

1. Introduction
The focus of this review is on the reactivity of cationic, tricoordinated boron electrophiles, and on recent applications that rely on intermediates having sextet boron and net positive charge. Older reviews have appeared that address early studies involving a variety of boron cations,1 or that emphasize the structural, bonding, and theoretical aspects of di-, tri-, and tetracoordinate boron cations (borinium, borenium, and boronium, respectively; Fig. 1).2 The latter review by Kölle and Nöth is a milestone in the field of boron cation chemistry. It systematizes a number of earlier observations, and defines the currently accepted nomenclature for boron-containing cations. A more recent (2005) review by Piers et al. updates the structural and bonding aspects in depth,3 discusses reactions of boron cations in the gas phase as well as in solution, and explores several other topics of special interest to the organoelement community. We will take a somewhat different approach to follow developments from the organic chemistry perspective, including selected historical aspects of reactivity, some of which have not appeared in prior reviews. Several topics will necessarily overlap with the excellent overview by Piers et al., but detailed coverage will be limited to the solution chemistry of tricoordinate boron cations, the reactive intermediates known as borenium ions according to Nöth’s classification.2 This terminology depends on the number of ligands at boron and avoids distinctions based on bond order or resonance contributions. Nöth’s treatment also recognizes the iso-electronic relationship between borenium and carbenium ions, both of which feature a formally vacant p-orbital at the central atom (boron and carbon, respectively) and the same overall positive charge. Consistent with the carbenium analogy, borenium ions have long been suspected as intermediates in classical nucleophilic substitution chemistry involving B–X bonds in tetracoordinate boron structures, although we will find that these suspicions were often unfounded. On the other hand, recent studies have revealed fascinating new roles for borenium intermediates, ranging from enantioselective catalysis and memory of chirality applications to hints of C–F activation and C–H insertion chemistry. It would be fair to say that these recent developments were slow to unfold, given the long history of borenium chemistry.
Fig. 1.

Boron cation nomenclature and structural representations in current use.
2. History of borenium ions: often considered, seldom confirmed
2.1 Suspected intermediates in B-N protonation
The first reports to our knowledge where cationic sextet boron was implicated are the two 1933 papers by Wiberg and Schuster that describe the reaction of dichloro(dimethylamino)borane 1 with hydrogen chloride.4 The experiment was said to produce the hydrochloride (chlorhydrat) of 1, drawn using the notation shown as 2a (eq.1). This representation does not specify connectivity, and nothing in the 1933 papers indicates whether 2a is the same or different compared to the borenium salt 2b. Whether the authors intended to distinguish 2a from representations such as 3a or 3b is also uncertain. On the other hand, a 1947 paper by Wiberg and Hertwig shows a similar reaction (R2BNHR + HCl, eq. 2) and provides two generalized drawings of a single ionic structure (5a and 5b) that is clearly identified as the unstable intermediate leading to an isolable covalent adduct, drawn by Wiberg and Hertwig in two unambiguous representations 6a and 6b.5 These drawings and appended comments leave no question about the structure of 5 and the greater stability of 6. However, they may have escaped the notice of subsequent authors, some of whom accepted the borenium structure 2b as a more stable alternative compared to 3b and attributed this conjecture to Wiberg.
2.2 Hypothetical isomers of Cl3B·NHMe2
In 1952, Brown and Osthoff argued that 2b is “probably correct”, and constitutes a better representation than 3b. As partial support for this assignment, they noted that the substance reacts rapidly with aqueous silver ion to precipitate AgCl under conditions where Cl3B·NMe3 is inert.6 Two years later, a study by Goubeau et al. reached the opposite conclusion in favor of 3b as the stable structure because a solution in chloroform does not conduct electric current, in contrast to diethylamine hydrochloride.7 Nevertheless, 2b appeared again in a subsequent report suggesting that the preferred structure may depend on the conditions used for the generation of 2/3.8 Such issues were not easily resolved using the instrumental methods available to most workers by 1960 (primarily, IR spectroscopy), and relevant 11B NMR data were not reported until much later. In 1975, Ryschkewitsch and Myers observed a 11B chemical shift of δ = 8.7 ppm (downfield vs. BF3·OEt2 at 0.0 ppm),9 a value that is consistent with tetracoordinate boron as in 3b and is well upfield of the range expected for 2b according to more recent studies of non-stabilized borenium species (δ >50 ppm).2,3 The former authors did not mention that there had been any disagreement regarding the structure (2b vs. 3b), nor did they make strong claims regarding their own NMR-based assignment of structure 3b.9 However, Ryschkewitsch and Meyer did raise the possibility that an equilibrium between 3b and the borenium salt 7 may explain the formation of a cationic byproduct 8 containing tetracoordinate boron (a boronium salt) when dimethylamine is reacted with BCl3 (eq. 3).
2.3 The first observable borenium salt
By 1970, Ryschkewitsch et al. had already encountered related chemistry starting from the adduct 9 of 4-picoline and BCl3 (eq. 4). Treatment of 9 with 2 equiv of aluminum chloride in dichloromethane gave an equilibrium mixture containing mostly (Keq = ca. 20) the tri-coordinated cation 10 according to the concentration-dependent 11B chemical shift (δ 47.3 = ppm).10 The solution of 10 reacted spontaneously with trimethylamine to form the boronium salt 11. In contrast to 10, the starting 9 did not react with trimethylamine under similar room temperature conditions in the absence of aluminum chloride. Thus, 9 does not equilibrate with the borenium chloride 12 spontaneously, and the role of aluminum chloride is to force the equilibrium to 10 by abstraction and binding of chloride. Finally, addition of Me4NCl to the solution of 10 resulted in reversion to 9, thereby confirming the lower stability of the borenium ion 12 compared to 9. This behavior is consistent with the presence of a mostly unoccupied orbital at boron in 10, an issue that will be revisited in a later section of this review. Structure 10 has stood the test of time,10b and has become a reference point for the spectroscopic characterization of labile borenium structures. It has also established a definitive precedent for generating transient borenium intermediates by B–X heterolysis in the presence of halophilic Lewis acids (BCl3, Al2Cl6, etc.).
2.4 Nucleophilic substitution of X3B·NR3 and Py·BF2X
Facile leaving group displacements are known with many amine-trihaloborane adducts,11–21 and rationales involving the SN1-like generation of borenium ions have been considered repeatedly.12–16 However, detailed investigations have found that bimolecular mechanisms are generally preferred. Persuasive evidence for SN1-like leaving group heterolysis has been reported in only one system, the reaction of the trimethylamine triiodoborane complex 13 with anionic nucleophiles (eq. 5).15 This process occurs in dichloromethane at 50 °C in the absence of added halophiles, and the qualitative observations implicate a borenium ion 14 as the intermediate in halide exchange leading to 15. On the other hand, halide exchange from the analogous adducts 16 (X= F, Br) with BCl3 occurs by a bimolecular process to afford 18 (eq. 6), among other products.15 The reaction is believed to take place via a bimolecular transition state such as 17, and not by SN1 heterolysis to a borenium ion. No exchange of halides occurs between various pairs of reactants 13 and 16 at 50 °C, and 16 does not undergo exchange with Et4NCl in the absence of BCl3, in contrast to the behavior of 13. Furthermore, isotopic label experiments using 10B-containing substrates 13 and 16 rule out competing B–N dissociation pathways under the reaction conditions. This evidence is consistent with the borenium pathway for halide exchange from 13, but not from 16.
In a more recent investigation, the conversion of the difluorohaloborane complex 19 to the boronium salt 21 was shown to occur by a bimolecular mechanism according to the classical test that rate depends on the reactivity of the nucleophile (eq. 7).16,17 Several qualitative reactivity comparisons were made, and the pyridine example (19, R= H) was found to procceed to 21 within minutes at room temperature, too fast for NMR monitoring. In contrast, the 2-picoline case (19, R= o-methyl) required 1–2 hours to achieve similar conversion to 22. The fluorine substituents in 19 were essential for good reactivity, and are believed to help stabilize a penta-coordinated transition state 20. The analogous BBr3 adduct did not react at room temperature.
By the same test of nucleophile-dependent reactivity, the trimethylamine iodoborane complex 23 also reacts by an SN2-like mechanism to form boronium salts 24 with various amines (eq. 8; R= H, 5 h at room temperature; R= CN, 5 h, 70 °C, both in benzene).22,23 The relative ease of this bimolecular reaction for R= H is interesting because 23 is isosteric with neopentyl iodide. Evidently, the differences in bond lengths (dative B–N > C–C) and electronegativity (B < C) result in a looser SN2 transition state that better tolerates the steric demands of the nucleophile. It is harder to say how the differences in electronegativity in 23 compared to neopentyl iodide would affect an SN1 heterolysis process, but there is no evidence that a primary borenium ion 25 would be energetically accessible. Certainly, the isoelectronic and isostructural neopentyl carbenium ion would not be regarded as a plausible intermediate in benzene at room temperature, and generation of 25 under these conditions seems unlikely. Several related nucleophilic substitution reactions of 23 under comparably mild conditions have also been reported. 24–27
Based on the evidence presented so far, simple borenium intermediates that lack stabilization from an adjacent electron donor such as the π system in 10 (eq. 4) should be invoked only after a careful evaluation of alternative mechanisms. There is some evidence that the combination of a weak B–I bond and increased substitution at boron may be sufficient to allow B–I heterolysis, as in the equilibrium between 13 and 14 (eq. 5), but this is an unusual environment. On the other hand, the presence of second row substituents (B–N, B–O) that can donate unshared electron pair density to boron is quite common, and promotes stabilization of borenium intermediates by lone pair (n) delocalization involving the boron 2p-orbital, as discussed below.
2.5 Aminoborenium ions; stabilization by n-delocalization
Nöth and Fritz considered the possible role of n-stabilized aminoborenium ions in the protonation of bis(dialkylamino)boranes during the 1960’s,28 but convincing evidence was not obtained until well-behaved substrates were encountered in the 1,3-diazaborolidine series (Scheme 1). 29 Thus, protonation of 26 with triflic acid generated the amine-stabilized cation 27 in solution (11B δ = 25.8 ppm). The same cation was obtained in crystalline form using an alternative method via nucleophilic displacement and internal proton transfer from 29, and the structure was confirmed by X-ray crystallography. These experiments indicate that 27 is more stable than the covalent adduct 28, although they do not exclude the possibility that 28 is present in equilibrium with the borenium ion 27. Chemical shift comparisons among a number of related (diamino)borenium salts show δ 11B values in the range of 24–31 ppm with counterions including bromide, triflate, and tetrachloroaluminate. The minimal anion dependence of chemical shifts is easily understood if tetracoordinated adducts such as 28 are minor components in equilibrium due to n-delocalization by the two amino groups. The n-delocalization effect is also responsible for the relatively high field 11B chemical shift values for 27 and related salts compared to tricoordinate boron cations lacking n-delocalization (δ > ca. 45 ppm). 2 On the other hand, neutral tetracoordinate boron species similar to 28 are observed at δ 11B values of ca. 0 ± 15 ppm.2,29
Scheme 1.

Electrophilic activation of 1,3-diazaborolidines by protonation or by Lewis acid catalysts29
The nucleophilic displacement method can also be used with hindered amines, as illustrated by the reaction of triflate 29 with 2,6-lutidine to afford the borenium salt 30 (Scheme 1). A similar borenium salt can be obtained starting from the 2-chloro-1,3-diazaborolidine 31, but the chloride is not sufficiently reactive unless it is converted into the isolable Lewis acid adduct 32 using AlCl3. Although 32 has no net charge, it does contain a borenium subunit that apparently facilitates nucleophilic attack below room temperature via the presumed intermediate 33 to give 34 (>90% isolated). An alternative mechanism has not been ruled out where 32 is converted into the transient borinium cation 35 prior to attack by the nucleophile. Related acyclic borinium cations are known, but they are stabilized by allenic delocalization. In a cyclic environment, similar delocalization would require a larger ring (as in the observable 36 with n = 4) to accommodate sp-hybridized dicoordinate boron,2 but this would not be feasible in the diazaborolidine environment (36 with n = 0). Many other examples of relevant aminoborinium and aminoborenium ions are discussed by Kölle and Nöth in their 1985 review2 that will not be repeated here. Nöth also addressed several methods for the generation of various other boron cations,30 including borenium ions that lack n-delocalization. The following paragraphs will focus on newer developments in methodology. Most of these studies were conducted to explore the limits for isolation of minimally stabilized, highly electrophilic borenium species (Sections 3, 4), but some of the work was stimulated by intended applications in organic synthesis that are discussed later (Sections 5–7).
3. Recent developments in the generation of observable borenium intermediates
3.1 Electrophilic activation by protonation or by Lewis acid catalysis
Protonation of aminoboranes is the oldest method for generating transient as well as stable borenium salts. It has not been used extensively in recent efforts to detect minimally stabilized borenium species, but interesting synthetic applications have appeared that exploit chiral borenium ions and analogues as enantioselective catalysts (see Section 6 for details). A specific example is shown in eq. 9 where the oxazaborolidine 37 is treated with TfOH to generate a mixture of equilibrating species including 38 and 39 according to 1H NMR considerations.31 Although no 11B NMR data were reported, the presence of the borenium ion 38 in the equilibrium mixture was supported by observing that the mixture functions as a potent chiral Lewis acid catalyst for the Diels-Alder reaction of 1,3-butadiene at −78 °C. If 38 is one of the two major species observed by 1H NMR, then substantial borenium cation stabilization by the oxygen electron pairs can be inferred.
Borenium salt 38 is structurally related to key intermediates in the enantioselective CBS reduction using the Corey-Itsuno catalysts 40 in combination with THF·BH3 (eq. 10).32 In the first stage of this fascinating process, borane is proposed to function as a Lewis acid that converts 40 into the activated intermediate 41. Although 41 has no net charge, it does contain a borenium subunit and can therefore function as a Lewis acid that binds the carbonyl oxygen of a ketone substrate to accelerate nucleophilic attack by internal hydride in a 6-center process. The proposed role of 41 is supported by extensive enantioselectivity data and analogies to be discussed further in Section 6.
The conversion from 40 to 41 resembles the activation of 31 by aluminum chloride that was discussed in connection with Scheme 1. Lewis acid activation should also be possible with borane derivatives having B–O, B–F, or B–Cl bonds instead of B–N bonds. In the broader sense, any Lewis acid that is capable of interacting with the electron pair of a boron–heteroatom bond can produce transient species having partial borenium ion character.
3.2 Halide abstraction by a halophile
The first method shown to generate an observable borenium ion 10 was discussed in Section 2 (eq. 4) and involved chloride abstraction from the boron trichloride complex 9 by aluminum trichloride. Improved versions of this general approach are featured in several recent studies involving the generation of minimally stabilized borenium ions (Scheme 2). Thus, the pyridine complex 42 of diphenylchloroborane was treated with the chlorophile SbCl5 in dichloromethane at rt to generate a dark yellow solution of the air-sensitive borenium salt 43 (11B δ = 58.2 ppm).33 This substance is of interest because it is isoelectronic with trityl cation, and because its properties help to clarify a long-standing mystery regarding the apparent generation of hypothetical dicoordinated diphenylborinium salts Ph2B+ ClO4− or Ph2B+ SbCl6−. Thus, treatment of 43 with 1 equiv of pyridine afforded the boronium salt 44 (11B δ = 8.6 ppm). The same salt had been obtained earlier by the reaction of Ph2BCl with SbCl5 in deuterated nitromethane, followed by the addition of pyridine. However, the initially formed cation generated from Ph2BCl and SbCl5 in nitromethane was found to have a 11B signal at δ = 19.8 ppm, far upfield of the 85.5 ppm value calculated for diphenylborinium cation, but consistent with the solvated (boronium) structure 45. Presumably, 45 then reacts with sequentially added pyridine via dissociation to the borenium intermediate 46. These findings suggest that earlier encounters with “Ph2B+ “ species in polar solvents more plausibly involved the corresponding solvated (tetracoordinate) boron cations (boronium salts). Furthermore, chemical shift calculations indicate that the perchlorate derivative depicted earlier as the ionic borinium salt Ph2B+ ClO4− prefers the covalent structure Ph2BOClO3 based on a 11B signal at δ = 46.0 ppm in nitromethane (calcd 47.0 ppm).34 If the computational level [B3LYP/6–31+G(d)] in the hypothetical gas phase environment reflects the situation in nitromethane as suggested by the chemical shift match, then the perchlorate is bound to boron in a monodentate arrangement, and the tricoordinated Ph2BOClO3 is not complexed to the moderately nucleophilic nitromethane.
Scheme 2.

