neuronal processes and may have neurotrophic functions. Cell staining with transfected green fluorescence protein (GFP) allows morphological identification of surviving neurons (Figure 2C). These experiments showed that the kinetics of neuronal survival of efnB-treated neurons is comparable to the kinetics of BDNF-dependent survival (Figure 2D). Use of efnB2-Fc constructs containing the extracellular sequence of efnB2, another member of the efnB family of ligands that binds EphB receptors and is highly homologous to efnB1, gave results identical to those obtained with efnB1-Fc (data not shown)
Figure 1. EfnB1 protects neurons from excitotoxic insults.
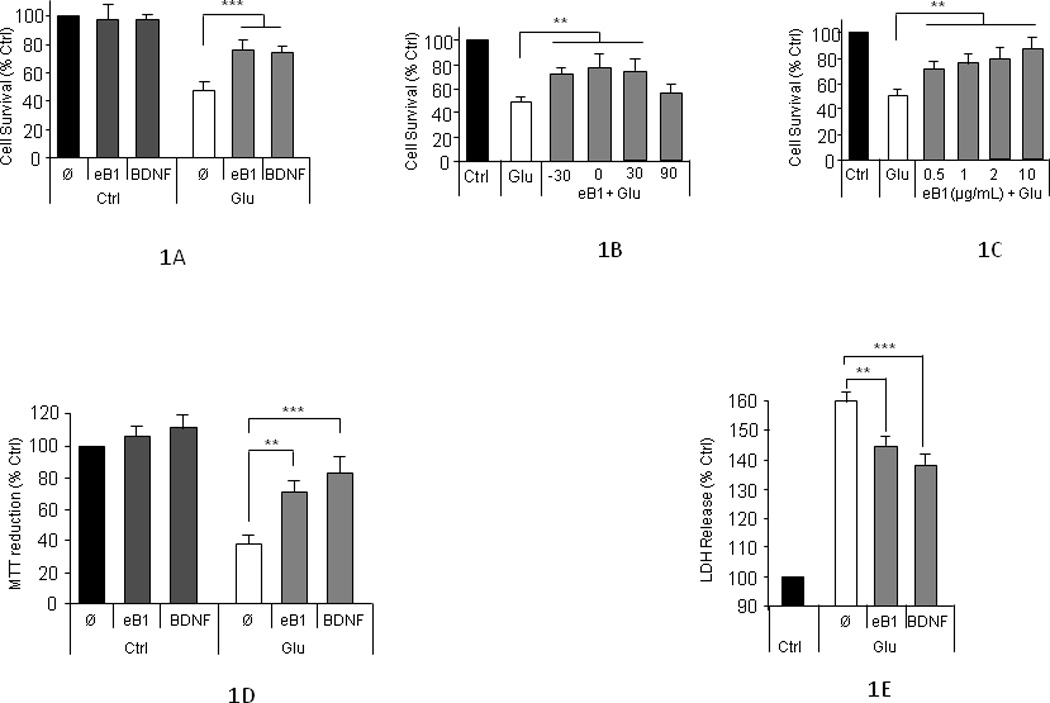
(A) Neuroprotection measured by counting cell nuclei: Viability of mature (8–13 DIV) rat cortical neuronal cultures was quantified by counting healthy nuclei stained with Hoechst kit 33342 (see Methods). Results are shown as percentage of untreated cultures (mean ± sem). Neuronal death is induced by treatment with 50µM glutamate for 3 hours. Pretreatment with efnB1-Fc (2µg/mL) for 30 minutes before glutamate addition is neuroprotective. Pretreatment with BDNF (50ng/mL) is used as positive control (paired t-test, n=6; p<0.005). Ø: no neuroprotective factors. (B) Kinetics of efnB1 neuroprotection: Rat cortical neuronal cultures as above were treated with 50µM glutamate (Glu) for 3 hours in the presence or absence of efnB1 (eB1) added 30 minutes before glutamate (−30), the same time as glutamate (0) or either 30 minutes (30) or 90 (90) minutes after glutamate. Neuronal viability was measured as in Fig. 1A. EfnB1 is unable to rescue neurons if added to the culture later than 30 minutes after glutamate addition. (n=5; p<0.01). (C) Dose-response effects of efnB1 neuroprotection: Primary neuronal cultures were treated with glutamate (Glu) and increasing concentrations of efnB1-Fc (eB1) indicated in figure. Cell survival was determined as above (n=3, p<0.05). (D) Neuroprotection by MTT metabolism: Rat cortical neuronal cultures were treated with efnB1-Fc, BDNF and/or glutamate as described above. Cell survival was measured using the MTT as described in Methods (n=5; **, p<0.01; ***, p<0.005). Ø: no neuroprotective factors. (E) Neuroprotection by LDH. Assay was performed using the Cytotoxicity Detection Kit Plus according to manufacturer’s instructions (LDH; Roche Applied Science)]. Treatment with efnB1-Fc, BDNF and glutamate were as above. (N=4; **, p<0.01; ***, p<0.005). Ø: no neuroprotective factors.
Figure 2. Neuroprotective activity of efnB1 by MAP2 staining and time-dependent morphological changes.