Many of the recent advances in the generation and isolation of highly electrophilic borenium salts have been made by using variations of the halide abstraction method (Scheme 2). Thus, Gabbai et al. employed an approach where the tetracoordinated substrates are formed in situ from hindered diarylfluoroboranes 47 and DMAP in the presence of trimethylsilyl triflate (TMSOTf) as the fluorophile.35 This approach gave sterically protected, crystalline borenium salts 48 or 49 that were sufficiently stable for X-ray structure determination. A similar approach allowed isolation of the N-heterocyclic carbene adduct 50 by using an in situ source of the nucleophilic carbene, 1,3-dimethylimidazolidene, in place of DMAP.36 A related activation mechanism via a borenium intermediate has been suggested for the conversion of 9-chloro-9-bora-9,10-dihydroanthracene into boronium salts upon treatment with silver tetrafluoroborate in the presence of bipyridines.37
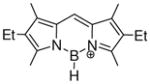
Scheme 2 also illustrates two halide abstraction examples involving extensively conjugated heterocyclic substrates. Both examples generate borenium cations (52 from 5138 and 55 from 5439) using triethylsilyl cation equivalents as exceptionally potent halophiles. In addition to their high reactivity, the triethylsilyl reagents serve as convenient sources of minimally interactive anions such as tetrakis(pentafluorophenyl)borate, (C6F5)4B−, and the brominated carborane CbBr6− that are required for isolation and X-ray characterization of the highly electrophilic, air-sensitive borenium ions 52 and 55.38,39a,b Another advantage of the triethylsilyl cation reagents is that the only byproduct of borenium ion generation is the volatile and easily removed triethylhalosilane. That key advantage was especially useful for the isolation of 55, a cation that has been implicated as an intermediate in the substitution chemistry of the subphthalocyanine 54, and also in the controlled ring expansion of 54 into phthalocyanines.39c In connection with the related preparation of 52, it is also worth noting that the (C6F5)4B− anion was sufficiently stable to survive under the conditions for reduction of 52 to 53 with di-isobutylaluminum hydride.38 Like its precursor, 53 was characterized by X-ray methods, and the substance constitutes a rare example of an observable borenium ion that contains a B–H bond.
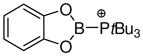
A somewhat different example of halide abstraction form a heterocyclic substrate is shown in eq. 11.40 In contrast to the examples of Scheme 2, the tetracoordinate 56 is converted to a borenium ion 57 by the moderately fluorophilic borontrifluoride etherate, evidence that 57 has lower fluoride affinity. One might question whether it is correct to regard 57 as a borenium ion since the heterocyclic cation is part of a conjugated 6π-aromatic system, but the borenium terminology is based only on the coordination number and net positive molecular charge, and not on bond order at boron.2 Nevertheless, it is clear that 57 and related aromatic cations of the general structure 58 belong in a very different stability category. In some cases, related cations survive if the counterion X- is triflate, or even the relatively nucleophilic chloride anion.40,41 Clearly, aromaticity is an important factor in this environment, and cations such as 57 and 58 constitute a special case. For that reason we will not comment more on their chemistry, although we will briefly revisit their electrophilicity in Section 4.
A special case at the lower end of the stability range is shown in eq. 12. Treatment of 59 with the strongly chlorophilic carborane salt Ag+ CbBr6− in the presence of 0.9 equiv of triethylphosphine oxide results in the formation of an isolable, crystalline borenium salt 60 that was characterized by X-ray crystallography.42 Formation of 60 may involve a typical chloride abstraction from the phosphine oxide adduct 61 that should be formed reversibly. However, chloride abstraction also occurs in the absence of the phosphine oxide in experiments where 59 is treated with the Et3Si+ CbBr6− reagent in aromatic solvents. These observations raise the intriguing possibility that a species equivalent to the borinium ion 62 may be accessible. No electrophilic boron species could be detected by spectroscopy in these experiments, but their generation is supported by the formation of electrophilic borylation products derived from the aromatic solvent as discussed further in Section 8. Although the prospects for generating dicoordinate boron cations such as 62 need to be evaluated with care in view of the “Ph2B+ “ problem discussed earlier (Scheme 2), 62 does have the potential added benefit of n-delocalization from oxygen. On the other hand, 62 should be more electrophilic compared to the hypothetical borinium intermediate 35 in the 1,3-diazaborolidine series where n-delocalization from nitrogen should provide greater stabilization (Scheme 1).
3.3 The borinium-borenium interface; gas phase oxyborenium ions
Oxygen-stabilized dicoordinate boron cations related to 62 have not been detected in solution, although there are gas phase precedents under flowing afterglow conditions (eq. 13). For example, the borinium ion 63 was generated from trimethyl borate, and the derived oxygen-stabilized borenium adduct 64 with trimethyl borate was identified by mass spectroscopy.43 This observation indicates that 63 is exceptionally electrophilic and interactive, and raises questions regarding the survival of “free” oxygen-substituted borinium species in solution. Other examples of gas phase generation of borenium ions from borinium species have been reviewed by Piers et al.3 while the generation of borenium species from nitrogen-stabilized borinium ions in solution was covered in depth by Kölle and Nöth.2 The latter review also discusses the role of ring size in n-delocalization. In brief summary, access to observable borinium ions requires delocalization by two sets of adjacent heteroatom lone pairs and results in an sp-hybridized, linear environment at boron. This may be feasible in an acyclic structure or in medium-sized rings, but is not expected in a 5-membered ring such as 62.
3.4 Borenium salt generation by hydride abstraction
3.4.1 Hydride abstraction by tris(pentafluorophenyl)borane
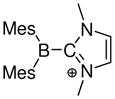
Oxygen-stabilized tricoordinate boron cations (borenium ions) have also been generated in solution using the method of hydride abstraction (Scheme 3). This approach resembles halide abstraction in the sense that survival of the resulting borenium salt depends on controlling the nucleophilicity of the counterion. In one intriguing recent approach, this was done by generating both the anion and the cation from non-ionic reactants.44 Thus, catechol borane 65 was treated with the electrophilic borane B(C6F5)3 in the presence of tri-tert-butylphosphine as the nucleophile. Due to steric hindrance, the phosphine does not bind irreversibly to B(C6F5)3 according to the frustrated lone pair concept of Stephan et al., thereby allowing the formation of the less hindered catechol borane adduct 66 in equilibrium. Subsequent hydride transfer to B(C6F5)3 occurs to afford the isolable borenium salt 67 (11B δ = 29.9 ppm for the cation; −25.4 ppm for the anion), structure confirmed by X-ray crystallography. Each step in this sequence is reversible, so the isolation of 67 in 96% yield indicates a clear thermodynamic advantage for the borenium salt 67 compared to the tetracoordinate 66. Because the B3H bonds in 66 and 67 probably have similar energies, the energy advantage for 67 reflects destabilization of 66 by steric congestion, and more important, stabilization of borenium salt 67 by n-delocalization. The B(C6F5)3 activation method has also been applied to pinacol borane 68 in combination with less hindered nucleophiles, although evidence for the formation of borenium salts is limited to 11B chemical shifts of minor solution species suggesting partial conversion to structures assigned as 69a (11B δ 22.2 = ppm) and 69b (11B δ = 26.4 ppm).45
Scheme 3.

3.4.2 Hydride abstracton by trityl salts
An older method for hydride abstraction using trityl cation as the “hydridophile” was explored and updated in our laboratory. The original procedure had reported the conversion of pyridine borane into the boronium salt by reaction with trityl tetrafluoroborate in the presence of pyridine.46 No intermediates in this process were mentioned, but the boronium salt [Py2BH2]+ BF4− was isolated and characterized. First attempts in our laboratory to perform an analogous reaction starting from a cyclic amine borane complex 70 (Scheme 3) utilized trityl tetrakis[(3,5-bistrifluoromethyl)phenyl]borate 71a in an attempt to generate the borenium salt 72a.47a No borenium species were detected, and the main product proved to be the unexpected fluoroborane complex 73, suggesting self-destruction by the highly reactive salt 72a. When a similar experiment was performed using the more robust trityl tetrakis(pentafluorophenyl)borate 71b as the electrophile, some evidence for the generation of borenium ions was indeed observed in the form of a transient NMR signal at 11B δ = 39 ppm, but the reaction was difficult to control and the evidence was not conclusive.47a A subsequent re-investigation using rigorously anhydrous conditions observed a more strongly deshielded borenium signal at 11B δ = 59 ppm, and demonstrated that this intermediate is the desired 72b by hydride quenching to re-generate 70.47b The more recent study also confirmed the conversion from 72b into a second borenium species having the previously observed signal at 11B δ = 39 ppm upon addition of one equivalent of water. Thus, initial experiments had detected 74 instead of 72b due to contamination by water. High reactivity for 72b is no surprise because the borenium subunit is stabilized only by the π-electrons of an appended benzene ring. As expected, 72b reacts readily with good nucleophiles including Bu4NBH4 (to form 70) or pyridine (to form the pyridine adduct boronium salt), but 72b also reacts with the usually robust tetrakis(pentafluorophenyl)borate anion in a self-destruct mode. The latter process is very slow at room temperature, but affords the B-pentafluorophenyl adduct 75 in 46% yield after 24 h in refluxing benzene, followed by quenching with Bu4NBH4.
3.4.3 Hydride bridged borenium salt dimers
Attempts to extend the hydride abstraction method to simple borane compexes 76 (L= amine or phosphine) have encountered a more complex situation (Scheme 4).48,49 In all cases explored to date using the trityl salt 71b, the hydride abstraction process is quite fast at low temperatures, and triphenylmethane is formed as expected.48 However, the reaction stops after consumption of 50 mol% of 71b due to the formation of an observable intermediate 78 (11B δ 0 to −3 ppm if L= tertiary amine or pyridine; ca. −27 ppm if L= tertiary phosphine) consisting of two borenium subunits linked by a hydride. This cationic structure features a symmetrical three-center two-electron (3c2e) B—H—B bond, and is formed by the interaction of 76 as an electron donor with the formally empty p-orbital of the hypothetical borenium ion 79 as the acceptor. Although mechanistic details of the hydride abstraction step have not been studied, initial interaction between 76 and 71b may involve a 3c2e intermediate or transition state 77. Nucleophilic attack by the starting borane complex (76) may then be feasible to displace triphenylmethane as the leaving group, resulting in a direct pathway to the symmetrical hydride-bridged “dimer” 78, but dissociation of 77 to borenium ion 79 is an alternative that has not been ruled out. Mechanistic ambiguities are also reflected in the reactivity of 78 with weak nucleophiles. For example, solutions of 78 (L = triethylamine) generated in dichloromethane decompose at rt over several hours to form a complex mixture of products after quenching with borohydride. The volatile products include toluene and diphenylmethane among several fragments derived from the triphenylmethane that is generated in the hydride abstraction step. Apparently, these products are the result of a Friedel-Crafts alkylation of triphenylmethane at the ipso-carbon by the boronium salt 80 to form benzyl chloride and diphenylmethyl cation 81, followed by hydride transfer. Thus, dichloromethane may be sufficiently nucleophilic to interact with 78, thereby generating an equilibrium concentration of 80. However, prior dissociation of 78 to the borenium ion 79 is not ruled out, and neither 79 nor 80 has been detected by spectroscopy. Similar questions arise in the conversion of 78 into observable tetracoordinated adducts 82 (complete conversion with X= OTf; partial conversion with X= NTf2). In the absence of decisive evidence, species such as 77, 78, or 80 were regarded as equivalents (and potential sources) of the presumably higher energy borenium ion 79,47b but direct involvement by 79 as a solution intermediate remains an open question. On the other hand, there can be no doubt regarding the relatively greater stability and structure of the hydride-bridged “dimers”, one of which (78 with L = NMe3) has been characterized by X-ray crystallography.50 Thus, the trityl cation method for hydride abstraction appears not to be suited for generating observable non-stabilized primary borenium ions.
Scheme 4.

Generation of borenium ion equivalents by hydride abstraction48–50
In an independent study,49a a variation of the hydride abstraction method was developed with similar substrates using a strong acid as the activating electrophile. Thus, treatment of ammonia borane (76 with L = NH3) with HOEt2+ B[C6H3(CF3)2]4− in ether resulted in hydrogen evolution and the formation of an isolable boronium salt 83. This substance was presumably formed by nucleophilic attack by the ether solvent on the intermediate borenium ion, analogous to the conversion from 79 to 80, and was characterized by the characteristic 11B chemical shift (δ = 0.21 ppm) as well as analytical data. Further conversion from 83 to hydrogen gas and oligomeric aminoboranes was rationalized via the formation of the hydride-bridged structures 84 and 85 as hypothetical intermediates, and DFT calculations were used to support the proposal that 84 corresponds to an energy minimum in this sequence.49a
3.5 Borenium ion generation by nucleophilic addition-heterolysis
One additional method for generation of borenium salts is considered in Scheme 5, involving a nucleophilic addition-heterolysis pathway from the reaction of a nucleophile and a trivalent boron substrate that contains a potent leaving group.1 Nöth has used the shorter descriptor “nucleophilic addition” for the same method, but we prefer the longer label because the process involves two stages, (1) reversible formation of a tetracoordinate boron intermediate, and (2) partial or total dissociation to release the borenium salt. Well-established examples of this approach involving amine-stabilized borenium ions were already discussed in connection with Scheme 1. However, examples where the borenium ion is not stabilized by n-electron donors may also have been encountered.51 Thus, the triflate derivative of 9-BBN (86) was treated with 2,6-lutidine in dichloromethane to afford a solution having two 11B signals at δ = 59 and 37 ppm (1:8 ratio). In contrast, pyridine reacted with 86 to afford the isolable tetracoordinate adduct 88a in 65% yield (11B δ = 13.4 ppm). This result implicates steric hindrance as the reason why 88b does not accumulate in solution. If the 59 ppm signal observed from 86 + 2,6-lutidine is due to a contaminant,51b then the 37 ppm signal may represent a time-averaged mixture containing the borenium salt 90, the adduct 88b, and the reactants in equilibrium.51a A similar reaction of Bu2BOTf with 2,6-lutidine gave a time averaged signal at 11B δ = 55 ppm, and ligand exchange with excess lutidine was demonstrated at room temperature. This indirect evidence was not discussed in depth, but more definitive data were obtained using the method of halide abstraction starting from the pyridine adduct 89 obtained from 9-BBN-Cl (87). Addition of the potent chlorophile gallium trichloride to 89 resulted in a solution having three signals in the 11B NMR spectrum at δ = 69.7, 58.5, and 6.1 ppm. The latter signal was attributed to unreacted 89, while the 69.7 ppm signal was assigned to the borenium tetrachlorogallate 91.51a Assuming a similar chemical shift for 90, the latter may be a minor component of the equilibrium reaction mixture from 86 and 2,6-lutidine. Borenium salts 90 and 91 are presumably stabilized to some extent by the pyridine π-system, and 90 should also benefit from the steric shielding effect of a 2,6-lutidine substituent. Nevertheless, these structures would rank among the least stabilized borenium salts to be detected by NMR methods.
Scheme 5.