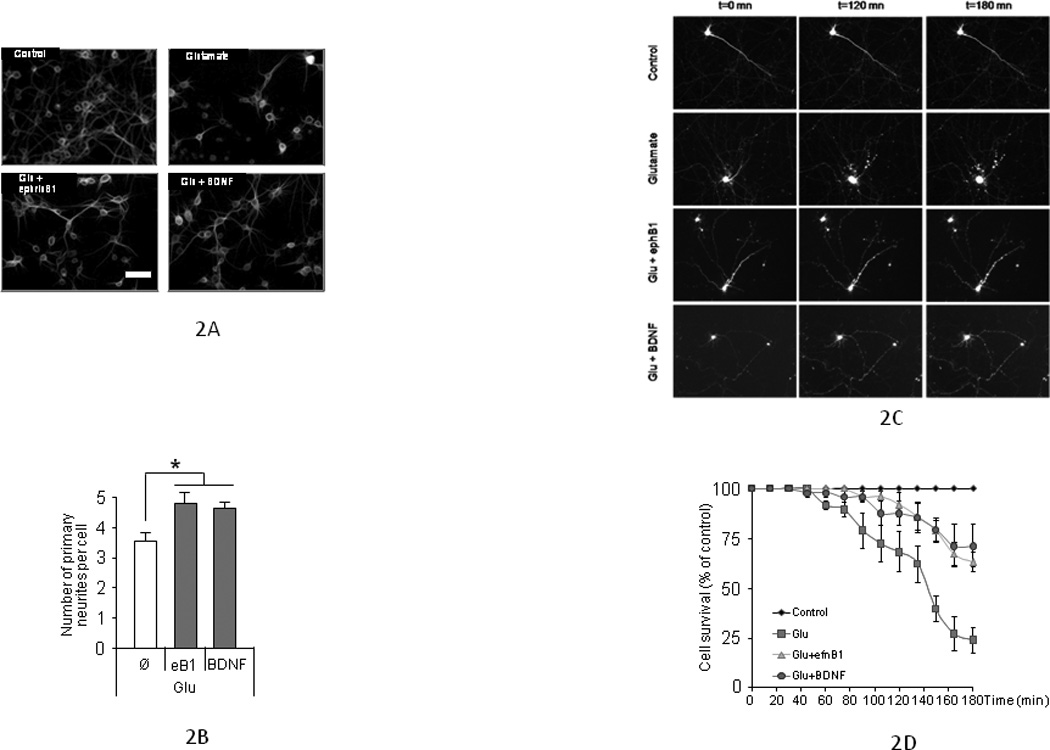
(A) Characteristic images of cortical neurons treated with glutamate in the presence or absence of either efnB1-Fc or BDNF and immunostained with anti-MAP2 antibodies. (B) Quantification of primary neurites of individual neurons from 4 independent cultures is shown. Neurites at least 3 times longer than the size of the soma were counted. Neurons treated with glutamate and either efnB1-Fc or BDNF have more neurites than neurons treated with glutamate alone (n=4, p<0.05). Ø: factor addition. (C) Time-lapse imaging of Green fluorescence protein (GFP) transfected neurons: Time-dependent morphological changes of GFP-transfected rat cortical neurons treated with glutamate (50µM; second row of panels); glutamate plus efnB1-Fc (2µg/mL; third row of panels) or glutamate plus BDNF (50ng/mL; fourth row of panels). Processes of cells treated with glutamate and either efnB1 or BDNF are preserved compared to cells treated with glutamate only. Cells with rounded soma and no extensions (second row, Glutamate alone) are considered not viable. (D) Quantification of neuronal survival by morphological analysis. GFP-transfected neurons were observed following glutamate treatment in the presence or absence of efnB1-Fc or BDNF. Cells were scored as either 1 if soma is intact with at least one extension or 0 if soma is either round with no extensions or not detected anymore. Results are presented as percent of cell survival over time (n=50 independent neurons from N=5 independent experiments). Ctrl: control cultures; Glu: addition of glutamate; eB1: addition of efnB1-Fc; BDNF: addition of BDNF.
3.2. EphB2 receptor mediates the efnB1-dependent neuroprotection
EfnB1 ligands exert their neuronal functions by binding neuronal EphB receptors, including EphB1, EphB2 and EphB3 (Lackmann and Boyd, 2008). To examine whether the neuroprotective effects of efnB are mediated by its cognate receptors, we used cortical neuronal cultures from embryonic brains of triple KO mice lacking the three EphB receptors expressed in neurons (Henkemeyer, et al., 2003). Figure 3A (left group of bars) shows that efnB1-Fc is unable to rescue these neurons from glutamate toxicity indicating the neuroprotective activities of efnB1 ligands are mediated by one or more EphB receptors. Since EphB2 is highly expressed in neurons (Litterst, et al., 2007; Sajjadi and Pasquale, 1993), we tested the neuroprotective activity of efnB1-Fc in neurons expressing EphB2 but lacking EphB1 and EphB3 (Henkemeyer, et al., 2003). Figure 3A (right group of bars) shows that efnB1-Fc rescues these neurons from glutamate toxicity indicating EphB2 mediates at least part of the neuroprotective functions of efnB ligands. Under our conditions, the efficacy of the efnB1 neuroprotection against glutamate toxicity was comparable to the rescuing activity of BDNF (Figures 1A,D,E and Fig 2). To further explore potential relations between the efnB1-dependent and BDNF-dependent neuprotection, we used TrkB receptor antagonist K525a which inhibits functions of the BDNF/TrkB system. Figure 3B shows that although this agent blocks the BDNF-dependent neuroprotection, it has no effect on the efnB1-Fc neuroprotection (right group of bars). Together, these data show that the neuroprotective activities of efnB ligands are mediated by EphB receptors but are independent of TrkB receptor that mediates BDNF neuroprotection.
Figure 3. EphB2 receptor mediates neuroprotective properties of efnB1.
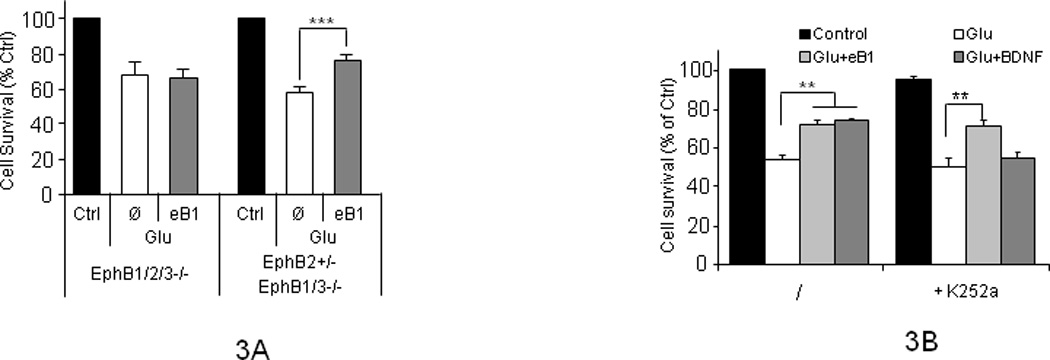
(A) Quantitation of intact neuronal nuclei in cultures treated (eB1) or untreated (Ø) with efnB1-Fc for 30 minutes followed by addition of glutamate for three hours as described in Figure 1. Left group: cortical neuronal cultures of 8 DIV prepared from brains of EphB1, EphB2 and EphB3 triple KO (EphB1−/−; EphB2−/−; EphB3−/−) mouse embryos are not protected from glutamate toxicity by efnB1. Right group: neuronal cultures prepared as above from embryos heterozygous KOs for EphB2 but homozygous KOs for both EphB1 and EphB3 (EphB2+/−; EphB1−/−; EphB3−/−) are protected by efnB1 from glutamate toxicity (n=5; p<0.005). EphB1/2/3 −/−, EphB triple KOs; EphB2+/−/EphB1/3−/−, mice heterozygous KOs for EphB2 and homozygous KOs for EphB1 and EphB3; Ctrl, control cultures. (B) EphrinB1 neuroprotection is independent of TrkB. Cortical neuronal cultures were treated as above with efnB1-Fc, BDNF or glutamate in the absence (left group of bars) or presence (right group of bars) of TrkB antagonist K252a (200 nM). In the presence of K252a, efnB1 but not BDNF rescues cells from death (p<0.01). Results are shown as percent of untreated cells. Ctrl, control cultures; Glu, addition of glutamate; Ø, no addition; eB1, addition of efnB1-Fc; BDNF, addition of BDNF.
3.3. The neuroprotective effect of efnB1 depends on presenilin
To examine whether PS1 plays a role in the neuroprotective properties of efnB1 we used cortical neuronal cultures prepared from PS1 knock-out (KO) mouse embryos (Baki, et al., 2004, Marambaud, et al., 2002). Figure 4A shows that efnB1-Fc rescues wild type (WT) neurons from glutamate toxicity but not neurons prepared from either PS1 heterozygous KO (PS1+/−) or PS1 homozygous KO (PS1−/−) mouse brains. It was unexpected that absence of one allele of PS1 (haploinsufficiency) resulted in severe loss of the efnB1-dependent neuroprotection indicating that both PS1 alleles are needed for full neuroprotective activity of efnB ligands. The effect of PS1 on the efnB neuroprotection was also evident by measuring neurite extensions per neuron using MAP2 immunostaining (Fig. 4B) and by released LDH (data not shown). Together, our data show that the neuroprotective activity of efnB ligands against glutamate excitotoxicity depends on PS1 and that absence of one PS1 allele significantly impairs the neuroprotective activity of the efnB/EphB system.
Figure 4. Neuroprotective function of efnB depends on PS1.
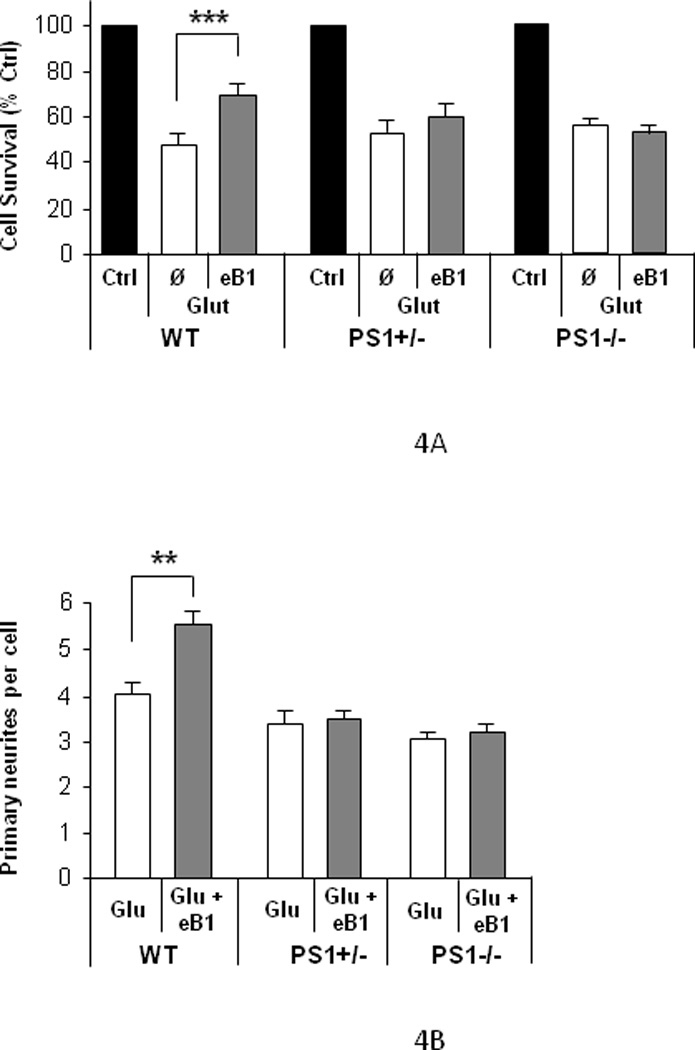
(A) Quantitation of intact nuclei of cortical neuronal cultures of 8 DIV prepared from wild type (WT), PS1 heterozygous (PS1+/−), or PS1 homozygous (PS1−/−) KO mouse embryos treated with glutamate (Glu) for 3 hours in the presence (eB1) or absence (Ø) of efnB1-Fc as described in Figure 1. EfnB1 shows significant neuroprotective effects only in WT neurons (n=7; p<0.005). (B) Quantitation of primary neuritis of individual neurons: efnB1-Fc protects WT, but neither PS1+/− nor PS1−/−, neurons from glutamate-induced fragmentation of neuronal processes (n=4; p<0.01).
3.4. PS1 mediates the neuroprotective effect of BDNF against glutamate-induced cell death
We then asked whether PS1 mediates activities of other factors known to protect cortical neurons from excitotoxicity including BDNF and progranulin (PGRN) a protein involved in the neurodegeneration of Frontotemporal dementia (FTD). Figure 5A shows that PS1 null (PS1−/−) neurons do not respond to BDNF neuroprotection while neurons expressing one PS1 allele (PS1+/−) are clearly impaired in their ability to respond to BDNF by inhibiting cell death compared to WT. Figure 5B however, shows that PGRN protects PS1 null neurons from glutamate toxicity with equal efficiency as it protects WT and heterozygous knock out (PS1+/−) neurons suggesting that the neuroprotective function of PRGN against excitotoxicity (Xu, et al., 2011) is independent of PS1. Together, our results indicate that PS1 mediates the neuroprotective activity of a limited number of factors including BDNF and efnB1.
Figure 5. PS1 mediates neuroprotective activities of BDNF but not PRGN.

(A) BDNF neuroprotection depends on PS1 genotype. Hoechst quantitation of intact nuclei of cortical neurons from WT, PS1 heterozygous (+/−), or PS1 homozygous (PS1−/−) mouse embryos prepared as in Fig. 3. Neurons were treated with glutamate for 3 hours in the presence (BDNF) or absence (Ø) of BDNF (50ng/ml) added 30 minutes before glutamate. BDNF is unable to protect PS1 null neurons from glutamate toxicity (n=5; ***, p<0.005; *, p<0.05). (B) PRGN neuroprotection is independent of PS1. Neurons were treated with glutamate for 3 hours in the presence (PRGN) or absence (Ø) of PRGN added 30 minutes before glutamate. Neuronal nuclei were counting as above. PRGN (15 ng/ml) protects both WT and PS1 null (PS1−/−) neurons from glutamate toxicity (n=4; p<0.01).