Minimally stabilized borenium ions by nucleophilic addition-heterolysis51,52
Recent experiments in our laboratory have extended the nucleophilic addition-heterolysis method to p-dimethylaminopyridine (DMAP) as the nucleophile.52 In contrast to the reaction of pyridine with 9-BBN triflate (86), the analogous reaction of DMAP with 9-BBN bistriflimide 92 afforded a solution having two 11B signals that do not interconvert on the NMR timescale, corresponding to the borenium salt 93 (11B δ = 66.5 ppm; minor) and the isolable boronium salt 94 (11B δ = 3.0 ppm; major). Support for structure 93 was obtained by generating the same δ = 66.5 ppm signal by an independent method via hydride abstraction from 95 with HNTf2, a process that gave 93 as the dominant solution species (>85% of 11B signal intensity). The chemical shift of 93 is consistent with the value reported for 91 (11B δ = 69.7 ppm),51a and provides additional support for the original interpretation of Narula and Nöth.
In view of the complications encountered in the pyridine series, it was surprising to find that the reaction of 92 with triethylamine is relatively simple (Scheme 5).52 A single dominant product, the borenium salt 97, was observed in solution according to chemical shift data (11B δ = 85.1 ppm) while the initially formed nucleophilic addition product 96 was not detected. Evidently, the heterolysis step from 96 is strongly favored by the resulting decrease in non-bonded interactions, and also by the stability of the bis-triflimide leaving group. Structure 97 is unique among all of the borenium salts that have been observed in solution to date because it contains neither n-electron nor π-electron donors capable of stabilizing boron. Its relatively facile generation undoubtedly is due to steric protection by the 9-BBN ring system, a factor that not only favors the heterolysis step from 96, but also prevents boronium adduct formation with unreacted triethylamine. On the other hand, the steric effect is not so large that boronium adduct formation with tertiary amines can be disregarded. Thus, 92 reacts readily with 1,8-bis(dimethylamino)naphthalene to give the isolable boronium adduct 99 (11B δ = 16.2 ppm). As discussed later (Section 7), 99 is unusually reactive compared to more typical boronium salts, suggesting facile equilibrium with the borenium salt 98 or with other potentially electrophilic species. However, the chemical shift and X-ray data support the tetracoordinate boron structure.
4. Borenium Lewis acidity vs. structure
4.1 Experimental evaluation of Lewis acidity
Borenium ions are stronger Lewis acids compared to typical boranes due the combined effect of the net positive charge and a formally vacant p-orbital at tricoordinate boron, but how much stronger are they? There is no simple way to answer this question. Judging from the experimental data presented in Sections 2 and 3, the degree of Lewis acidity of borenium ions spans a rather wide range, depending on the nature of substituents attached to boron. Thus, if the ability to form covalent bonds to counterions is used as a rough measure of Lewis acidity, then the two representative limiting cases would be (1) the aromatic cation 58 which resists complexation with the relatively nucleophilic chloride anion,40,41 and (2) the powerful electrophile H2BNMe3+ (25) that covalently binds even to the weakly coordinating bistriflimidate anion to form the neutral adduct 82 (X = NTf2; Scheme 4).50 However, the Lewis acidity of borenium ions depends on the steric as well as the electronic availability of the vacant p-orbital on boron. Thus, the borenium cation 97 (Scheme 5) does not bind the bistriflimidate counterion even though it lacks stabilizing π or n electron donors, nor does it bind excess added triethylamine.52 The more highly substituted 97 would have additional stabilizing hyperconjugative interactions with proximal σ-bonds compared to 25, but severe steric crowding is a major factor that makes 97 a weaker Lewis acid than 25. In contrast, the relatively unhindered borenium cation 58 (eq. 11) is unlikely to interfere sterically with an incoming nucleophile such as chloride ion, but 58 does not undergo conversion to the tetracoordinate boron adduct because the boron p-orbital is strongly populated by involvement in the aromatic π-system. In this case, electronic factors are dominant.
While most reports on borenium ion chemistry contain qualitative descriptors of Lewis acidity (such as the ability to coordinate counterions or other Lewis bases), comparisons of thermodynamic data that would help to rank the Lewis acidities for a broad range of borenium species have been rare. Such comparisons would be informative if performed using the same reference Lewis base to probe the equilibrium between tricoordinate and tetracoordinate boron species, but the literature data are very limited, while data obtained using spectroscopic evaluation of tetracoordinate boron adducts are also limited and rather difficult to compare. Estimates of Lewis acidity for borenium ions based on NMR methods have been reported in some cases. Thus, Piers et al. investigated the effect of complexation by borenium species 52 and 53 (Scheme 2) on the 1H chemical shift for the β-proton of crotonaldehyde (Child’s test), and concluded that the Lewis acidities of 52 and 53 are mutually close and comparable to those of Et2AlCl and BF3.38 In a somewhat different test involving non-equilibrium conditions, Ingleson et al. assessed the Lewis acidity of the hypothetical cation 62 (eq. 12) by measuring the 31P chemical shift of the Et3PO adduct 60 (δ = 106.9 ppm) and comparing this chemical shift with the corresponding values for the Et3PO adducts of several reference Lewis acids including (C6F5)3B (δ = 76.6 ppm), AlCl3 (δ = 80.3 ppm), and BBr3 (δ = 91.2 ppm).42 The chemical shifts for the three reference Lewis acids increased in the same order as their Lewis acidity determined from earlier studies using Child’s test. This correlation suggests that the non-equilibrium 31P chemical shifts may also be used to estimate relative Lewis acidities. Unfortunately, the standard Child’s test (complex formation with crotonaldehyde in CD2Cl2) could not be performed with 62 due to its incompatibility with the solvent. This problem was avoided in C6D6 solution, but the crotonaldehyde chemical shift data obtained for 62 in C6D6 did not show a simple correlation with the reference data in CD2Cl2, and correlation data for the same solvent were not reported.
The simpler test based on the 31P chemical shift of 60 is consistent with the notion that 62 is a very powerful Lewis acid that is probably more potent than the neutral boron Lewis acids B(C6F5)3 and BBr3. On the other hand, Lewis acidity estimates based on the 31P chemical shift criterion would be influenced by other factors resulting from structural changes near the boron subunit. Thus, the 31P chemical shifts for the Et3PO adducts corresponding to the hypothetical, non-stabilized borenium cation H2BNMe3+ (25) and the observable π-stabilized “borabenzylic” cation 72b (Scheme 3) happen to be identical (δ = 85.7 ppm in CD2Cl2) despite the apparent difference in stabilization.50 In view of these findings, more definitive experimental methods to probe the Lewis acidity of borenium species are needed, and remain to be developed.
4.2 Computational evaluation of Borenium Lewis acidity
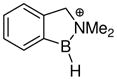
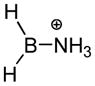
In an early computational attempt to gain insight regarding the factors that influence the bonding, stability, and Lewis acidity of boron cations, Nöth, Bursten et al. studied a series of cations R2BL+ (R = H, NH2; L = H2O, pyridine, NH3, etc.) using ab initio methods.53 Two different geometries were compared for the L = pyridine case, and the fully planar structure was found to be favored by 12.6 kcal/mol compared to the geometry having the pyridine ring turned perpendicular to the plane of the boron σ-bonds. This energy difference was associated with a π-delocalization effect, the same stabilizing interaction involving delocalization between the pyridine ring with the boron p-orbital that had been deduced in earlier studies.2 The ab initio investigation also explored the dissociation of the borenium cation H2BNH3+ (100) into the simple components (the borinium ion H2B+ and ammonia).53 This conversion was discussed in the context of heterolytic bond dissociation, a process that reflects the enthalpic component for B–N bond formation in the reverse reaction (ammonia + H2B+ as the Lewis acid), corresponding to the ammonia affinity of H2B+. Subject to the usual caveats regarding the evaluation of condensed phase phenomena using computed gas phase energies, this general approach offers a potential way to estimate NH3 affinities of other boron cations, including labile borenium ions that are difficult to compare under standardized solution conditions.
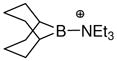
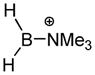
An extrapolation from the above precedent has been performed with the goal of ranking a series of borenium ions according to their gas phase NH3 affinities.50 The range of borenium ions studied (Table 1) includes several structures of synthetic interest (Lewis acid catalysts; electrophilic borylating agents). It also represents various bonding environments for boron, and includes most borenium examples characterized by X-ray crystallography during the past decade to allow the comparison of computed and experimental geometries. The sterically congested structures of observable borenium ions often pose obstacles for computational modeling due to the presence of significant non-bonding interactions that can be problematic for the most familiar DFT methods. On the other hand, Truhlar’s parametrized M06-2X functional54 has been found to perform well for very hindered boronium salts52 and various amine borane complexes,55a and thus was chosen for the NH3 affinity calculations. While the calculations disregard additional factors such as solvation, ion pairing, and conformational effects, a qualitative comparison of borenium Lewis acidities should still be possible. The data presented in Table 1 show that in the cases where reliable X-ray crystallographic data are available, the calculated bond lengths are in reasonable agreement with the experiment. Only qualitative enthalpies can be obtained from these computational results, but some trends deserve attention. Thus, Piers’ borenium ions 52 and 53 are predicted to have comparable Lewis acidities (entries 7,8), well in accord with the reported data.38 Moreover, the NH3 affinities of 52 and 53 are close to the NH3 affinity of BF3 calculated using the same method (ΔH = −20.4 kcal/mol), consistent with the Lewis acidities determined by Piers using Child’s test.38 As expected, borenium ions 25 or 100 having the highest NH3 affinity are sterically unhindered, and experience relatively little electronic stabilization by the ligands due to the absence of n- or π-donors (entries 13,14). The 9-BBN-derived cation 97 also lacks n- or π-donors, but the calculated NH3 affinity is rather modest (entry 4). The explanation for this contrast lies partly in the greater degree of boron substitution in 97 and the resulting hyperconjugative stabilization compared to 25, but steric hindrance may be even more important. One consequence of the highly hindered environment is apparent in the substantially longer B–NEt3 bond compared to the B–NH3 bond in the ammonia adduct, evidence that the larger ligand encounters severe steric strain. More generally, the above example reflects the elongation of bonds to the boron atom that occurs upon complexation with the Lewis base, and the pronounced moderating effect of steric hindrance on borenium NH3 affinity.
Table 1.
| entry | structure |
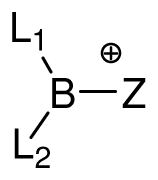
|
borenium cation | NH3 adduct L1L2B(Z)NH3 (99) | ΔHc | |||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| B-L1, Å | B-L2, Å | B-Z, Å | B-L1, Å | B-L2, Å | B-Z, Å | B-NH3, Å | ||||
| 1 | 2729 |
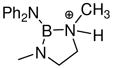
|
1.408 1.412 |
1.395 1.386 |
1.576 1.547 |
1.489 | 1.469 | 1.659 | 1.706 | −2.3 |
| 2 | 4935 |
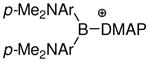
|
1.541 1.550 |
1.541 1.532 |
1.533 1.501 |
1.616 | 1.620 | 1.619 | 1.689 | −9.2 |
| 3 | 4835 |
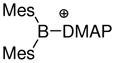
|
1.553 1.560 |
1.553 1.570 |
1.517 1.480 |
1.626 | 1.623 | 1.610 | 1.682 | −13.3 |
| 4 | 9750 |
|
1.559 | 1.555 | 1.571 | 1.618 | 1.621 | 1.712 | 1.660 | −13.6 |
| 5 | 5036 |
|
1.551 1.562 |
1.551 1.560 |
1.602 1.579 |
1.636 | 1.631 | 1.648 | 1.688 | −15.1 |
| 6 | 6744 |
|
1.368 1.369 |
1.369 1.373 |
1.945 1.933 |
1.438 | 1.440 | 2.015 | 1.665 | −17.4 |
| 7 | 5338 |
|
1.438 1.404d |
1.181 - |
1.438 1.402d |
1.522 | 1.196 | 1.520 | 1.664 | −18.8 |
| 8 | 5238 |
|
1.439 1.429d |
1.323 1.414d |
1.439 1.430d |
1.520 | 1.379 | 1.520 | 1.659 | −18.9 |
| 9 | 6042 |
|
1.377 1.381 |
1.360 1.372 |
1.386 1.374 |
1.443 | 1.438 | 1.474 | 1.631 | −23.6 |
| 10 | 3831 |
|
1.534 | 1.340 | 1.576 | 1.592 | 1.423 | 1.664 | 1.659 | −25.5 |
| 11 | 5539 |
|
1.385 1.380 |
1.385 1.380 |
1.385 1.380 |
1.466 | 1.466 | 1.466 | 1.650 | −27.6 |
| 12 | 7246 |
|
1.504 | 1.183 | 1.559 | 1.587 | 1.199 | 1.627 | 1.633 | −34.8 |
| 13 | 2548 |
|
1.181 | 1.181 | 1.528 | 1.198 | 1.198 | 1.604 | 1.620 | −48.8 |
| 14 | 10050 |
|
1.178 | 1.178 | 1.554 | 1.194 | 1.194 | 1.615 | 1.615 | −55.6 |
Counterpoise-corrected heterolytic association enthalpies (298.15 K) determined using the M06-2X/6-311++G(3df,2p)//M06-2X/6-31+G(d,p) level of theory;50, 54 solvents and counterions were not considered in the computation.
Bold numbers indicate bond lengths taken from X-ray crystal structure determination as cited.
ΔH corresponds to the enthalpy for [(cation + NH3)−(ammonia adduct)] in kcal/mol
X-ray data for 52 and 53 may be imprecise due to disorder as reported by Piers et al.38
Nitrogen n-donor substituents at boron substantially decrease the NH3 affinity of borenium ions, as exemplified by structure 27 (entry 1). Related borenium cations possessing oxygen n-donors such as cation 67 (entry 6) or Corey’s Diels-Alder catalyst 38 (entry 10) are somewhat more Lewis acidic, apparently due to the increased electronegativity of oxygen compared to nitrogen. The substantial difference between 67 and 38 in terms of NH3 affinity remains unexplained, although a reviewer has suggested a plausible rationale that 67 should be a weaker Lewis acid because it is part of a delocalized 10 π (aromatic) electron system. In any case, 38 clearly is a potent Lewis acid, as also expected from its catalytic reactivity. Similar oxygen electronegativity effects on NH3 binding energies were noted for dicoordinate boron cations (borinium ions) by Nöth, Bursten et al. in their computational study,53 and the general trends were recognized in earlier experimental work.2
Increased NH3 affinity also correlates with the presence of a larger number of electronegative atoms in the conjugated π-systems attached to boron (entries 7, 8, 11), but other factors are difficult to evaluate in these more complex examples. Thus, cations 52 and 53 (entries 7,8) are formally antiaromatic 12π electron systems, a factor that should also enhance NH3 affinity. However, Piers et al. found no evidence for substantial antiaromaticity using the nucleus-independent chemical shift criterion NICS(1),38 precluding the unambiguous assessment of antiaromaticity and electronegativity vs. simple n-delocalization from the nitrogen substituent attached to boron in cations 52 and 53.
The calculated NH3 affinities are generally consistent with the empirical comparisons of borenium Lewis acidities as discussed in Sections 1–3. To a first approximation, the ordering of calculated NH3 affinities may reflect the Lewis acidities of borenium ions toward other nucleophiles, provided that they are relatively unhindered. However, the structure of the test nucleophile is certainly important. One telling observation is the fact that 97 (Table 1, entry 4) does not form an adduct with triethylamine. If triethylamine had been selected as the test nucleophile for Table 1, then 97 would have to be classified as a weak Lewis acid, a ranking that would be somewhat at odds with reactivity trends to be discussed in Section 8.
The most striking feature of Table 1 is the broad range of ΔH values, differing by >50 kcal/mol from the weakest to the strongest Lewis acid (from entry 1 to entry 14). By comparison, the NH3 affinities for simple borane derivatives (tricoordinate boron lacking any formally positive substituent) span a much narrower range. Thus, the same computational approach gave the following NH3 affinities for several boranes: BMe3 (ΔH = −14.3 kcal/mol), BF3 (ΔH = −20.4 kcal/mol), BCl3 (ΔH = −25.3 kcal/mol), BH3 (ΔH = −27.9 kcal/mol), values that are in good agreement with earlier computational and experimental studies.55 Because the inherent NH3 affinities are much larger for borenium ions compared to boranes, the moderating effect of stabilizing substituents is also larger, and this trend is evident in Table 1.
5. Borenium ions and stereogenic boron
5.1 Racemization by heterolysis
The preceding sections establish correlations between empirical, computational, and spectroscopic data pertaining to the relative stabilities of borenium salts and the isomeric (tetra-coordinated) amine boranes. With very few exceptions, the amine borane isomers have proved to be more stable. The computational evidence provides a qualitative basis to answer the question “How much more stable?”, and there are also some answers to this question from experimental evidence. Thus, a classical study by Ryschkewitsch and Garrett succeeded in the first resolution of a tetra-coordinated, stereogenic boron cation using crystallization methods starting from 102, the halogenation product of the boronium salt 101 (Scheme 6).56 Challenging aspects of this work included controlling the homogeneity and solubility of the intermediate boronium salts involving the manipulation of up to three different anions after halogenation, depending on the halogen source (Z = I−, ICl2−, PF6−, Br3−, Br−). Solubility is especially important in the key anion metathesis from racemic 102 (Z = Br) and the chiral tris(catecholato)arsenate anion As(C6H4O2)3−, leading eventually to the single enantiomer of the boronium hexafluorophosphate 103 (arbitrary absolute configuration shown). Survival of 103a or 103b in the solid state and in solution without racemization rules out the heterolysis events leading to achiral borenium cations 104 (by trimethylamine departure), 105 (by pyridine departure), or the increasingly improbable dication 106 (by bromide departure). In the most extreme test of configurational stability, the non-racemic B-chloro analog 103a was recovered unchanged from boiling HCl, but racemization did occur upon warming with pyridine. The mechanistic aspects of racemization by pyridine were not discussed, but plausible rationales might include reversible nucleophilic attack by pyridine at tetra-coordinate boron.
Scheme 6.