3.5. PS1 mediates neuroprotection from glutamate-induced necrotic death
To explore the cell death pathways affected by PS1, BDNF and efnB, we examined the presence of necrotic and apoptotic death markers in our cultures. Both pathways have been implicated in glutamate-induced neuronal cell death; their relative contribution depending on culture conditions and glutamate treatment (Bonfoco, et al., 1995; Cheung, et al., 1998; Gwag, et al., 1999). Morphological inspection of glutamate treated neuronal cultures stained with Hoechst dye (see Methods) indicated a predominantly necrotic cell death as nuclei were marked by condensation but little or no nuclear fragmentation (Figure 6A). Annexin V and propidium iodide staining, markers of apoptotic and necrotic cell death respectively (Baki, et al., 2008; Kain and Ma, 1999; Sato-Matsumura, et al., 2000), also indicated necrosis as the predominant cell death mechanism (Figure 6A). This conclusion was verified by the glutamate-induced robust increase of the 150kDa spectrin down break product (SDBP), a marker of necrotic pathways produced by calpain processing of spectrin. In contrast, there was no significant increase of the 120 kDa SDBP, a marker of apoptotic cell death produced by the caspace-3 processing of spectrin (Figure 6B, upper panel) (Nath, et al., 2000; Siman, et al., 1989). Absence of significant apoptotic cell death was also indicated by the absence of increased activation of caspase-3, a key executor of the apoptotic pathway (Figure 6B, lower panel). Finally z-VAD, a pan-caspase inhibitor widely used to block apoptotic cell death (Chou, et al., 1995; Degterev, et al., 2005), did not prevent glutamate-induced neuronal death (Figure 6C). In contrast, necrostatin-1 (nec-1), a recently described agent that blocks necroptosis, a form of necrotic cell death (Degterev, et al., 2005), strongly suppressed glutamate-induced neuronal death in all cultures (Figure 6D). Together, our data show that glutamate induced a predominantly necrotic cell death in our cultures and that PS1 is necessary for the neuroprotective effects of efnB and BDNF against necrotic cell death. In addition, the antinecrotic activity of nec-1 is independent of PS1.
Figure 6. Glutamate induces necrotic cell death.
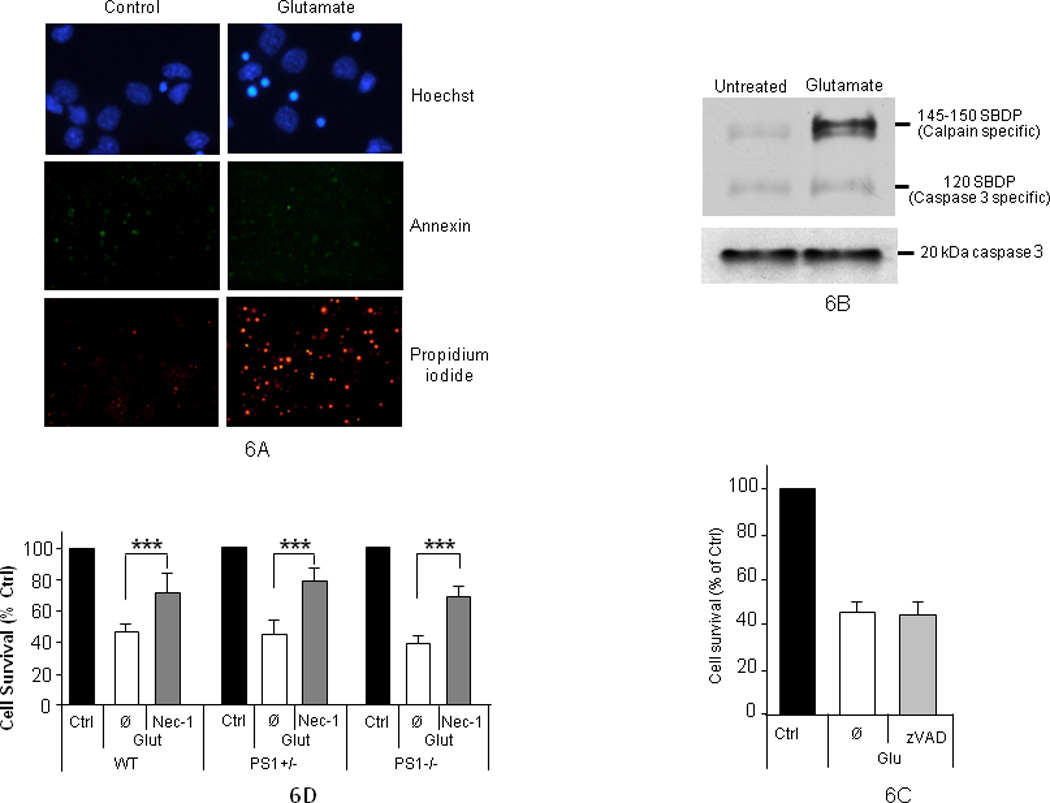
(A) Control (untreated) or glutamate-treated (50µM; 3 hours) mouse neuronal cultures of 8 DIV were stained with Hoechst, Annexin and propidium iodide. Images indicate glutamate induces mostly necrotic death. For clarity, the Hoechst picture is at higher magnification than the Annexin and propidium stainings. (B), Upper panel: neuronal cultures as above were treated with glutamate and extracts were prepared in RIPA buffer. Spectrin break down products (SBDP) were detected on WBs of 6% Tris-glycine gels using anti-spectrin antibodies. Glutamate stimulates calpain but not caspase-3 cleavages of spectrin. Lower panel: activated caspase-3 was detected using 10–20% gradient Tris-glycine gels using anti-caspase antibodies. (C) Apoptosis inhibitor z-VAD does not reduce glutamate-induced neuronal death. Rat cortical neurons were treated with glutamate in the presence (zVAD) or absence (Ø) of 25µM zVAD added 30 minutes before glutamate. Neuronal viability was measured by Hoechst staining. Glu: Glutamate-treated cultures; Ctrl: non-treated control cultures (D) Necrostatin-1 (Nec-1) protects neurons from glutamate toxicity independent of PS1. Intact nuclei were counted as above. Nec-1 (25 µM) protects WT, PS1 heterozygous (+/−) and PS1-null (PS1−/−) neurons from glutamate toxicity (n=4; p<0.005). (Ø), no Nec-1
3.6. Cell surface expression and degradation of neuronal EphB2 and TrkB are impaired in the absence of PS1
Binding of ligands to cognate receptors at the cell surface stimulates internalization and degradation of receptors, a common consequence of ligand-receptor interaction known to mediate important biological functions (Chen, et al., 2005; Grimes, et al., 1996; Zwang and Yarden, 2009). Accordingly, ligand-induced Trk receptor endocytosis plays crucial roles in neurotrophin-dependent neuronal survival (Heerssen, et al., 2004; Kuruvilla, et al., 2004; Zheng, et al., 2008). Since efnB ligands also stimulate endocytosis and metabolism of EphB2 receptor (Litterst, et al., 2007), we asked whether impairment of the neuroprotective activities of efnB and BDNF in PS1 null neurons correlates with any abnormalities in the cell surface expression and ligand-induced degradation of their cognate receptors EphB2 and TrkB respectively. Figure 7A shows that in PS1 KO neurons, the ligand-induced degradation of cellular EphB2 and TrkB proteins is reduced compared to WT neurons suggesting PS1 is necessary for efficient ligand-induced degradation of these receptors. Importantly, both PS1 alleles are needed for efficient receptor degradation as absence of one allele results in reduced ligand-dependent degradation (Figure 7A). To examine whether these observations reflect changes in the cell surface expression of receptors, a site where they first react with cognate ligands, we biotinylated surface proteins of WT, PS1 heterozygous and PS1 homozygous KO neurons and isolated surface TrkB and EphB2 receptors. Figure 7B (lanes 1, 3 and 5) shows that compared to WT neurons (lane 1), constitutive cell surface expression of these receptors is decreased in the absence of PS1 (lanes 3 and 5) although total cellular amounts of TrkB and EphB are not reduced in PS1 KO neurons (Figure 7A, lanes 1, 3 and 5). Together, these data indicate that in the absence of PS1, TrkB and EphB2 receptors are retained in intracellular compartments indicating that transport of these receptors to plasma membrane may be defective. This suggestion is in agreement with literature that PS1 is involved in protein transport (Naruse, et al., 1998) and that PS1 regulates cell surface expression of proteins including integrins and lipoprotein receptor (Tamboli, et al., 2008; Zou, et al., 2008). As expected (Litterst, et al., 2007; Zheng, et al., 2008), treatment of WT (PS1+/+) neurons with either efnB or BDNF resulted in a pronounced reduction of the cell surface levels of their cognate receptors compared to untreated control neurons (Figure 7B, lanes 1 and 2). The degree of this reduction however, depends on the genotype of PS1. Indeed, compared to WT neurons, receptor reduction is less pronounced in heterozygous KO (PS1+/−) neurons and it is undetectable in homozygous PS1 KO (PS1−/−) neurons as treatment with BDNF or efnB1 fails to decrease the levels of cell surface TrkB or EphB receptors relative to the amounts of untreated PS1−/− neurons (Figure 7B, lanes 5 and 6). Thus, our data show that ligand-induced internalization/degradation of receptors is blocked in the absence of PS1. The ligand-induced phosphorylation of both EphB2 and TrkB however, determined at 30 minutes following ligand treatment, were comparable between WT and PS1-KO neurons (Figures 7C and D). Together, these data show that ligand-induced internalization/degradation, but not ligand-induced phosphorylation of receptors is regulated by PS1.