Stereogenic boron structures containing potential leaving groups56,57
A systematic investigation of potentially relevant mechanistic issues was reported many years later for a different chiral substrate.57 Toyota et al. were able to separate boronium salt enantiomers 107 and ent-107 using hplc methods (Scheme 6). Racemization did not occur at room temperature, but warming in heptane resulted in enantiomer interconversion. In the temperature range from 73–93 °C, the process was characterized by activation parameters ΔH≠ = 146 kj/mol and ΔS≠ = 84 J/mol-deg. The authors argued that a positive entropy of activation is in good accord with a mechanism involving reversible B–N dissociation to form the less constrained intermediate 108. The alternative formation of a borenium ion 109 would result in increased ionic character and (presumably) would lead to entropically unfavorable solvent ordering, resulting in a negative activation entropy. In a related system (107 with fluoride in place of fluoropropionate), they also found no significant rate difference when the racemization was studied in tetrachloroethane or in the more polar solvent nitrobenzene. These observations are consistent with the B–N dissociation pathway for racemization and suggest that the borenium ion 109 is not involved.
5.2 Asymmetric memory at stereogenic boron
Beginning in the 1990’s, reports from our laboratory described asymmetric memory applications for stereogenic boron that encounter similar questions about the relative ease of B–N dissociation vs. generation of borenium intermediates.58 Thus, reaction of the amidine carboxylate 110 with KBF3/TMSCl afforded a mixture of diastereomeric oxazaborolidinones 111 and 112 (Scheme 7). The initial isomer ratio varied with the experiment, but slow removal of solvent consistently gave a crystalline mass that was highly enriched in the trans diastereomer 112. Subsequent enolization of recrystallized 112 with potassium tert-butoxide followed by alkylation afforded products with high enantiomeric purity, and in some cases, with excellent diastereoselectivity as illustrated by the conversion to 114. This result requires that boron configuration is retained throughout the sequence from the reactant 112 to the intermediate enolate 113 and the final product 114.
Scheme 7.

Asymmetric memory at stereogenic boron58
The conversion from the diastereomer mixture 111 + 112 into nearly pure 112 is an example of crystallization-induced asymmetric transformation (also known as “second order” asymmetric transformation), a phenomenon that can result in a near-total preference for that diastereomer having the higher crystal lattice stability. While the crystallization process is fascinating, the current discussion is concerned more with the mechanism for interconversion of 111 and 112 in solution. Reversible B–N bond dissociation provides a simple explanation, although the increased electronegativity of substituents at boron in 111/112 compared to the cyclic amine borane 107 (Scheme 6) should work against this pathway by increasing the B–N bond energy. The alternative involvement of borenium intermediates such as 115 was therefore considered, but decisive evidence for or against this possibility was not obtained due to the limited solubility of 111/112 and the potential involvement of subtle racemization pathways.
Added mechanistic insight required a more robust and more soluble oxazaborolidinone substrate that would resist epimerization at room temperature, and that would also be easier to assay (Scheme 8). Increased stability for tetra-coordinated boron species was demonstrated by R. W. Chapman upon incorporation of an electronegative fluorine substituent into the aryl group Ar*, a modification that allowed chromatographic separation of both diastereomeric oxazaborolidinones 117 and 118.59 To facilitate the assay of equilibrium events, the structures were further modified by incorporating a chiral reporter group into the aryl substituent, remote from the stereogenic boron. This feature allows monitoring changes of configuration at oxazaborolidinone carbon as well as at boron because all of the resulting stereoisomers are diastereomers that can be distinguished by simple NMR methods. Thus, it was shown that the initially formed 4:1 ratio of cis:trans diastereomers 117 and 118 is converted into an 8:1 trans:cis ratio by epimerization at boron in refluxing toluene. The same equilibrium ratio (8:1 trans:cis, or 118:117) was obtained at room temperature upon treatment with TMSCl as catalyst, again via exclusive epimerization at boron. The catalyzed epimerization at room temperature probably involves reversible formation of borenium intermediates (119 when TMSCl acts as a fluorophile; 120 when TMSCl acts as an oxophile), while the thermal equilibration is more likely to result from reversible B–N dissociation. Both 119 and 120 would be stabilized by n-electron donation from adjacent heteroatoms (O and F, respectively), so their involvement as transient species is consistent with the stability comparisons discussed in Section 4.
Scheme 8.

Epimerization at boron and asymmetric transformation59
The base-induced equilibration of 117 using DBU in CDCl3 was also studied (Scheme 8), and gave a new trans diastereomer 121 (8:1 trans:cis = 121:117; Scheme 8).59 Separation of 121 followed by hydrolytic cleavage of the heterocycle and the amidine 123 gave phenylglycine 124 with >99.5% ee and overall inversion of carbon configuration compared to the starting materials 116 and 117. This sequence proves that the base-catalyzed equilibration starting from 117 does not affect boron configuration, and therefore does not involve borenium intermediates. Simple base-induced enolization via the chiral enolate 122 is the pathway that equilibrates the diastereomers 117 and 121.
5.3 Chiral salicylaldimine boronate complexes
The examples of Scheme 8 show that the configurational stability of stereogenic, tetra-coordinated boron is increased by an electron-withdrawing substituent (fluorine) attached to the Ar* group. This factor decreases the rate of all heterolysis events at boron, and protects against borenium ion pair formation as well as dissociation of the dative B–N bond to form neutral fragments (tri-coordinated borane and amine). Another way to stabilize tetra-coordinated boron structures is by incorporating additional fused rings that include boron and one or more of the boron ligands. This structural variation has been explored in salicylaldimine-derived boron complexes, some of which are discussed in connection with Scheme 9, below.
Scheme 9.

Epimerization in chiral salicylaldimine complexes60
Activation of KPhBF3 with TMSCl as fluorophile in the presence of the salicylaldimine 125 was found to produce diastereomeric salicylaldimine complexes 126 (trans methyl and phenyl) and 127 (cis methyl and phenyl).60 Conventional room temperature crystallization afforded 126 in 73% yield, but crystallization-induced asymmetric transformation did not occur upon slow solvent removal at room temperature, as expected if epimerization at boron is negligibly slow. On the other hand, treatment of purified 126 with 10 mol % of TMSCl in dichloromethane produced an equilibrium ratio of 5.8: 1 126: 127 after three days at room temperature. When this experiment was repeated under conditions of slow solvent evaporation, the resulting crystalline solid was found to have a ratio of 26: 1 126: 127 (96% yield from the 5.8: 1 mixture of diastereomers). This result indicates that interconversion of boron epimers is catalyzed by TMSCl, and promotes asymmetric transformation during the crystallization. Although the mechanism of epimerization at boron is not firmly established, a role for the borenium intermediate 128 is implicated in view of the observed catalysis by the oxophilic Lewis acid TMSCl. The alternative of silicon-assisted heterolysis at the phenolic B–O bond is not ruled out, but access to the relevant oxygen electron pairs adjacent to the quaternary boron would encounter steric repulsions. On the other hand, B–O heterolysis to 129 might play some role in the thermal equilibration of diastereomers observed when 126 was heated in refluxing toluene. In this case, the high energy of a borenium-like species 129 may be partially offset by delocalization involving the neutral quinoid resonance form 130. However, the alternative possibility for uncatalyzed (thermal) epimerization at boron involving B–N heterolysis must also be considered, as discussed below in a related context.
Several examples discussed in Schemes 8 and 9 demonstrate asymmetric memory (retention of configuration) at stereogenic boron in structures where no other stereogenic atom is present, as in the enolates 113 and 122. A somewhat different example of the asymmetric memory phenomenon was encountered in a related context by Hutton et al.61 Thus, warming the amino phenol 131 with phenylboronic acid and glyoxylic acid in DMF afforded a single dominant enantiomer and diastereomer of the imine complex 133 (Scheme 10; 97% isolated; > 99% ee). Because 133 no longer contains a stereogenic carbon atom, the boron configuration must already be set at the stage of an intermediate 132. Furthermore, the proton shift that converts 132 into 133 must occur much faster than potentially competing heterolysis events involving the B–O or B–N bonds. Once formed, 133 is very stable and survives heating in refluxing toluene (24h) without appreciable (<0.5%) racemization, in contrast to the analogous salicylaldimine complex 126 (Scheme 9) which does undergo thermal equilibration with the diastereomer 127 under the same conditions.
Scheme 10.

Configurational stability, conformational stability, and stereogenic boron61,62
The above results are easy to explain if thermal epimerization at boron in 126 occurs by B–N dissociation. Hutton et al. have noted that the corresponding process in 133 is more difficult because it would require not only the B–N dissociation, but also the conformational changes needed to re-orient the B–Ph and C–Ph subunits in a sterically demanding medium ring environment 134 (Scheme 10). The medium ring intermediate may also have been encountered in the somewhat more labile complex 135 studied earlier by Braun et al.62 Individual enantiomers of 135 were separated by hplc methods, and were shown to undergo thermal racemization at 65 °C in decane (ΔG≠ 109 kJ/mol). The authors did not explicitly invoke the medium ring 136 as the intermediate responsible for racemization, but did note that racemization is somewhat slower in an analogoue having a shorter, and therefore stronger, B–N bond (replace Cl in 135 by NO2; ΔG≠ 115 kJ/mol). Braun et al. also provide references to a number of simpler examples of chiral amine boranes that contain stereogenic boron. So far, there is general agreement that loss of configuration at tetra-coordinated boron in the chiral amine boranes under uncatalyzed (thermal) conditions occurs by B–N dissociation.63 On the other hand, heterolysis to generate borenium intermediates is implicated in reactions conducted in the presence of Lewis acid catalysts (Schemes 8, 9).
6. Enantioselective catalysis and chiral borenium ions
The preceding section considered chiral amine boranes containing stereogenic tetracoordinate boron. Configurational stability at boron was demonstrated in a number of examples under ambient conditions, and was optimized by adjusting electronic factors or steric constraints to a level sufficient to prevent the spontaneous generation of borenium ions. This allowed the stoichiometric use of chiral amine boranes in asymmetric synthesis applications that require memory of chirality. In a somewhat different approach to asymmetric synthesis, chiral amine boranes can also be used in a catalytic mode. This scenario typically involves the reversible generation of Lewis acidic borenium ions by leaving group departure from tetracoordinate boron in the starting amine complex. In contrast to the stoichiometric examples of Section 5, the catalytic applications require that borenium ions are generated spontaneously. Because the borenium catalyst must bind reversibly to all potential nucleophiles in the system, the substituents must be optimized to provide the desired level of stabilization. As briefly discussed in Section 3.1 and Section 4, this can be done by incorporating one or more n-electron donors as ligands at boron to moderate electrophilicity. A more detailed look at the structural requirements as well as some applications of this concept are presented in the context of enantioselective catalysis by chiral borenium sources in the following sections.
6.1 The Dual Function of Oxazaborolidines in the CBS Reduction
The first encounters with a chiral catalyst containing a borenium subunit were reported in 1983, well before the nature of the key catalytic event was understood (Scheme 11). In the course of studies directed toward enantioselective ketone reduction, Itsuno et al. reported that a reagent derived from diphenylvalinol (137) and BH3·THF gave highly enantioselective reduction of aryl ketones (≥ 94% ee).64 The 2:1 stoichiometry of BH3·THF to 137 was noted as an important variable, but the role of the second equivalent of borane was not known at the time of the initial report. During the subsequent optimization as reported in 1987, Itsuno et al. found that pretreatment of 137 with one equiv of BH3·THF at 0 °C allowed isolation of a chiral complex, but the structure of the complex was not reported.64b This complex could be used catalytically with BH3·THF for the enantioselective reduction of ketones and O-methyloximes, but the authors did not comment on the mechanism of borane activation or the nature of the hydride donor.
Scheme 11.

By 1987, Corey et al. had observed similar catalysis using preformed borane catalysts with BH3·THF for ketone reduction.65a The previously unidentified complex was assigned as the oxazaborolidine 138 (Scheme 11), based in part on the 11B NMR shift at δ = 28.1 ppm, in the range expected for trivalent boron containing nitrogen and oxygen substituents. The role of the borane adduct 139 in ketone reduction was recognized, and the related oxazaborolidine 141 (generated from 140 and BH3·THF) was shown to be a superior catalyst. This complex was effective at catalyst loadings as low as 5 mol%, and reduced ketones within 1 min at rt using BH3·THF at a reagent loading of 60 mol% relative to the substrate.
The structure of 141 as the major species in solution was supported by the 11B NMR signal at δ = 28.3 ppm, while a small signal at 7.6 ppm was assigned to a dimeric structure. When a solution of 141 in THF was treated with excess BH3·THF, two new upfield 11B signals were observed at δ = 3.2 and −19.4 ppm, values that are consistent with the presence of tetracoordinate boron. A subsequent communication from Corey et al. described 142 (11B δ = 33.5 ppm), the B-methyl analogue of 141.65b In contrast to 141, oxazaborolidine 142 reacted with BH3·THF to gene rate a new downfield signal (δ 11B = 36.5 ppm), consistent with the presence of an endocyclic tricoordinated boron nucleus as in 144. An upfield-shifted signal for the exocyclic N–BH3 resonance was also observed (δ11B = −15.4 ppm), similar to one of the signals from the borane adduct of 141. Apparently, the combination of oxygen n-delocalization as well as the steric and electronic effects of the B-methyl substituent in 144 is sufficient to favor tricoordinate boron, while the B–H analogue 143 is more electrophilic and exists mostly as the tetracoordinated THF adduct in the equilibrium mixture. Although 143 is net neutral and is not technically a borenium salt, the endocyclic boron is part of a borenium subunit that is strongly electrophilic compared to 141.
Corey et al. proposed that oxazaborolidine catalysts related to 141 play a dual activation role in the ketone reductions. Borane complexation enhances the Lewis acidity of 141 by generating 143, and promotes ketone activation via the Lewis acid-Lewis base adduct 145. However, the formation of the tetracoordinate boron species 143 and 145 simultaneously activates the borane as a hydride donor, resulting in the facile intramolecular (6-center) hydride transfer from 145 (Scheme 11). This process is feasible only if the ketone coordinates from the less hindered, convex face of the bicyclic borenium core, cis to the complexed BH3 subunit as shown in 145, and leads to 146 as the initial product of hydride transfer. Like 143, structure 146 contains a borenium subunit (the exocyclic tricoordinate BH2 next to formally positive nitrogen), but in contrast to 143, 146 has no stabilizing interaction between the ring boron with the oxygen n-electron donor ligands. On the other hand, 146 may gain some benefit from complexation between the exocyclic tricoordinate BH2 boron with alkoxide oxygen n-electrons in a 4-center arrangement. Further interaction with the BH3·THF reagent is then believed to occur via a 6-center process that eventually leads to expulsion of the reduced alkoxyborane fragment as shown. The latter incorporates a new B–O bond at the expense of the exocyclic B–O bond in 146, while transfer of hydrogen from BH3·THF to the tricoordinate boron results in regeneration of the catalyst 143.
The favored configuration of the ketone in 145 places the larger substituent (RL) exo with respect to the bicyclic core to minize steric interactions with the oxazaborolidine, and accounts for exceptional enantioselectivity over a broad range of substituents. Subsequent work has extended this methodology to allow facial discrimination even for ketones with sterically similar substituents, and a number of applications in total synthesis are also reported.32,65b These findings have been reviewed in depth32 and will not be discussed here, except to note that the CBS reduction (Corey, Bakshi and Shibata65) has become one of the most powerful methods for enantioselective ketone reduction.66 An interesting mechanistic analogy for the dual activation process has also been encountered in the oxazaborolidine-catalyzed enantioselective ethynylation of aldehydes by alkynyldimethylboranes.67 The overall events are similar, including borane complexation to oxazaborolidine nitrogen, carbonyl binding at the Lewis acidic borenium subunit, and 6-center transfer of the B-ethynyl group to the activated aldehyde carbonyl.
6.2 The role of borenium species in Diels-Alder catalysis
Chiral boranes activated conventionally by electronegative substituents have been used as catalysts for the Diels-Alder reaction, but applications are largely limited to relatively reactive cyclic dienes.68a To improve reactivity, Corey et al. explored a family of catalysts that generate borenium salts in situ.68b Thus, a bicyclic oxazaborinane 148 was formed in solution from the protected chiral amino alcohol 147 by treatment with 0.9–1.0 equiv of BBr3 at −78 °C (eq. 14).69 Indirect evidence for an equilibrium between 148 and the borenium salt 149 was provided by the observation that this mixture (“reagent A”) is a potent Diels-Alder catalyst for the reaction of acrolein derivatives with cyclopentadiene at −94 °C. However, reagent A did not catalyze the Diels-Alder reaction with less reactive acyclic dienes at −94 °C, and could not be used at temperatures above −60 °C due to decomposition. For access to more potent borenium catalysts, 148 was treated with BBr3 or AgBAr4 at low temperatures according to the method of halogen abstraction (Ar = C6H3(CF3)2; see Section 3.2 for analogies). In the BBr3 experiment, direct evidence for a shift in equilibrium toward the presumed borenium salt 150 was obtained by observing the 11B NMR signal of BBr4− anion, but no mention was made of the 11B signals for the borenium subunit of either 150 or the analogous 151 generated using the AgBAr4 procedure. The lack of direct 11B NMR evidence for tricoordinate boron is also a recurring issue with most of the oxazaborolidine-derived borenium catalysts to be discussed in the next section. This problem may well be a consequence of the low-temperature enhancement of quadrupolar relaxation that effectively quenches the 11B NMR signals of unsymmetrical boron species, a complication that should be less severe for the highly symmetrical (observable) BBr4− anion in the case of 150.70,71 The in situ generation of the borenium salt 151 (reagent B) allowed catalysis of representative Diels-Alder reactions within 1 h at temperatures as low as −94 °C, and expanded substrate scope to include the moderately reactive dienes 1,3-butadiene and 1,3-cyclohexadiene. Using α-bromoacrolein as the dienophile and 151 generated in situ, the corresponding cycloadducts 152 and 153 were obtained in high yields and with 93–94 % ee. However, the temperature requirement for experimentation at −60 °C or below somewhat limits potential applications for catalysts 150 or 151.
Eventually, the problems of borenium catalyst stability and scope were overcome by developing optimal activation procedures in the readily accessible proline-derived oxazaborolidine environment.31,72–77 The potential for Lewis acidity and carbonyl complexation had already been recognized in the context of CBS reduction using catalysts 141 or 142 (Scheme 11), but the method of activation had to be modified to avoid interference by the B–H bonds present in reagents and intermediates. At an early stage of catalyst development, Corey et al. reported that activation of 37 with the strong Brønsted acid CF3SO3H (triflic acid, TfOH) generates a powerful catalyst for the Diels-Alder reaction.31 As briefly mentioned in Section 3.1 in the context of electrophilic activation of B–N bonds by protonation, the catalytically active borenium cation 38 is present in equilibrium with the tetracoordinate boron complex 39 (Scheme 12). To gain more insight regarding the key equilibrium, attempts were made to perform a similar activation with methansulfonic acid (MsOH), but this did not afford a catalytically active source of borenium intermediates (Scheme 12). Corey et al. interpreted the outcome based on the lower acidity of MsOH compared to TfOH, and on the relatively low basicity of oxazaborolidine nitrogen resulting from the donation of n-electrons from N to B. Even if protonation had occurred, there is the added concern that increased nucleophilicity of MsO− compared to TfO− would decrease the equilibrium concentration of the catalytically active borenium salt (38 adjusted for X = OMs) relative to the covalent, tetracoordinated amine borane adduct 39 (X = OMs). Similar equilibrium considerations apply to all methods for catalytic generation of chiral, formally cationic borenium intermediates as shown in Scheme 12, including oxazaborolidine B–N protonation of 37 (to 38/39 using TfOH;31 to 155/156 using Tf2NH72) or 154 (to 157/158 using TfOH31), and protonolysis of the borohydride B–H bond of 160 using Tf2NH to give 161/162.74 However, attempts to generate 161 directly from 37 by methylation were not successful due to the low nucleophilicity at nitrogen that results from n-electron donation to boron.
Scheme 12.

Chiral oxazaborolidines as sources of borenium salts31,72,74,77
It is of considerable interest to characterize the reactive intermediates in Scheme 12 and to assay the relevant equilibria, but this has proven to be a challenging task. Among the borenium intermediates proposed, only 161 has been characterized by 11B NMR (δ = 34 ppm),74 a value that falls within the expected range given the presence of oxygen as an n-electron donor, and is shifted significantly downfield compared to the precursor 160 (δ 11B = 7.7 ppm). No NMR signals were reported for the corresponding tetravalent triflimide adduct 162, suggesting that the equilibrium strongly favors the borenium salt 161 as the only species observed. In contrast to 161/162, the nature of the other intermediates shown in Scheme 12 has been deduced almost entirely from proton NMR chemical shift evidence (Table 2), and from relative reactivity comparisons. To help interpret the NMR data, Scheme 12 and Table 2 also include the somewhat distinctive case of electrophilic B–N activation from 37 by AlBr3 as the Lewis acid.75 The resulting adduct 163 has no net charge, but contains a borenium subunit and is a highly effective Diels-Alder catalyst.
Table 2.
1H NMR characterization of oxazaborolidine borenium equilibria
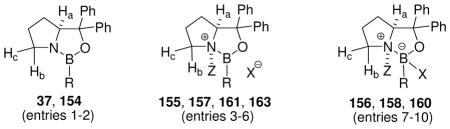
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | Structure | B-coord | R | Z | X | Ha | Hb | Hc | Ref |
| 1 | 37 | 3 | o-Tol | Lp | -- | 4.54 | 3.40 | 3.24 | 31 |
| 2 | 154 | 3 | Me | Lp | -- | 4.35 | 3.40 | 3.05 | 31 |
| 3 | 155 | 3 | o-Tol | H | NTf2 | 5.44 | NA | NA | 72 |
| 4 | 157 | 3 | Me | H | OTf | 4.85 | 3.40 | 3.20 | 31 |
| 5 | 161 | 3 | o-Tol | Me | NTf2 | 4.95 | NA | NA | 74 |
| 6 | 163 | 3 | o-Tol | Br3Al− | -- | 5.26 | 3.95 | 3.51 | 75 |
| 7 | 156 | 4 | o-Tol | H | NTf2 | 5.01,4.90b | NA | NA | 72 |
| 8 | 158 | 4 | Me | H | OTf | 4.75 | 3.15 | 3.00 | 31 |
| 9 | 160 | 4 | o-Tol | Me | H | 4.38 | 3.46 | 3.02 | 74 |
| 10 | 157·DMF | 4 | Me | H | DMF+ | 5.00 | 3.50 | 3.00 | 31 |
1H NMR data in ppm, CD2Cl2
Ha values assigned to two diastereomers
Most of the chemical shift data in Table 2 were obtained from equilibrating mixtures containing activated tetracoordinate boron species (Scheme 12) that serve as borenium precursors according to the test of exceptional catalytic reactivity in the Diels-Alder reaction. Data are presented in three categories, starting with the parent oxazaborolidines (entries 1, 2), followed by the presumed tricoordinated borenium species (entries 3–6), two of the corresponding tetracoordinated structures (entries 7–8), and two related tetracoordinated complexes for comparison (entries 9, 10; amine borane 160 and the DMF adduct 157·DMF). Because 11B NMR data were not available for most of these structures, the borenium assignments (entries 3–5) were proposed by comparing chemical shifts for Ha from the observable species in equilibrium. Assuming that the higher field shift for Ha indicates tetracoordinate boron (for example, 158, entry 8), the lower field shift would then be assigned to the corresponding borenium structure (157, entry 4).31 For the specific pair of equilibrating tricoordinate and tetracoordinate boron structures 157 vs. 158, the chemical shifts of Hb and Hc also follow the same pattern (entries 4 and 8, respectively), suggesting that the combination of formally positive nitrogen and tricoordinate boron is responsible for deshielding protons at adjacent carbons. However, in one case (entry 7; equilibrium of 155 with 156), a total of three Ha signals were observed, including two that were assigned to a tetracoordinated structure 156 and attributed to the formation of two diastereomers.72 By analogy, two diastereomers of 158 might also be present in the equilibrium with borenium salt 157. Given the similarity of chemical shifts for signals assigned to 157 and the signals of 158 (Δδ ≤ 0.2 ppm; entries 4 and 8),31 the possibility remains that both sets of signals for Ha given in entry 8 were due to diastereomers of 158. If this conjecture is correct, then the concentration of 157 may have been too low to detect. Nevertheless, 157 would still be present in equilibrium, and its intrinsic catalytic reactivity could be sufficiently large to catalyze the Diels-Alder reaction as observed.
Entries 9 and 10 are included in Table 2 to allow a broader range of 1H NMR chemical shift comparisons for tetracoordinate with tricoordinate boron in the oxazaborolidine environment. Inspection of the 1H data for all of the entries suggests that the absolute chemical shift values for Ha reflect the presence of electronegative groups at boron as much as they reflect coordination number, and there is some overlap in the range of values assigned to tricoordinate vs. tetracoordinate boron. Entry 3 (δ = 5.44 ppm) stands out as the most likely case of an observable borenium salt (155, tricoordinate B) because the chemical shift difference vs. entry 7 (156, tetracoordinate B) is relatively large (0.4–5 ppm). Also interesting is the Ha chemical shift (δ = 5.26 ppm) for the AlBr3-complexed catalyst 163. This value is close to the Ha chemical shift of 155 even though 163 has no net charge and might plausibly be involved in equilibrium with a hypothetical structure 164 (Scheme 12) where bromine is shared between boron and aluminum. According to the absolute values of chemical shifts for Ha, the assignment of a borenium structure 161 for entry 5 might be questioned because the observed Ha chemical shift of δ = 4.95 ppm is so similar to the Ha chemical shifts in entries 7 and 10. However, it is important to note that 161 has an N-methyl substituent, and that Ha in the best available comparison structure having the same substitution pattern (160, entry 9) appears well upfield (δ = 4.38 ppm).
6.3 Reactivity and scope of oxazaborolidine-derived Diels-Alder catalysts
The NMR evidence considered above supports the assignment of borenium structures for 155, 161, 163 in solution, but another important criterion is catalytic reactivity. Initial comparisons had shown that the triflate-activated catalyst 38/39 promotes the Diels-Alder reaction with modestly reactive dienes such as 1,3-butadiene, isoprene, or 1,3-cyclohexadiene, but this required the relatively potent dienophiles 2-bromoacrolein or 2-methacrolein (eq. 15, 16). Furthermore, full conversion in the cyclohexadiene/methacrolein example required 24h at −78 °C.31 According to these observations, the reactivity of the equilibrium pair 38/39 as catalyst ranks between that of 148/149 (no reaction with either 1,3-butadiene and 1,3-cyclohexadiene) and that of the tetraarylborate salt 151 (both reactions complete within 2h at −94 °C).
Comparisons of oxazaborolidine catalysts reported by Corey et al. are summarized in Table 3 for the catalyzed reactions of cyclopentadiene with several representative dienophiles (methacrolein; diethyl fumarate; cyclopentenone; 2,5-dimethylquinone), arranged in approximate order of Diels-Alder reactivity for each catalyst with the more demanding fumarate or cyclopentenone dienophiles (compare entries 3, 5, 7, 12 for fumarate; or entries 4, 8, 11 for cyclopentenone). Given the differences in diene stoichiometry, dienophile concentration, and catalyst loading, the temperature/time data provide at best a very qualitative picture of reactivity. However, it is clear that the bistriflimide-activated catalysts 170/171 or 161/162 are more reactive than the triflate catalysts 38/39 or 168/169. Overall, the fastest reactions are observed using the bistriflimide-activated 161/162, or the AlBr3-activated 163 (N-methyloxazaborolidine substrate). The latter catalyst was highly effective for promoting Diels-Alder cycloaddition with the less reactive substrates using catalyst loadings as low as 4 mol%,75 while 10–20 mol% loading was needed for similar applications using other oxazaborolidine catalysts. The improved turnover for 163 was attributed to the greater bulk of the AlBr3 Lewis acid, a factor that may prevent product inhibition of the catalyst.75 The reactivity trends are also reflected in Table 4 where a broader range of dienes and dienophiles is summarized. From the combined data of Tables 3 and 4, it is clear that the chiral oxazaborolidine catalysts provide excellent yields and enantioselectivities in many Diels-Alder reactions, and that the best catalysts offer remarkably broad scope for synthetic applications. These practical advantages for catalysts 38/39, 155/156, and 163 are reflected by the commercial availability of the precursor 37.76
Table 3.
Reactivity of catalytic borenium sources with cyclopentadiene and selected dienophiles
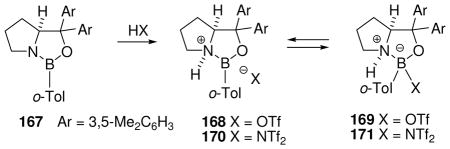
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | Catalyst | mol% | Dienophile | conc | T | time | Yield | eea | Ref |
| 1 | 38/39 | 6 | methacrolein | 0.2Mb | −95 °C | 1h | 99% | 91% | 31 |
| 2 | 168/169 | 6 | methacrolein | 0.2Mb | −95 °C | 1h | 97% | 96% | 31 |
| 3 | 168/169 | 20 | di-Et fumarate | 0.2Mc | −35 °C | 1.5h | 99% | 98% | 73 |
| 4 | 168/169 | 20 | cyclopentenone | 0.2Mc | −20 °C | 14h | 99% | 92% | 73 |
| 5 | 155/156 | 20 | di-Et Fumarate | 0.2Md | −60 °C | 2h | 99% | 99% | 72 |
| 6 | 155/156 | 20 | di-Me-quinone | 0.2Md | −95 °C | 2h | 99% | >99% | 72 |
| 7 | 161/162 | 10 | di-Et fumarate | 0.25Mc | −60 °C | 8h | 99% | 96% | 74 |
| 8 | 161/162 | 10 | cyclopentenone | 0.25Mc | −50 °C | 8h | 99% | 92% | 74 |
| 9 | 161/162 | 10 | di-Me-quinone | 0.2Mc | −78 °C | 1h | 97% | 98% | 74 |
| 10 | 163 | 4 | methacrolein | 0.5Mc | −78 °C | 2h | 99% | 93% | 75 |
| 11 | 163 | 4 | cyclopentenone | 2.0Mc | −40 °C | 1h | 95% | 92% | 75 |
| 12 | 163 | 4 | di-Et fumarate | 2.0Mc | −78 °C | 6h | 95% | 98% | 75 |
| 13 | 163 | 4 | di-Me-quinone | 0.2Mc | −78 °C | 0.5h | 99% | 98% | 75 |
ee’s refer to major cycloadduct
10:1 diene:dienophile
5:1 diene:dienophile
diene:dienophile not given; a ratio of 1.1:1 is given for a related reaction.
Table 4.
Simple Diels-Alder reactions catalyzed by oxazaborolidine-derived borenium sourcesa
| entry | Catalyst | mol% | diene | Dienophile | T °C | time | yield | eea | Ref |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 168/169 | 20 | butadieneb | Methacrolein | −78 °C | 24h | 85% | 94% | 31 |
| 2 | 168/169 | 6 | isopreneb | Methacrolein | −78 °C | 13h | 96% | 97% | 31 |
| 3 | 168/169 | 20 | c-hexadieneb | Methacrolein | −78 °C | 24h | 58% | 92% | 31 |
| 4 | 168/169 | 20 | c-pentadienec | Et Acrylate | −20 °C | 16h | 96% | >99% | 73 |
| 5 | 168/169 | 13 | c-pentadienec | Et Crotonate | 4 °C | 72h | 46% | >98% | 73 |
| 6 | 168/169 | 20 | c-pentadienec | Butenolide | −20 °C | 64h | 94% | 90% | 73 |
| 7 | 170/171 | 20 | c-pentadienec | Et Crotonate | 20 °C | 16h | 94% | 97% | 72 |
| 8 | 170/171 | 20 | butadienec | di-Et Fumarate | 20 °C | 16h | 92% | 93% | 72 |
| 9 | 170/171 | 20 | butadienec | di-Et Fumarate | 20 °C | 16h | 92% | 93% | 72 |
| 10 | 155/156 | 20 | c-pentadienec | TFE Acrylate | −60 °C | 2h | 97% | >99% | 72 |
| 11 | 163 | 4 | c-pentadienec | TFE Acrylated | −78 °C | 8h | 98% | 99% | 75 |
| 12 | 163 | 4 | isoprenec | Methacroleine | −78 °C | 16h | 99% | 97% | 75 |
| 13 | 161/162 | 10 | c-pentadienec | TFE Acrylatef | −78 °C | 1.5h | 98% | 97% | 74 |
reactions conducted in toluene or dichloromethane at a concentration of 0.2M unless noted; ee’s refer to the major isomer
10:1 diene:dienophile
5:1 diene:dienophile
concentration 2M
concentration 0.5M
concentration 0.25M
The oxazaborolidine-derived borenium source 168/169 (bis(3,5-dimethylphenyl) series; triflate anion) is an effective catalysts for modest dienophiles including α,β-unsaturated esters and ketones (Table 4, entries 4–6). However, the improved dienophile scope of catalyst 168/169 was demonstrated only for the highly reactive cyclopentadiene because catalyst decomposition was already problematic at 0 °C. On the other hand, the corresponding HNTf2 activated catalyst 170/171 was stable even at rt, allowing Diels-Alder reaction of unsaturated lactones, cyclic enones, and quinones with the less reactive acyclic dienes (Table 4, entries 8–9; see also Scheme 13).72 These findings have largely solved the problem of catalyst scope and reactivity.
Scheme 13.