Figure 7. PS1 regulates ligand-induced degradation and cell surface expression of neuronal EphB2 and TrkB receptors.
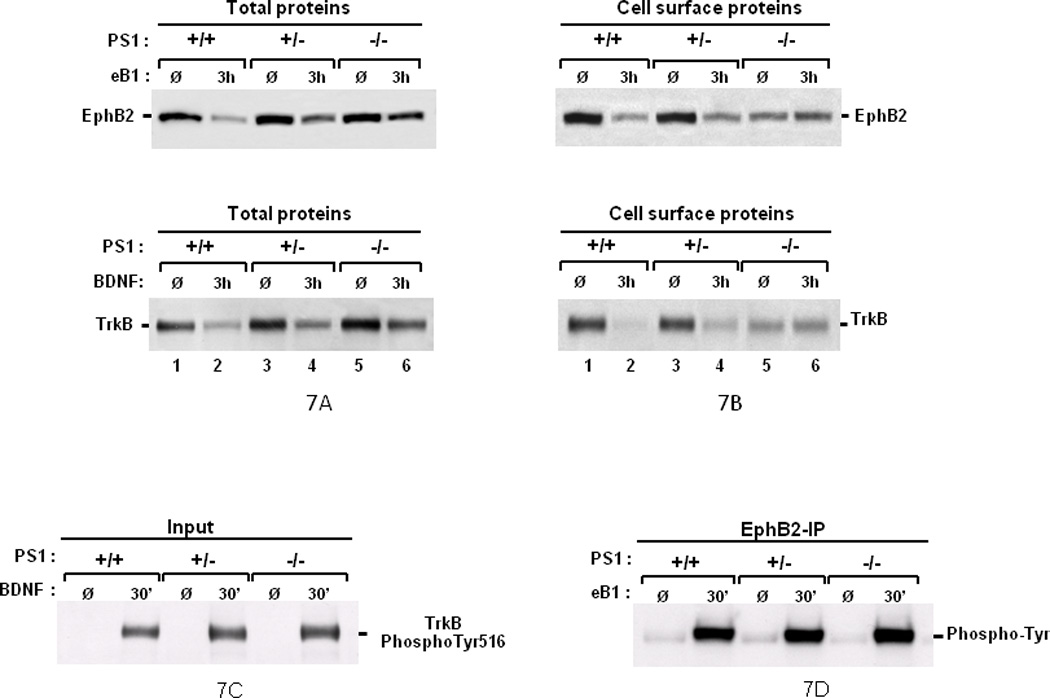
(A) Absence of PS1 decreases the ligand-induced degradation of EphB2 and TrkB. Cortical neuronal cultures prepared as in Figure 3 from WT (+/+), PS1 heterozygous (+/−), or PS1 homozygous (−/−) KO mouse embryonic brains were untreated (Ø) or treated for 3 hours (3h) with either efnB1-Fc (eB1, upper panels) or BDNF (BDNF, lower panels) and total protein extract was probed on WBs with anti-EphB2 (EphB2) or anti-TrkB (TrkB) antibodies respectively. Absence of PS1 results in decreased ligand-induced degradation of both receptors (lanes 2, 4 and 6). (B) PS1 promotes surface expression and ligand-induced internalization of EphB2 and TrkB. Neuronal cultures from WT (+/+), PS1 heterozygous (+/−), or PS1 homozygous (−/−) KO mice were treated as above with efnB1-Fc (upper panels) or BDNF (lower panels). Cell surface protein was then labeled with NHS-Biotin for 1 hour at 4°C before protein extraction and strepavidin pull-down. Obtained protein samples were analyzed on WBs as above. Absence of PS1 results in decreased constitutive surface expression of EphB2 and TrkB (Ø lanes) as well as in defective ligand-induced internalization (3h lanes). (C and D) Absence of PS1 does not affect the ligand-induced phosphorylation of EphB2 and TrkB. WT or PS1-KO mouse neuronal cultures at 8 DIV were treated for 30 min with BDNF and then lysed in RIPA buffer. Total protein was separated by WB and probed directly with anti-TrkB phospho-Tyr516 antibodies (C). Mouse neurons as above were treated for 30 min with efnB1 and lysed in RIPA. One mg of protein was IPed with an anti-EphB2 antibody R407 and IPs were analyzed on WBs with anti-phosphotyrosine antibodies (D). Each experiment in this figure was repeated at least three times with similar results. (Ø), no treatment
3.7. EfnB1 and BDNF neuroprotection is independent of γ-secretase
Recent work revealed that the PS1/γ-secretase system participates in the downstream processing of efnB-induced metabolism of EphB receptors by degrading the EphB2 derivative EphB2/CTF1. In addition, γ-secretase inhibitors (GSIs) block the cleavage of EphB2/CTF1 by γ-secretase in both, cell lines (Litterst, et al., 2007) and in primary cortical neuronal cultures (data not shown). Since the efnB/EphB system regulates neuroprotection, we used GSIs to ask whether γ-secretase activity is involved. Surprisingly, however, these inhibitors had no effect on the efnB-dependent neuroprotection (Fig. 8A). Similarly, GSIs had no effect on the BDNF-dependent neuroprotection (Fig. 8B). Mouse neuronal cultures gave results similar to those obtained with rat cultures and GSI DAPT gave results similar to those obtained with GSI L685,458 (data not shown). Together, these data suggest that the γ-secretase function of PS1 is not involved in the BDNF- and efnB1-dependent neuroprotection against glutamate-induced excitotoxicity.