Enantioselective Diels-Alder reactions catalyzed by oxazaborolidines72,74,77
The oxazaborolidine catalysts also have considerable potential for the control of regiochemistry (Scheme 13). Thus, the Diels-Alder reaction of trifluoroethyl acrylate with isoprene afforded the para isomer 172 with excellent regiocontrol using 170/171 as catalyst. However, the more challenging reaction of 2,5-dimethyl-1,4-benzoquinone 173 with isoprene produced a ca. 2:1 mixture of 174:175 as a consequence of poor discrimination by the activated dienophile complex.72 Fortunately, the corresponding iodoquinone 176 reacted selectively to give a 13:1 ratio of 177:178. This regioselectivity was attributed to preferential activation of the carbonyl group remote from iodine by the chiral Lewis acid 170 and subsequent cycloaddition at the less hindered double bond. If desired, adduct 177 can be converted to 174 by reductive deiodination, and 178 can also be used as a convenient partner in palladium-catalyzed coupling chemistry.
Excellent regiocontrol as well as useful enantioselectivity was observed in the oxazaborolidine-catalyzed 2+4 cycloadditions of diene 179 (Scheme 13). Thus, reaction with 2-methylcyclopentenone in the presence of ent-161/162 afforded 180 as the exclusive product with 82% ee.74 Adduct 180 was upgraded to 99% ee by simple crystallization, and was coverted into estrone methyl ether in several steps. In an alternative approach, the modified oxazaborolidine 180 was used to catalyze the Diels-Alder reaction of 179 with the enal ester 181 as the dienophile. In this case, the product 182 was obtained in 92% yield (94% ee), and was converted to estrone in seven steps.77 Coverage of other important total synthesis applications of oxazaborolidine-catalyzed Diels-Alder reactions appears in a recent review, along with models to explain catalyst enantioselectivity.68b
One last topic in the Diels-Alder area will be mentioned in the context of catalyst reactivity. Thus, Yamamoto et al. studied the activation of a monocyclic oxazaborolidine 183 by protonation with several acids, including MsOH, TfOH, Tf2NH, and the exceptionally strong carbon-based acid HC(C6F5)Tf2 (186, Eq. 17).78 No spectroscopic data were reported for the activated intermediates 184 or 185, but catalytic reactivity was observed in the Diels-Alder reaction of cyclopentadiene with ethyl acrylate for all acids except MsOH (Table 5, entries 1–4). The reactivity pattern was interpreted based on the generation of borenium cations 184, presumably in equilibrium with the tetracoordinate boron structures 185 as usual. In all cases where Diels-Alder reaction occurred with these catalysts, high enantioselectivity was observed, and the best ee as well as the highest yield were obtained using the highly hindered carbon acid 186 for activation.
Table 5.
Activation of oxazaborolidine 183 for Diels-Alder reaction with cyclopentadiene
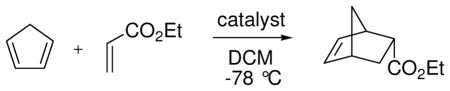
| |||||||
|---|---|---|---|---|---|---|---|
| entry | activator | mol% | dienophile | time | yield | ee | ref |
| 1 | MsOH | 5% | Et acrylateb | 1h | 0% | -- | 78 |
| 2 | TfOH | 5% | Et acrylateb | 1h | 30% | 97% | 78 |
| 3 | Tf2NH | 5% | Et acrylateb | 1h | 43% | 97% | 78 |
| 4 | 186 | 5% | Et acrylateb | 1h | 73% | >99% | 78 |
| 5 | SnCl4 | 1% | methacroleinc | 2h | >99% | 95% | 81 |
| 6 | SnCl4 | 10% | Et acrylatec | 16h | 96% | 95% | 81 |
| 7 | SnCl4 | 10% | di-Me-quinonec | 1.5h | 94% | 99% | 81 |
experiments were conducted in DCM, [0.2M] in dienophile at −78 °C
5:1 diene to dienophile
3:1 diene:dienophile
The yields of Diels-Alder products obtained under standardized conditions (Table 5, entries 1–4) were found to increase as the coordinating ability of anion X− derived from the HX used in the activation step decreases. This order of catalyst reactivity follows the same general trend as deduced by Corey et al. from a combination of NMR evidence and optimization data. Assuming that Tf2NH is already strong enough to mostly protonate the oxazaborolidine substrates as indicated by the NMR data for Corey’s bicyclic oxazaborolidine 161, then greater Diels-Alder reactivity using Yamamoto’s carbon acid HC(C6F5)Tf2 (186) for activation can be taken as evidence that the hindered anion of 184d (−C(C6F5)Tf2) is less coordinating compared to bis-triflimidate79 (Tf2N−) and more resistant to formation of the covalent adduct 185d. In any event, the excellent reactivity of Yamamoto’s catalyst can be exploited for demanding Diels-Alder applications, including the regioselective cycloaddition of 2-alkylcyclopentadienes starting with mixtures that contain the 1-alkylcyclopentadiene isomer,78 and also the catalyzed cycloaddition of sensitive α,β-acetylenic ketones with acyclic 1,3-dienes.80
Yamamoto et al. have also explored the electrophilic activation of 183 using SnCl4 (Table 5 entries 5–7; eq. 18).81 In the best experiments, the presumed Lewis acid-Lewis base adduct 187 was an effective catalyst at a loading of 1% relative to the dienophile. Catalysis was also observed using a deficiency of SnCl4 relative to 183, even at ratios as low as 1:4 SnCl4:183. This finding prompted a study of the effect of Lewis basic impurities on the catalytic efficiency of 187. Little change in yield or ee was observed upon addition of 5 mol% of water, iPrOH, EtOAc or even DMF to the reaction mixture, and practical results were obtained by performing the reaction in unpurified DCM at −78 °C. The high reactivity of 187 is reminiscent of the behavior of 163 in the bicyclic oxazaborolidine series. Several other Lewis acids such as AlCl3 or TiCl4 could also be used, but the choice of Lewis acid is important. Thus, the BF3·OEt2 adduct was not an effective catalyst starting from oxazaborolidine 183, and a similar observation was reported in the bicyclic series (188) in the prior study.75
6.4 Miscellaneous uses of chiral borenium Lewis acid catalysts
The Diels-Alder applications considered in Sections 6.2–6.3 rely on Lewis acid (borenium) complexation at dienophile carbonyl oxygen. The same phenomenon has been exploited in several other Lewis acid catalyzed reactions where the borenium cation serves as an oxophilic activating agent. This topic will be considered only briefly because most of these synthetic applications have not raised new structural or characterization issues that directly reflect on the properties of borenium catalysts. Nevertheless, some of the examples reveal interesting subtleties about the details of Lewis acid catalysis. For example, the Tf2NH-activated catalyst 155/156 has been used to catalyze the Mukaiyama-Michael reaction of a ketene trimethylsilyl acetal 189 (eq. 19),82 resulting in the expected 1,4-addition product 191 with good enantioselectivity. On the other hand, ketone-derived enol silanes 192 were shown to react with trifluoroethyl acrylate in the presence of catalyst 163 to give the cyclobutane product 193 (98 % ee; eq. 20).83 This latter reaction has been described as an asynchronous 2+2 cycloaddition, while the related process of eq. 19 is believed to involve an ionic intermediate 190 resulting from Lewis acid-catalyzed 1,4-addition, followed by silyl transfer. However, catalyst 155/156 could not be used in the 2+2 cycloaddition (eq. 20) due to decomposition involving the silyl enol ether.
A mechanistically related synthesis of α-iodo-α,β-unsaturated esters 196 from benzaldehyde and acetylenic ester 194 has been reported using catalyst 170/171 in combination with TMSI (eq. 21). The process involves formation of a nucleophilic silyl ketene acetal 195 in situ, followed by Mukaiyama aldol reaction with the catalyst-benzaldehyde complex.84
Oxazaborolidinium triflimidate catalyst 170/171 has also been used for the enantioselective cyanosilylation of aliphatic as well as aromatic aldehydes, while the analogous triflate catalyst 168/169 gave better results for the cyanosilylation of ketones (eq. 22).85 This application relies on an unusual nucleophilic cyanide equivalent, the isonitrile 197, generated in situ from Me3SiCN and methyldiphenylphosphine oxide. Because 197 is thought to be in equilibrium with its precursors, it is remarkable that the borenium catalyst 168/169 is an effective Lewis acid for carbonyl activation despite the presence of nucleophilic phosphine oxide as well as the isontrile 197. The cyanosilylation of ketones was quite slow, and required many days at temperatures in the range of 25–45 °C. Nevertheless, useful enantioselectivity was observed for the product 198a obtained from acetophenone, and especially for the triflate analogue 198b (95% ee). Details of the enantioselective transition state geometry are beyond the scope of this review, but can be found in the full paper along with a number of related cyanosilylation examples.85b The triflate substituent of 198b is uniquely suited for palladium catalyzed coupling applications, so the sequence of Eq. 22 potentially provides access to a range of non-racemic p-substituted benzene derivatives containing a quaternary carbon.
Some years earlier, Corey and Cimprich had reported a procedure for the enantioselective ethynylation of aldehydes using a monocyclic oxazaborolidine catalyst 199 (eq. 23).67 Although the conversion implies some similarity to the cyanosilylation of eq. 22 because both reactions involve an sp-hybridized nucleophile and an oxazaborolidine Lewis acid for carbonyl activation, the proposed mechanism for eq. 23 is quite different and proceeds by a fascinating dual activation sequence reminiscent of the CBS reductions (Section 6.1). Thus, a Lewis acid-Lewis base interaction between the alkynylborane and the oxazaborolidine 199 generates the exocyclic B–N bond and borenium subunit, followed by aldehyde complexation to form the crucial intermediate 200. The activated, exocyclic tetracoordinate boron then delivers nucleophilic alkyne for attack at the activated, boron-complexed carbonyl group to afford the propargylic alcohol after aqueous workup.
7. Miscellaneous applications of borenium Lewis acids
7.1 B–O activation
Apart from the enantioselective applications using the powerful oxazaborolidine-derived catalysts presented in the previous sections, there have been relatively few studies addressing boron cations as Lewis acid catalysts. In one informative example, protonation of the neutral, salicylaldimine-derived complex 201 with TfOH/THF resulted in an isolable product (Scheme 14).86 The 11B NMR signal at δ = 3.9 ppm was consistent with tetracoordinate boron, and narrowed the choice of plausible solution species to 203 or 204. However, 1H NMR data revealed the presence of bound THF, confirmed by elemental analysis, thus ruling out the simpler structure 204. Presumably, the tricoordinate (borenium) salt 202 is formed from 201 by protonation and heterolysis of the B–OCH3 group, followed by solvation to give the THF adduct 203. Formally, 203 is a boronium salt because it contains tetracoordinate boron in the cationic subunit, but facile heterolysis of the exocyclic B–O bond would be expected to release the weakly bound THF in solution, resulting in facile equilibrium with the higher energy borenium salt 202. Therefore, 203 functions as an in situ source of 202, and can be used as a Lewis acid catalyst. One example has been reported using 203 as pre-catalyst to induce the polymerization of propylene oxide. The authors proposed initiation via 205, the Lewis acid-Lewis base adduct of 202 and propylene oxide. The subsequent propagation steps were not investigated in detail, but the authors considered possible explanations involving ligand B–O or B–N cleavage. Another rationale to consider for the propagation steps is a conventional sequence of SN2 attack by monomer (propylene oxide) at the less substituted oxiranium C(3) carbon in 205, followed by repetitive SN2 attack by propylene oxide on the oxiranium terminus C(3) in a growing chain.
Scheme 14.

Generation of borenium salt 201 and polymerization catalysis86
Other Lewis acid catalyzed reactions have been rationalized by invoking intermediates that contain a borenium subunit in a neutral structure.87–90 Thus, Evans et al. observed that samarium (III) iodide catalyzes the hydroboration of 3-pentenol (206) with catecholborane (65), resulting in an 11:1 ratio of the diols 207:208 after oxidative workup (eq. 24). Among other mechanisms considered for the activation of 65, Evans et al. noted that formation of the borenium-like Lewis acid-Lewis base complex 209 should increase reactivity at the boron center. However, the alternative possibility of net hydroboration via intervention of metal hydride species was not ruled out.
In a somewhat different application of Lewis acid B–O activation, Hall et al. evaluated Sc(OTf)3 as a catalyst for the crotylboration of aldehydes with the (Z)- or (E)-crotylboronates 210 (Scheme 15).88 Formation of the rearranged products 212 was strongly accelerated compared to the uncatalyzed reactions and proceeded with high diastereospecificity (98:2 syn:anti from Z-210; 2:98 syn:anti from E-210), the classical test for crotylboration via a cyclic (closed) transition state such as 211.88b To support the proposal that Sc(OTf)3 accelerates the reaction by bonding to boronate and not carbonyl oxygen, comparisons were made between the prenyl derivative 213 and the 9-BBN analogue 214 in the reaction with hydrocinnamaldehyde. The boronate 213 reacted >100 times faster under identical catalyzed conditions (dichloromethane, −78 °C), while the 9-BBN-derived 214 reacted with no appreciable rate increase over the background (uncatalyzed) process. This evidence argues against carbonyl activation by the Lewis acidic Sc(OTf)3, and is consistent with an important role for the borenium-like transition state 211. Other oxophilic Lewis acids such as AlCl389 or BF390 have been used to catalyze similar aldehyde allylations or crotylations with boronate reagents.
Scheme 15.

Synthetic applications involving alkoxyborenium intermediates88
Attempts to react the ester-containing allylic boronate 215 (Scheme 15) with benzaldehyde in toluene via the Sc(OTf)3 activated intermediate 216 encountered a slow reaction at 0 °C (<5% conversion, 16 h). On the other hand, the analogous experiment with Brønsted acid catalysts resulted in a large rate enhancement.91 The initially formed product 218 undergoes lactonization to 219 under the acid-catalyzed conditions. Although the mechanistic details remain uncertain, the borenium intermediate 217 was proposed as the species responsible for accelerating the reaction. Hall et al. have also developed effective catalysts for enantioselective crotylboration that may function via a related mechanism, although the use of a chiral diol additive as well as a Lewis acid raises additional mechanistic possibilities.92
7.2 Lewis acidity and π-conjugated borenium analogues
The Lewis formal charge representation of borenium cations (Section 1, Fig. 1) places no specific restrictions on the distance between boron and the formal plus charge. On the other hand, the dative bond representation of Fig. 1 (L→BR2+) is more explicit and treats the ligand L as a typical Lewis base, presumably one that is capable of independent existence. No distinction between the more inclusive Lewis formal charge representation and the dative representation has been needed so far because nearly all of the borenium salts discussed in prior sections easily fit both representations. However, structure 5036 (Scheme 2) does raise some questions. In this example, the ligand in the L→BR2+ representation corresponds to 1,3-dimethylimidazolidene as the Lewis base. There is no problem with terminology if L can be a transient nucleophilic carbene as well as a Lewis base, as in the preparation of 50, resulting in a formal charge that is placed farther from boron. On the other hand, the boronopyridinium salt 220b (Scheme 16) is a case where the dative bond representation 220a is technically correct for the “nucleophilic carbene ligand” of a borenium salt, but appears somewhat artificial. We prefer to classify 220 as a π-conjugated borenium analogue based on the simple criterion that its reactivity suggests enhanced electrophilicity at tricoordinate boron in the cationic environment. Several other examples are included in Schemes 16 and 17 where a positively charged substituent is present in a conjugated π-system connected to tricoordinate boron, and where borenium-like Lewis acidity is the only other criterion for regarding these structures as π-conjugated borenium analogues.
Scheme 16.

Scheme 17.

The Lewis acidity of 220 has not been evaluated directly, but enhanced electron demand at boron is implicit in the use of 220 and related substances as catalysts for the thermal condensation of amines and carboxylic acids to form amides.93 These reactions are believed to involve the generation of an acyloxyborane 221 as the key intermediate,93a presumably formed by nucleophilic attack of carboxylate at boron in 220. Catalysis by the cationic N-methylpyridinium salt was considerably more effective than by the parent pyridine, suggesting that attack by the nucleophile is accelerated by the remote positive charge.
Added insight regarding the effect of a cationic substituent on Lewis acidity at boron was provided in a study of the conjugated phosphonium borane 222 by Kawashima et al. (Scheme 16).94 Surprisingly, the spectroscopic properties of this substance were found to have a pronounced temperature dependence. Thus, 11B NMR spectroscopy above 273 K revealed a broad signal at δ = 56 ppm, consistent with trivalent boron in an aromatic environment. However, cooling the sample resulted in signal broadening, and eventually (< 230 K) gave a new broad maximum near δ = 0 ppm, a value that indicates conversion to a tetracoordinate boron structure 223 where the boron p-orbital is no longer part of a conjugated π-system. The UV spectrum was also temperature dependent, and decreased absorbance was observed for the 300 nm π-π* band upon cooling the sample. Treatment of the equilibrium mixture of 222/223 with ionic fluoride or chloride resulted in conversion to the corresponding product of halide exchange (224).
In another definitive study, Stephan et al. were able to generate several isolable conjugated borenium salts 226 using the method of hydride abstraction from the borohydrides 225 (Scheme 16).95 The tricoordinate boron could not be detected using 11B NMR methods, presumably due to quadrupolar relaxation,70,71 but enhanced Lewis acidity was confirmed by other methods, including the Gutmann-Beckett test. This procedure (comparison of 31P chemical shifts for the triethylphos-phine oxide adducts 227 with reference structures), resulted in Gutmann acceptor numbers of 78.1 for (C6F5)3B and 80.2–85.6 for 226 (R,R′ = alkyl or H), evidence for increased electron demand resulting from replacement of the C(4) fluoride by formally positive phosphorus.
7.3 π-Conjugated borenium analogues as selective anion sensors
A variety of conjugated borenium salts containing cationic aryl or heteroaryl boranes have been studied by Gabbaï et al. in connection with efforts to develop selective anion sensors. This topic has been reviewed in depth,96 so only a few highlights will be mentioned. Most of the work has focused on the relatively simple, but very hindered (and therefore, isolable) borenium structures 228–233 (Scheme 17). All of these salts react with nucleophilic anions to form tetracoordinate boron species in organic solvents, but the challenge has been to develop anion sensors that might respond selectively in the presence of water. Fluoride or cyanide detection using boron-based reagents is a logical choice due to their relatively strong bonds and compact nature, but there are a number of hurdles to overcome, mostly related to the aqueous environment. The reagent needs sufficient anion affinity to overcome hydration enthalpy, especially in the fluoride case where the anion is stabilized by strong hydrogen bonds with water. High sensitivity also requires Lewis acid selectivity for the anion over water or other Lewis bases, while practical applications require reagents that are stable under relevant assay conditions. Anion binding was expected to benefit from Coulombic attractions with the cationic substituent Z, while the LUMO energy-lowering effect of the cation would promote bonding with all nucleophiles. Indeed, the cationic phosphonium borane 228 (X = I) was shown to bind fluoride in water (10% methanol added for solubility).97 The ortho-isomer 229 was found to have a higher fluoride binding constant in methanol by orders of magnitude (K > 106 M−1 for 229 vs K ~ 400 M−1 for 228),97b but its limited stability in water was problematic.
The ammonium boranes 230 and 231 (X = OTf) proved to be sufficiently stable in aqueous media for detailed study, and both isomers formed cyanide as well as fluoride adducts in organic solvents.98 According to assay by UV spectroscopy (suppression of the maximum at 320 nm), the ortho-isomer 231 retained fluoride affinity in 95:5 water:DMSO (K = 910 M−1) to give 235, but did not bind cyanide ion under aqueous conditions, presumably due to increased steric interactions involving cyanide and the bulky BMes2 and NMe3 groups. In contrast, the para-isomer 230 displayed negligible fluoride affinity in the presence of water because of the lower activating effect of the para vs. ortho trimethylammonium groups, but 230 bound cyanide quite strongly (K = 3.9×108 M−1 at pH 7, 3:2 water:DMSO) to form 234. Neither isomer (230 nor 231) interacted with aqueous chloride, bromide, acetate, or nitrate. Thus, selective assay for fluoride (with 231) or cyanide (with 230) is possible using these reagents and their analogues. Overall, the fluoride binding affinities of 228–231 in aqueous systems may be too low for some applications, but substantially higher fluoride binding constants were observed in the phosphonium borane series for analogs of 228 having increasingly hydrophobic phosphorus substituents.97c
In yet another contrast in selectivity, Gabbaï et al. have reported that the ortho-dimethyl-sulfonium borane 232 (Scheme 17) does bind cyanide ion, and does so with high affinity.99a The 1,8-disubstituted naphthalene analog 233 was ca. 100-fold less effective. Binding constants were not reported, but a fluorescence quenching technique was used to detect the binding of 232 with aqueous cyanide at levels as low as 50 ppb. With 0.1 ppm of cyanide, the fluorescence quenching effect was large enough for simple visual evaluation. The origin of strong cyanide binding with 232 to give 236 was probed by DFT methods and NBO (Natural Bond Orbital) analysis. Stabilization was found not only by a donor-acceptor π(CN)→σ* (S–C) interaction, but also by a back-bonding effect, lp(S)→π*(CN). These energy contributions were estimated to total 4.1 kcal/mol for 236, and may explain why 232 is a much better binding agent for cyanide compared to the ammonium analogue 231.
One remaining problem with the anion sensors is that the reagents discussed so far report anion binding by decreased absorption or decreased fluorescence. This is the predicted outcome of anion binding to sp2 boron in a conjugated π-system to form the less conjugating (tetracoordinate) sp3 boron adduct, a factor that compromises sensitivity. Gabbaï et al. have reported a potential solution to this problem using reagent 240a (eq. 25), a boronium salt, an example of a BODIPY dye (see also structures 52, 53, Section 3.2), and a sensor having a “turn-on” response under specialized conditions.99b
Access to 240a was achieved using the method of halide abstraction from 237 with TMSOTf to generate a tetracoordinate boron complex 239 (eq. 25). By analogy to Scheme 2, this conversion may involve a borenium intermediate 238, although no mechanistic proposal was invoked by the authors.99b Subsequent addition of DMAP afforded the air-stable tetracoordinate boronium triflate 240a (11B NMR, δ = 0.91 ppm). Both 237 and 240a were strongly fluorescent in solution, but addition of 10 equiv of tetrabutylammonium iodide to 240a suppressed most of the fluorescence, apparently due to a heavy element effect resulting from anion exchange to form the boronium iodide ion pair 240b. However, addition of an equivalent of fluoride ion to the solution containing 240b gave a five-fold increase in fluorescent emission intensity, amounting to a “turn-on” response by 240b as a fluoride sensor. The basis for restoration of fluorescence upon addition of fluoride was proposed to be the reaction of 240b with fluoride to form the highly fluorescent 237, and similar conversion to 237 was also observed from 240a. The mechanism for these conversions is not known, although generation of borenium salts similar to 238 via SN1-like DMAP dissociation followed by fluoride trapping might be considered. On the other hand, the boronium triflate 240a does not react with chloride, bromide, or iodide ion. This observation raises questions about an SN1-like heterolysis. A reviewer has suggested that a pentacoordinate boron transition state 241 may be involved in the nucleophilic displacement from 240 to 237, by analogy to the pyridine-initiated bromide displacement discussed in connection with eq. 7 (Section 2.4), but this interesting alternative remains to be evaluated.
7.4 Tricoordinate boron in structures containing metal cations
Cyanide and fluoride affinity has also been probed using other cationic borane reagents,100–102 some of which incorporate cationic transition metal substituents placed at a considerable distance from the boron center.102 The latter structures can be included within the broadest representations of borenium salts suggested in Fig. 1 (Section 1), although there is no compelling reason to do so. In general, Lewis acidity at boron increases with proximity to the cationic site. Qualitatively, the activating effect increases as the number of conjugated cationic substituents increases.101 Cationic structures are also known where a transition metal is directly connected to tricoordinate boron.103 Although some of these structures can be potent Lewis acids and resemble more typical borenium salts in their ability to bind Lewis bases, the transition metal containing structures are beyond the scope of this review and will not be discussed beyond the inclusion of leading recent references.100–103
8. Borenium reagents for C-B bond formation
8.1 Borenium ion equivalents
The preceding sections describe methods that generate transient borenium intermediates or, rarely, that produce directly observable borenium salts in solution. Many of these techniques differ in the details of boron activation, but share the common feature that a tetracoordinate boron intermediate is in equilibrium with the borenium salt (tricoordinate boron). This rather simple statement deserves closer scrutiny when some of the relevant structures already discussed are considered side by side (Fig. 2), a comparison that raises questions about the importance of the coordination number at boron and the relevance of the borinium/borenium/boronium terminology. For example, the observable tetracoordinate boron cations 45, 83, and 203 should be classified as boronium salts, but all three can also be regarded as solvated borenium salts that may (or may not) dissociate in solution, while 45 could also be classified as a bis-nitromethane solvated borinium ion. If these complex cations do undergo dissociation, there is the added subtlety that solvated tight ion pairs or solvent-separated ion pairs may be formed. Such nuances were often discussed during the classical era of carbenium ion chemistry, but related questions have not been studied systematically for the analogous borenium ions.
Fig. 2.

Borenium equivalents
Another issue is that borenium cations at the higher end of the NH3 affinity range (Table 1) are likely to share electrons with solvent whether or not the complexes are observable. In the case of dichloromethane, this interaction can lead to solvent (and reagent) decomposition via n-electron sharing (see 80, Fig. 2), while in aromatic solvents, the interaction can generate π-complexes that may or may not undergo further reactions depending on circumstances (see Section 8.3). Electron sharing can also involve unreacted precursor molecules, as in the hydride-bridged “dimers” 78 or 84 where the B–H bond of the starting amine borane serves as a σ-donor leading to the 3c2e bond. It would be no surprise to encounter similar examples where n-electrons are shared loosely between a borenium ion and its precursor B–X: bond (X = Cl, O, N) in solution, similar to the gas phase adduct 64 (formally, a borenium ion that corresponds to a dimethoxyborinium ion bound to the precursor trimethyl borate as n-electron donor). In many cases where these issues arise, the only experimental evidence regarding boron coordination number is likely to be the 11B chemical shift for the dominant observable species with no guarantee regarding its relevance to the reactive species. Added insight may be gained from DFT calculations that address the transient intermediates, but at the current level of development, we will not know with certainty whether free borenium ions, ion pairs, bridged dimers, or solvates are responsible for a given transformation.
In some cases, the species in solution may be characterized rather well (eq. 26), but their classification is still not entirely straightforward. Thus, structure 242 (from 241 + SbCl5) has a tetracoordinate boron (formally, boronium) environment due to internal oxygen complexation according to its 11B chemical shift of δ = 8.7 ppm in CD2Cl2, but in nitromethane, the observed chemical shift is δ = 16.2 ppm.104 This latter value was interpreted as evidence for an equilibrium involving a borenium salt 243 as well as a boronium salt 244, either or both of which might also be regarded as solvated borinium species, depending on the (unknown) B–O bond lengths. By the same logic, 242 could be regarded as an internally complexed borinium ion. Similar issues were considered in Scheme 5, in connection with the boronium salt 99 (Fig. 2), obtained form 9-BBN-NTf2 and 1,8-dimethylaminonaphthalene, where unusually long B–N bonds could be taken as reason to classify 99 as a diamine complex of the corresponding borinium ion.
For all of the above reasons, it is simpler to think in terms of borenium ion equivalents in the reactions of high energy boron cations. This deliberately vague terminology includes all permutations of tetracoordinate (or partly tetracoordinate) boron structures that are potentially capable of dissociating in solution to give tricoordinate boron cations, and it also includes various activated structures such as the Lewis acid adducts discussed in Sections 6 and 7 where a borenium subunit is present in a neutral molecule. The following sections describe the reactivity of the higher energy borenium equivalents as reagents for the formation of C–B bonds. It is fair to say that the underlying mechanisms are not well known in these examples and that relevant studies are at an early stage, but substantial preparative potential is already clear.
8.2 Do borenium ions participate in hydroboration chemistry?
Studies in our laboratory have encountered olefin hydroboration using activated amine boranes that might be regarded as borenium equivalents. For example, pyridine iodoborane 245 converted β-methylstyrene into the iodoborane adduct 248 and treatment with pinacol in the presence of base afforded the pinacolboronate 249 in excellent yield (eq. 27).105 According to most indications, related hydroborations do not typically involve a leaving group heterolysis event prior to the step that generates the alkene π-complex 246, and are best regarded as nucleophilic displacements where the alkene attacks the iodoborane reagent to give 246 directly, followed by bond reorganization via the ion pair transition state 247. The evidence in favor of the SN2-like mechanism is mostly circumstantial, but it is reasonably satisfying. Thus, the rate of reaction is sensitive to the nature of the alkene as well as the pyridine ligand, suggesting that both reactants are involved in the rate-determining step. Furthermore, the reagent 245 belongs to the same family of activated pyridines studied many years ago and shown to resist spontaneous generation of borenium ions via leaving group heterolysis at room temperature (Section 2.4). Moreover, the hydroboration process works in toluene solution, a solvent where initial generation of a borenium intermediate would be suspect. Intramolecular versions of the hydroboration process using tethered, unsaturated amine iodoborane complexes work especially well, but these reactions are also believed to take place without dissociation to borenium species.106
Hydroborations also take place using the high energy activation procedures. Little of this chemistry has been investigated so far, and only isolated examples can be mentioned. In one case, the C3-symmetric chiral phosphine borane 250 (eq. 28) was treated with the trityl cation reagent Tr+B[C6F5]4− (71b) under conditions that should generate a hydride-bridged “dimer” from the phosphine borane.48 When the activation was performed using a 3:1 ratio of 250:71b in the presence of α-methylstyrene (toluene, rt) followed by conventional oxidative workup, the chiral alcohol 251 was obtained with 25% ee (190% yield relative to 71b as limiting reagent).107 We did not fail to notice that reagent 250 had been prepared starting from 3 equiv of commercial 251 (>95% ee) while the hydroboration produced 1.9 equiv of 251 with 25% ee.
Another attempt to access enantioselective hydroborating agents was made starting from the chiral pyridine borane 252 (eq. 29).107,108 Activation of 252 with 50 mol% of the trityl salt 71b at −78 °C followed by warming to −15 °C gave characteristic NMR signals indicating that 254 had been formed. In a similar experiment conducted in the presence of β-methylstyrene, subsequent quenching and oxidative workup gave only 20% of the desired alcohol 255 (14 % ee) while activation at room temperature produced racemic 255. Activation at rt was also evaluated in the absence of the alkene, and a 11B NMR signal was observed indicating that diborane had been formed, apparently via some process equivalent to pyridine transfer from 252 to the trityl cation in competition with the desired hydride transfer. This observation indicates that part (or all) of the racemic product product is formed without the direct involvement of 253 or 254, but the 14% ee at lower temperatures supports at least some participation by a chiral B–H source with the B–N bond intact.
In a simpler context, activation of triethylamine borane was investigated using the trityl salt 71b in the presence af an allylic silane (eq. 30).50 This experiment was designed to test whether an electrophilic borylation/de-silylation sequence would compete with the hydroboration process, but oxidative workup gave the primary alcohol 258 in 87% yield based on triethylamine borane. Evidently, the simple hydroboration mechanism is dominant, and the hydride-bridged borenium source 256 is an effective hydroborating agent. Related reagents hold promise for specialized applications in work that is ongoing.
8.3 Borenium equivalents in electrophilic aromatic borylation
8.3.1 Intramolecular borylation
Electrophilic borylation of benzene derivatives has long been known. Nearly all of the early reports relied on drastic Friedel Crafts conditions in the temperature range from 100–200 °C, including several intramolecular borylations that produced boron-containing heterocycles.109 However, in the striking example of 259 in the presence of Al2Cl6, the borylation to give 260 occurred within 30 min at 0 °C (Eq. 31).110 A similar experiment using the more soluble Al2Br6 was monitored by 1H NMR spectroscopy at −90 °C, and produced an intermediate assigned as the borenium ion 261 based on the presence of a doublet for the N-methyl group and an ABX pattern for the benzylic CH2-N protons. When this solution was allowed to warm to 0 °C, conversion to a product assigned as 262 occurred. The presumed N-protonation was attributed to protic acid contaminants in the non-purified aluminum halide reagents. The structures of 261 and 262 were not confirmed by 11B NMR spectroscopy, leaving open the possibility that the observable low temperature intermediate may contain tetracoordinate boron. Nevertheless, the facile cyclization at 0 °C does suggest the involvement of an unusually reactive electrophile such as the presumed borenium ion 261 or a related borenium equivalent.
Some years later, an analogous cyclization approach was investigated in our laboratory using amine borane activation under conditions that had been developed to access borenium equivalents in simpler systems (Scheme 18).47a Thus, N,N-dimethylbenzylamine borane 263 was treated with 50 mol% of the trityl salt 71b to generate the H-bridged dimer 264 in bromobenzene solution. No cyclization was observed at 20 °C (or after > 24h heating at 50 °C in benzene), but further addition of 40 mol% of 71b (or the use of 90 mol% 71b from the outset) resulted in the formation of cyclic products within four hours at 20 °C. These findings suggest that 264 requires further activation, but it is also possible that 71b scavenges unknown nucleophilic species that inhibit the cyclization. The main cyclization product was the previously described labile borenium salt 72b,47b but quenching with Bu4NBH4 afforded the stable amine borane 70 (72% overall). A catalytic procedure for the conversion from 263 to 70 was also demonstrated using 5 mol% 71b. This required considerably higher temperatures (160 °C in toluene, sealed tube) to overcome the activation barrier and to maintain a catalytic cycle at a convenient rate, but the procedure was efficient and gave 70 in 90% isolated yield. The stoichiometric activation method at 20 °C has been extended to prepare several other cyclic amine boranes including 265–268.47b
Scheme 18.