Figure 8. EfnB1 and BDNF induced neuroprotection is independent of γ-secretase.
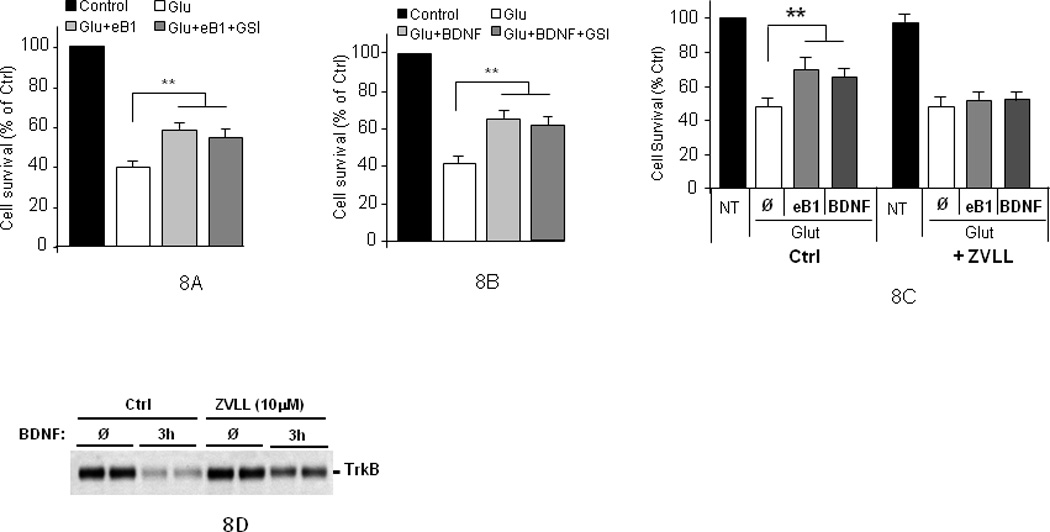
Rat neuronal cultures pre-treated with GSI L685,458 for one hour before they were treated with efnB1 (A) or BDNF (B) for 30 minutes and then with glutamate for three hours. Neuronal viability was measured by Hoechst staining (n=4; p<0.01). Similar data were obtained when GSI DAPT was used instead of L685,458 (not shown). (C) Effects of protease inhibitor ZVLL (10µM) on the efnB-Fc and BDNF neuroprotection from glutamate toxicity. Cultures were pre-treated for one hour with ZVLL (10µM) then efnB or BDNF was added for 30 minutes and then glutamate was added for 3 hours (n=4; p<0.01). NT: no treatment. (Ø): no protective factor (D) WT cortical neuronal cultures prepared as above were untreated (Ø) or treated for 3 hours (3h) with BDNF and total protein extract was probed on WBs with anti-TrkB antibodies (duplicate samples are shown).
Since absence of PS1 reduces the ligand-stimulated metabolism of EphB2, we used protease inhibitor ZVLL to further examine whether reduction of this metabolism by other factors will also affect ligand-dependent neuroprotection. ZVLL has been shown to specifically block the ligand-induced degradation of EphB2 but it has no effect on the constitutive levels of this receptor or on its ionomycin-induced degradation (Litterst, et al., 2007). Figure 8C shows that although ZVLL has no effects on the survival of control cultures, it inhibits the efnB-dependent neuronal survival. We obtained data that, similar to its effects on EphB2 receptor, ZVLL has no effects on the constitutive levels of TrkB (Figure 8D, lanes marked Ø) but reduces both, the BDNF-induced TrkB metabolism (Figure 8D, lanes marked 3h) and the BDNF-dependent neuroprotection (Fig. 8C). These data suggest that ligand-induced receptor degradation may play important roles in neuroprotection, a suggestion also supported by recent literature (Chen, et al., 2005).
4. Discussion
Excitotoxicity, a form of neuronal damage due to excessive activation of glutamate receptors, is a neurotoxic mechanism implicated in the pathogenesis of many neurodegenerative disorders including AD and stroke (Greenamyre and Young, 1989; Mattson, et al., 2000). As a result, factors protecting neurons from excitotoxic insults are considered potential treatments of neurodegenerative disorders (Choi, 1996) and Memantine, a drug currently used to treat AD, is an antagonist of NMDA receptor. Here we report the novel observation that efnB ligands function as neuroprotective factors able to rescue cortical neurons from glutamate excitotoxicity and that the neuroprotective activity of efnB depends on EphB receptors. We also show that PS1 is necessary for the neuroprotective activity of the efnB/EphB system. Surprisingly, neurons expressing one PS1 allele (PS1+/−) did not display significantly higher rates of survival than PS1 null neurons, suggesting that both PS1 alleles are needed for full expression of the neuroprotection activities of the efnB/EphB system against glutamate toxicity. These observations prompted us to ask whether PS1 may mediate neuroprotective functions of other ligand-receptor systems including BDNF/TrkB. This system is known to protect brain cortical neurons from toxicities, including excitotoxicity and oxidative stress, and has been proposed as a therapeutic for neurodegenerative disorders (Nagahara and Tuszynski, 2011). Our data show that BDNF rescues WT cortical neurons from glutamate toxicity, an effect blocked by TrkB antagonists. In the absence of PS1 however, BDNF is unable to protect neurons from glutamate suggesting that PS1 mediates the neuroprotective functions of this neurotrophin against excitotoxicity. Interestingly, our data reveals a PS1 gene dosage effect on BDNF-dependent neuroprotection because full neuroprotective activity by BDNF requires both alleles of PS1. Thus, absence of even one PS1 allele (haploinsufficiency) reduces the neuroprotective effects of both BDNF and efnB albeit to different degrees. Recent reports show that PRGN, a protein that plays important roles in FTD, a brain neurodegenerative disorder distinct from AD, protects cortical neurons from glutamate-induced death (Xu, et al., 2011). The neuroprotective activity of PRGN however, is independent of PS1 as glutamate-treated cultures showed similar rates of survival regardless of PS1 genotype. These data indicate that the neuroprotective functions of PRGN involve different mechanisms than the mechanisms that involve PS1. Thus, PS1 mediates neuroprotective functions of a specific subset of cellular factors.
Depending on culture conditions, including glutamate concentration, excitotoxicity-associated neuronal cell death can be mediated by distinct cell death pathways including apoptosis and necrosis (Bonfoco, et al., 1995; Gwag, et al., 1999). Using markers specific to each pathway (see Results), we determined that in our cultures glutamate induces predominantly the necrotic cell death pathway, a conclusion supported by data that nec-1, a recently described agent that blocks necrotic cell death (Degterev, et al., 2005), suppressed glutamate-induced neuronal death. In contrast, z-VAD, a suppressor of apoptotic cell death (Chou, et al., 1995; Degterev, et al., 2005), had no significant effects on the survival of glutamate-treated cultures indicating this pathway does not contribute significantly to the neuronal death measured in our cultures. Together, our observations show that efnB and BDNF rescue neurons from necrotic death induced by excitotoxicty and that PS1 is necessary for the neuroprotective effect of these factors against necrosis. Interestingly, the neuroprotective activity of nec-1 is independent of PS1 suggesting nec-1 acts downstream of PS1, efnB and BDNF in the necrotic cascade.
PS1 is an important component of the γ-secretase complex that participates in the downstream processing of cell surface proteins including EphB2 receptor, a process inhibited by GSIs (Litterst, et al., 2007). Our data however, show that GSIs have no effect on the neuroprotective function of efnB. Similarly, inhibition of γ-secretase had no effect on the BDNF-dependent neuroprotection, a result not unexpected as there is no evidence that γ-secretase participate in the metabolism of TrkB. We conclude that the role of PS1 in neuroprotection is independent of its function in γ-secretase, a conclusion consistent with literature that PS1 has γ-secretase independent functions (Baki, et al., 2004; Lee, et al., 2010; Tu, et al., 2006; Zhang, et al., 2009).
Binding of ligands to cognate receptors at the cell surface is often followed by receptor endocytosis and metabolism, a process important to ligand-induced receptor functions including Trk receptor-mediated neuronal survival (Grimes, et al., 1996; Heerssen, et al., 2004; Kuruvilla, et al., 2004; Zheng, et al., 2008). Our data show that absence of PS1 affects both, the levels of EphB2 and TrkB at neuronal cell surface and the ligand-induced metabolism of surface EphB2 and TrkB. Thus, under non-stimulated (constitutive) conditions, absence of PS1 results in reduced expression of cell surface TrkB and EphB2 although the total amount of each receptor was similar in PS1 KO and WT neurons. Furthermore, absence of PS1 impairs the ligand-induced degradation of cell surface EphB2 and TrkB. Thus, PS1 may promote ligand-dependent neuroprotection by regulating surface expression and ligand-induced endocytosis and metabolism of cognate neuronal receptors. This hypothesis is supported by our data that ZVLL, an agent that inhibits the ligand-induced degradation of EphB2 and TrkB, also blocks the efnB- and BDNF-dependent neuroprotection. Our data however, do not exclude the possibility that other PS1 functions (Pimplikar, et al., 2010) are also involved in the neuroprotective roles of PS1. Interestingly, absence of PS1 had no effects on the ligand-induced phosphorylation of TrkB and EphB2 detected within 30 minutes following ligand treatment. This result indicates that receptor phosphorylation is not sufficient to support the neuroprotective activities of efnB and BDNF. In contrast, our observations highlight the importance of PS1 and receptor internalization and metabolism on the neuroprotective functions of the efnB-EphB and BDNF-TrkB systems, a conclusion consistent with data that receptor endocytosis is crucial to neuronal survival (Kuruvilla, et al., 2004).
It is noteworthy that loss of one allele of PS1 results in a severe reduction of the neuroprotective activities of both efnB and BDNF. These data support the hypothesis that inactivation of the neuroprotective activity of even one PS1 allele by genetic legions will decrease neuronal protection from toxic insults in vivo resulting in increased rates of neuronal cell death, a process that over many years may lead to dementia. This observation is analogous to FTD where loss or inactivation of one allele of PRGN by genetic mutations (haploinsufficiency) results in dominant transmission of neurodegeneration and FTD (Goedert and Spillantini, 2006) and raises the possibility that loss of PS function may be involved in FAD neurodegeneration caused by PS mutants (Pimplikar, et al., 2010; Shen and Kelleher, 2007). Together, our data reveal a new PS function in neuroprotection that links PS directly to neuronal survival, a parameter of crucial importance to dementia. Finally, our data that neurons deficient in PS1 are rescued from toxic insults by agents that act independent of PS1, such as PRGN and nec-1, suggests that these agents may be considered therapeutically in neurodegenerative conditions caused by loss of PS1 function.
Acknowledgements
We are grateful to Dr. Junying Yuan and members of her lab for useful discussions on Necrostatin-1 and to Dr Michael Parides of the Mount Sinai Biostatistics Center for helpful discussions on the statistical analysis of our data. This work was supported by NIH grants AG-17926, AG-08200, NS 047229 and Alzheimer's Association grant IIRG-10-174237. JD was partially supported by a grant from “Fondation pour la Recherche Médicale”.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. Embo J. 2004;23(13):2586–2596. doi: 10.1038/sj.emboj.7600251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baki L, Neve RL, Shao Z, Shioi J, Georgakopoulos A, Robakis NK. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J Neurosci. 2008;28(2):483–490. doi: 10.1523/JNEUROSCI.4067-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci U S A. 1995;92(16):7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZY, Ieraci A, Tanowitz M, Lee FS. A novel endocytic recycling signal distinguishes biological responses of Trk neurotrophin receptors. Mol Biol Cell. 2005;16(12):5761–5772. doi: 10.1091/mbc.E05-07-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung NS, Pascoe CJ, Giardina SF, John CA, Beart PM. Micromolar L-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology. 1998;37(10–11):1419–1429. doi: 10.1016/s0028-3908(98)00123-3. [DOI] [PubMed] [Google Scholar]
- Choi DW. Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol. 1996;6(5):667–672. doi: 10.1016/s0959-4388(96)80101-2. [DOI] [PubMed] [Google Scholar]
- Chou CC, Lam CY, Yung BY. Intracellular ATP is required for actinomycin D-induced apoptotic cell death in HeLa cells. Cancer Lett. 1995;96(2):181–187. doi: 10.1016/0304-3835(95)03927-o. [DOI] [PubMed] [Google Scholar]
- Cisse M, Halabisky B, Harris J, Devidze N, Dubal DB, Sun B, Orr A, Lotz G, Kim DH, Hamto P, Ho K, Yu GQ, Mucke L. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469(7328):47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103(6):945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266(5186):816–819. doi: 10.1126/science.7973638. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Egea J, Klein R. Bidirectional Eph-ephrin signaling during axon guidance. Trends Cell Biol. 2007;17(5):230–238. doi: 10.1016/j.tcb.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. Frontotemporal lobar degeneration through loss of progranulin function. Brain. 2006;129(Pt 11):2808–2810. doi: 10.1093/brain/awl291. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Young AB. Excitatory amino acids and Alzheimer's disease. Neurobiol Aging. 1989;10(5):593–602. doi: 10.1016/0197-4580(89)90143-7. [DOI] [PubMed] [Google Scholar]
- Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, Mobley WC. Endocytosis of activated TrkA: evidence that nerve growth factor induces formation of signaling endosomes. J Neurosci. 1996;16(24):7950–7964. doi: 10.1523/JNEUROSCI.16-24-07950.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwag BJ, Canzoniero LM, Sensi SL, Demaro JA, Koh JY, Goldberg MP, Jacquin M, Choi DW. Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neuroscience. 1999;90(4):1339–1348. doi: 10.1016/s0306-4522(98)00508-9. [DOI] [PubMed] [Google Scholar]
- Heerssen HM, Pazyra MF, Segal RA. Dynein motors transport activated Trks to promote survival of target-dependent neurons. Nat Neurosci. 2004;7(6):596–604. doi: 10.1038/nn1242. [DOI] [PubMed] [Google Scholar]
- Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J Cell Biol. 2003;163(6):1313–1326. doi: 10.1083/jcb.200306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kain SR, Ma JT. Early detection of apoptosis with annexin V-enhanced green fluorescent protein. Methods Enzymol. 1999;302:38–43. doi: 10.1016/s0076-6879(99)02007-8. [DOI] [PubMed] [Google Scholar]
- Kuruvilla R, Zweifel LS, Glebova NO, Lonze BE, Valdez G, Ye H, Ginty DD. A neurotrophin signaling cascade coordinates sympathetic neuron development through differential control of TrkA trafficking and retrograde signaling. Cell. 2004;118(2):243–255. doi: 10.1016/j.cell.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Lackmann M, Boyd AW. Eph, a protein family coming of age: more confusion, insight, or complexity? Sci Signal. 2008;1(15):re2. doi: 10.1126/stke.115re2. [DOI] [PubMed] [Google Scholar]
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leem JY, Saura CA, Pietrzik C, Christianson J, Wanamaker C, King LT, Veselits ML, Tomita T, Gasparini L, Iwatsubo T, Xu H, Green WN, Koo EH, Thinakaran G. A role for presenilin 1 in regulating the delivery of amyloid precursor protein to the cell surface. Neurobiol Dis. 2002;11(1):64–82. doi: 10.1006/nbdi.2002.0546. [DOI] [PubMed] [Google Scholar]
- Litterst C, Georgakopoulos A, Shioi J, Ghersi E, Wisniewski T, Wang R, Ludwig A, Robakis NK. Ligand binding and calcium influx induce distinct ectodomain/gamma-secretase-processing pathways of EphB2 receptor. J Biol Chem. 2007;282(22):16155–16163. doi: 10.1074/jbc.M611449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. Embo J. 2002;21(8):1948–1956. doi: 10.1093/emboj/21.8.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Soriano E. Functions of ephrin/Eph interactions in the development of the nervous system: emphasis on the hippocampal system. Brain Res Brain Res Rev. 2005;49(2):211–226. doi: 10.1016/j.brainresrev.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev. 1997;77(4):1081–1132. doi: 10.1152/physrev.1997.77.4.1081. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Zhu H, Yu J, Kindy MS. Presenilin-1 mutation increases neuronal vulnerability to focal ischemia in vivo and to hypoxia and glucose deprivation in cell culture: involvement of perturbed calcium homeostasis. J Neurosci. 2000;20(4):1358–1364. doi: 10.1523/JNEUROSCI.20-04-01358.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 2003;3(2):65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- Morgan DM. Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol Biol. 1998;79:179–183. doi: 10.1385/0-89603-448-8:179. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10(3):209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21(5):1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Nath R, Davis M, Probert AW, Kupina NC, Ren X, Schielke GP, Wang KK. Processing of cdk5 activator p35 to its truncated form (p25) by calpain in acutely injured neuronal cells. Biochem Biophys Res Commun. 2000;274(1):16–21. doi: 10.1006/bbrc.2000.3070. [DOI] [PubMed] [Google Scholar]
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133(1):38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Pimplikar SW, Nixon RA, Robakis NK, Shen J, Tsai LH. Amyloid-independent mechanisms in Alzheimer's disease pathogenesis. J Neurosci. 2010;30(45):14946–14954. doi: 10.1523/JNEUROSCI.4305-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitulescu ME, Adams RH. Eph/ephrin molecules--a hub for signaling and endocytosis. Genes Dev. 2010;24(22):2480–2492. doi: 10.1101/gad.1973910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robakis NK. Mechanisms of AD neurodegeneration may be independent of Abeta and its derivatives. Neurobiol Aging. 2011;32(3):372–379. doi: 10.1016/j.neurobiolaging.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajjadi FG, Pasquale EB. Five novel avian Eph-related tyrosine kinases are differentially expressed. Oncogene. 1993;8(7):1807–1813. [PubMed] [Google Scholar]
- Sato-Matsumura KC, Matsumura T, Nakamura H, Sawa H, Nagashima K, Koizumi H. Membrane expression of annexin I is enhanced by calcium and TPA in cultured human keratinocytes. Arch Dermatol Res. 2000;292(10):496–499. doi: 10.1007/s004030000172. [DOI] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104(2):403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9(5):1579–1590. doi: 10.1523/JNEUROSCI.09-05-01579.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamboli IY, Prager K, Thal DR, Thelen KM, Dewachter I, Pietrzik CU, St George-Hyslop P, Sisodia SS, De Strooper B, Heneka MT, Filippov MA, Muller U, van Leuven F, Lutjohann D, Walter J. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J Neurosci. 2008;28(46):12097–12106. doi: 10.1523/JNEUROSCI.2635-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126(5):981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xilouri M, Bruban J, Shioi J, Shao Z, Papazoglou I, Vekrellis K, Robakis NK. Extracellular progranulin protects cortical neurons from toxic insults by activating survival signaling. Neurobiol Aging. 2011;32(12):2326 e5–2326 e16. doi: 10.1016/j.neurobiolaging.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460(7255):632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Shen WH, Lu TJ, Zhou Y, Chen Q, Wang Z, Xiang T, Zhu YC, Zhang C, Duan S, Xiong ZQ. Clathrin-dependent endocytosis is required for TrkB-dependent Akt-mediated neuronal protection and dendritic growth. J Biol Chem. 2008;283(19):13280–13288. doi: 10.1074/jbc.M709930200. [DOI] [PubMed] [Google Scholar]
- Zou K, Hosono T, Nakamura T, Shiraishi H, Maeda T, Komano H, Yanagisawa K, Michikawa M. Novel role of presenilins in maturation and transport of integrin beta 1. Biochemistry. 2008;47(11):3370–3378. doi: 10.1021/bi7014508. [DOI] [PubMed] [Google Scholar]
- Zwang Y, Yarden Y. Systems biology of growth factor-induced receptor endocytosis. Traffic. 2009;10(4):349–363. doi: 10.1111/j.1600-0854.2008.00870.x. [DOI] [PubMed] [Google Scholar]