Intramolecular nitrogen-directed aromatic borylation47
In the course of studies aimed at clarifying the mechanism of the nitrogen-directed borylation of Scheme 18, the ortho-deuterated benzylamine borane 269 was explored (Eq. 32).47b Two isotopomeric products 270 and 271 were formed, corresponding to a deuterium isotope effect kH/kD = 2.8 for the product-determining step. This KIE result argues against rate-determining cyclization from the borenium equivalents 264 or 272 to a Wheland intermediate 273 (Fig. 3) followed by rapid deprotonation. However, it would be consistent with slow (product-determining) proton removal from 273, or with mechanisms where C–H cleavage occurs in the transition state leading to cyclic products. One such possibility is suggested in Fig 3 starting from the hydride-bridged dimer 264. According to DFT calculations, the borenium π-complex 272 and the Wheland intermediate 273 have nearly identical energies and very similar geometries. The lowest barrier found from these intermediates to cyclized products involves the transition structure 274, corresponding to a C–H insertion mechanism that leads directly to the observed 72b and hydrogen.
Fig. 3.

B3LYP/6-31G* energies for cationic structures from 264 to 72b
The available experimental evidence does not prove whether 72b is the initial cyclization product from 272/273, or whether 72b forms via 273 and deprotonation to give 70, followed by the known hydride abstraction (Scheme 18).47b The need for >50 mol% of the trityl salt 71b to promote cyclization from 264 at room temperature is also not clear. A number of rationales remain to be evaluated, the simplest of which assumes that 71b converts 264 to the borenium ion 272 by abstracting hydride from 263 to drive the unfavorable dissociation equilibrium from 264. Alternative explanations might invoke a bonding 3c2e interaction between 71b with a B–H bond in 264 to provide additional electron demand. Although this would require the interaction of two cationic species (71b + 264), it may facilitate the dissociation of 264 to 272, or the conversion of 264 into a new (currently unidentified) intermediate that is capable of borylation.
Several carbon- or heteroatom-tethered phosphorus analogies (277 to 278) for the cyclization from 263 to 70 have also been investigated briefly using the activating procedure with TrB(C6F5)4 (71b), 90 mol% in bromobenzene at room temperature (Eq. 33).107 After reductive workup with Bu4NBH4, the cyclized products 278 were obtained in somewhat lower yield compared to the amine borane examples. In the case of 278c, the 30 % yield also reflects partial hydrolysis during chromatography. The same cyclization was demonstrated using activation with 1.1 equiv of the strong acid HN(SO2CF3)2 in place of 71b, and gave 67% conversion from 277c to 278c after 24 h at 100 °C according to NMR assay. Presumably, all of these cyclizations involve the generation of borenium equivalents, but the intermediates were not investigated in depth.
8.3.2 Intermolecular borylation
Some of the borenium equivalents discussed in earlier sections are of interest as electrophilic reagents for intermolecular borylation. One unusual example has already been discussed briefly in a different context, involving the thermal decomposition of borenium salt 72b to give 75 at 80 °C (Scheme 3).47b Although the mechanism of this reaction is not known, one possibility involves electrophilic ipso attack by the borenium cation on the tetraarylborate counterion, followed by fragmentation. Similar decomposition of borenium tetrakis(pentafluorophenyl)-borate salts has been noted by Ingleson et al.42 These are not typical intermolecular borylations because they involve bond formation between the two components of an ion pair, but the bonding events are mechanistically similar.
Recently, electrophilic borylation has been demonstrated using catechol borane-derived reagents. Relevant structures were already mentioned in Section 3 in the context of the hydride or halide abstraction methods for preparation of observable borenium cations (Scheme 3), and indirect evidence was discussed regarding the possible generation of a transient borinium cation 62 from 59 using AgCbBr6 (eq. 12). In a similar experiment, Ingleson et al. treated 59 or 279 with the triethylsilyl cation source 280 as the halophile in benzene solution (Scheme 19), and observed rapid conversion to the B-phenyl catecholboronate 283 under stoichiometric conditions at room temperature.42 The reactive (transient) electrophilic intermediate was represented by the formula 281 and abbreviated as CatB+. Despite considerable effort, cationic boron species were not detected and the specific structure of 281 was not established definitively. However, the borinium salt 62 was shown as one possibility, and the authors favored an electrophilic borylation mechanism via 282 that fits most simply if 281 is either the same as, or acts as a source of, the borinium salt 62. In short, 281 can be taken to represent a borinium equivalent. Borenium salts 284 were also considered as potential intermediates (L = nucleophilic halide from the Et3SiCl byproduct, or oxygen from catechol-containing species) and were discussed in connection with an alternative mechanism involving C-H insertion that was not favored by the authors. A third possibility that 284 may be capable of participating in a typical stepwise electrophilic aromatic substitution process was implied by noting that a tetracoordinate borylation transition state [CatB(arene)L]+ may be viable (L = a weak nucleophile).42
Scheme 19.

Intermolecular aromatic borylation using catechol-derived borenium salts42
An interesting catalytic version of the borylation was also developed using CatBH (65) as the stoichiometric boron source (Scheme 19b), with the reagents CatBBr (279) + Et3SiCbBr6 (280) serving in the role of pre-catalyst (arene solution, 80–100 °C).42 The catalytic cycle depends on regeneration of 281 from CatBH (65) and “H+” during or after aromatization of the Wheland intermediate 282. As the most potent known electrophile, “H+” exists in solution as 285, where ligand L = the best available electron pair donor. Therefore, the ligand is also involved in the proton removal step from 282, and could be part of the product-determining transition state leading from 282 to 283 + 285. The aromatic substrate is one of the possible choices for L, in which case 285 could be identical to the π-complex (not drawn) or to the corresponding Wheland intermediate 282.
The catalytic process worked well with benzene as the substrate at 80 °C and gave 283 in high yield. The catalytic borylation was also demonstrated with toluene (15 h at 100 °C; 93% isolated yield of a 1.2:1 meta:para mixture of borylation products),42,111 and with o-dichlorobenzene (10 h at 100 °C, exclusive borylation at C4, 99%). In the case of ethylbenzene, the product mixtures were considerably more complex due to alkyl migration under the superacidic conditions, including intermolecular ethyl transfer among the reactants and products, resulting in ca. 1.7 ethyls per product boronate. Multiple products were also obtained using meta or para xylenes.
More recent studies of intermolecular borylation have explored reagents that are better defined structurally, but questions remain regarding the key mechanistic aspects.52,112 Thus, Ingleson et al. described borylating agents prepared by the method of halide abstraction from CatB-Cl (59) or Cl4CatB-Cl (286) in combination with Et3N and AlCl3 (Scheme 20).112 Formation of the triethylamine adducts 287 was followed by AlCl3-induced heterolysis to give the borenium tetrachloroaluminates 288 and 289. The Cl4CatB reagent 289 was characterized in the solid state by X-ray crystallography, and the solution structures of 288 and 289 were supported by 11B NMR spectroscopy (δ = 27.9 and 28.1 ppm, respectively). These values are in good accord with those of the closely related and previously characterized borenium salts 67and 69a (Scheme 3).44,45 The new reagents were shown to react with electron-rich aromatic and heteroaromatic substrates Ar-H at room temperature to afford the corresponding borylation products 290 with high efficiency. However, the electron rich environment facilitated protodeboronation in a number of cases, so the initially formed catecholboronates were converted into the more stable pinacolboronates by trans-esterification with pinacol in the presence of triethylamine. The resulting 291 were obtained in good to excellent yield and with high regioselectivity, as shown for selected examples in Scheme 20 to illustrate the trends. In examples 291a and 291b, the regioselectivities were shown to complement the sterically controlled, meta-selective iridium-catalyzed borylations.112,113 In general, the more electrophilic reagent 289 (Cl4CatB environment) was considerably more reactive compared to 288, as seen in the shorter reaction time leading to product 291c. Attempts were also made to prepare 291 directly using the known borenium salt 69a as well as the 2,6-lutidine analogue 292, but these reagents were not sufficiently reactive to borylate the most reactive substrates dimethylaniline or N-methylpyrrole. The lower reactivity was attributed to better delocalization between boron and the oxygen n-electrons in 292 compared to the catechol analogues 288 or 289. Ingleson et al. did not comment further on the mechanism of the highly effective borylations using borenium salts 288 and 289 beyond noting the analogies with potential borylating agents considered in Scheme 19.
Scheme 20.

Electrophilic borylation of electron-rich aromatic and heteroaromatic substrates112
On a similar timescale, the intermolecular borylation of electron-rich aromatic substrates was reported from our laboratory using reagents containing the 9-BBN core (structures 92, 97, and 99) that were mentioned in Scheme 5, and are discussed in the borylation context in Scheme 21.52 Both of the cationic reagents 97 and 99 were found to borylate N-methylindole to afford 293a at convenient rates in dichloromethane at 50 °C (thick-walled Schlenk tube). Surprisingly, the reagent 99 having formally tetracoordinate boron proved to have comparable reactivity compared to the borenium salt 97. Because 99 is also more convenient to handle as the crystalline solid, it was used to prepare several other borylated products 293b–d as shown in Scheme 21 under similar conditions with reaction times on the order of hours. The neutral borane reagent 92 did not convert the aromatic substrates into products 293 using the same procedure at 50 °C. However, when 92 was used in combination with the hindered pyridine base 294, the borylation of N-methylindole proceeded within seconds at room temperature. Even though 92 + 294 is the most reactive reagent in the 9-BBN-derived series, it is far more difficult to handle compared to 99, and was therefore not evaluated for the borylation of other aromatic substrates.
Scheme 21.

Intermolecular borylation using 9-BBN-derived borenium equivalents52
The intermolecular borylation studies from our laboratory52 and from the independent work of Ingleson et al.112 were reported in adjacent papers, and described the borylations of very similar electron-rich substrates. Although the featured reagents in Schemes 20 and 21 were markedly different, there was one other similarity worth noting. Thus, both studies described isolable, well-characterized reagents having structures established by X-ray crystallography. However, this additional information has not helped to narrow the mechanistic choices. For purposes of illustrating the mechanistic dilemmas, several (speculative) alternatives are considered briefly in the context of Scheme 21. The first dilemma is that all of the reagents 92, 97, and 99 are potential sources of one and the same borinium ion 295 by variants of B–N bond heterolysis. Therefore, each reagent is also a potential source of various borinium equivalents 296 (L = weak nucleophile) that may produce analogues of the borenium ion 97 by dissociation of a single ligand. Any one of these dicoordinate or tricoordinate boron cations, or the neutral borane 92, may serve in the role of the key electrophile responsible for borylation. Even the isolable 99 may already qualify as a reactive borinium equivalent due to its unusually long B–N bonds.
A second complication is that the role of base (external base with 92; internal base with 97b or 99) has not been defined. It is not known whether the base serves only to scavenge the HNTf2 byproduct after borylation, thereby preventing the reverse reaction (reversible protodeboronation of 293), or whether the presence of base results in a lower activation barrier for borylation. In the special case of 99, a transient borenium intermediate 297 is especially appealing because it contains strongly electrophilic boron as well as an internal base that might participate in the transition state if proton transfer is rate-determining. The final dilemma is that each of the potential electrophiles may be capable of reacting via at least three mechanisms, (1) electrophilic aromatic substitution (EAS) with rate-determining formation of the Wheland intermediate, (2) EAS with rate-determining deprotonation, or (3) an intermolecular variant of the C-H insertion mechanism discussed in Section 8.3.1 (Fig. 3) for intermolecular borylation. Such mechanistic nuances are notoriously difficult to distinguish.
Another example of aromatic ring borylation is presented to underscore the subtle interplay of structure and mechanism in these reactions using an activated form of triethylamine borane. As mention in Section 8.2, activation of Et3N3BH3 with the trityl cation source 71b (50 mol %) generated the hydride-bridged “dimer” 256 (Eq. 30), but further hydride abstraction did not occur using excess 71b. Activation with a deficiency of Tf2NH also gave the corresponding “dimer” 299 according to NMR evidence (Scheme 22). However, addition of a full equivalent of the strong acid afforded the amine borane complex 298 exclusively, judging from the 11B NMR chemical shift (two maxima, δ = 0.6 and −7.4 ppm in CD2Cl2, assigned to O-bound and N-bound covalent bistriflimidate).50 This evidence is consistent with reversible formation of 298 and the starting Et3N·BH3 from 299, followed by protonation of the Et3N·BH3 and eventual conversion to the borenium salt 298. Alternatively, direct protonation of the hydride-bridged dimer 299 may be feasible as a way to force the conversion to 298.
Scheme 22.

Activation of triethylamine borane for intermolecular borylation50
The activated reagent 298 was not explored in depth, but proved sufficiently reactive to borylate N-methylindole over hours at room temperature to give an initial product tentatively assigned as 301, along with HNEt3+ Tf2N− (Scheme 22). Definitive evidence for 301 was not obtained, but treatment with disodium iminodiacetate in methanol gave the isolable complex 302 (72%). The initial conversion from 298 to 301 was attributed to electrophilic borylation, followed by internal (4-center) proton transfer via transition state 300, a pathway that forms the ammonium salt HNEt3+ Tf2N− directly, without releasing the strongly acidic Tf2NH. This pathway contrasts with the intramolecular borylation of Fig. 3, where the products consist of molecular hydrogen and a borenium cation (72b) resulting from an alternative 4-center process in the final step.
8.3.3 Recent developments in Muetterties borylation (BX3 plus proton scavenger)
Mechanisms based on borenium intermediates may be involved in typical aromatic borylations using BCl3/Al, a classical procedure that allows the preparation of arylboron dichlorides from benzene or other simple arenes using readily available reagents.114–117 The aluminum additive is important in two roles: (1) it scavenges the HCl byproduct of borylation, thereby preventing reversible protodeborylation of the product, and (2) it forms AlCl3 in situ as a chlorophilic Lewis acid that accelerates the borylation. In the first mechanism proposed for this reaction, Muetterties suggested initial formation of “active species BCl2+ or ArH·BCl2+ AlCl4−”, and eventually described the latter structure as the arene π-complex according to UV evidence.114 The simpler cation (BCl2+) is a dichloroborinium ion according to current terminology, while the cationic π-complex can be regarded either as a borinium equivalent, or as a borenium ion Cl2B− L+ where the ligand L is the arene substrate. Also interesting is a subsequent paper by Muetterties where footnote 10 mentions that “solvation of BCl2+ is a necessity”, a statement that might constitute the first, albeit indirect, suggestion of a borenium mechanism if the author intended to invoke Cl2B–L+ (L = solvent) as the electrophile for aromatic borylation.116 Another relevant structure Cl2B–(Cl+)–AlCl3− for the activated electrophilic intermediate has been considered by Olah.118 This potential intermediate contains a borenium subunit within a neutral structure. The identity of the key electrophile in these borylations remains uncertain, even though a borenium connection is plausible. Cautious interpretation is warranted, given that Muetterties borylations were often performed at elevated temperatures and that even diborane is a sufficiently strong electrophile for the borylation of benzene at ca. 100 °C.119
A more explicit connection with borenium intermediates was suggested in a recent modification of the Muetterties approach for use in the intramolecular borylation of 2-arylpyridines (Scheme 23).120 Thus, treatment of 2-phenylpyridine 303 with 3 equiv of BBr3 and Et2NiPr as HBr scavenger in dichloromethane gave the cyclic product 307 (89%) at room temperature. The proposed sequence involves formation of a complex 304 and subsequent halide abstraction by the excess BBr3 to give a transient borenium salt 305, followed by electrophilic borylation via a Wheland intermediate 306. The same procedure was effective for the N-directed borylation of several substituted or annulated derivatives of 303, and also for the corresponding conversion from the benzothiophenyl analogue 308 to 309 (87%). The borenium intermediates were not observed directly in this study.
Scheme 23.

Intramolecular borylation using BBr3/Et2NiPr120
A modified Muetterties procedure has also been developed for the intermolecular borylation of electron-rich aromatic substrates by Ingelson et al. (Scheme 24).10b The ingredients include BCl3 and AlCl3, the same species that are present under the original Muetterties conditions (BCl3/Al), along with a basic amine additive such as 2,6-lutidine or 4-dimethylaminotoluene (Me2NTol). Not only does the amine function as an acid scavenger, but it also serves as the key ligand L that forms the BCl3 complex 310, the immediate precursor of a borenium salt 311 in situ. Using 2,6-lutidine as the base, it was possible to isolate the corresponding salt 311a as a crystalline solid that was fully characterized by X-ray and spectroscopic methods (11B δ = 46.9 ppm). A closely related 4-picoline analogue 10 was mentioned in Section 2.4 as the first observable borenium salt to be characterized by 11B NMR spectroscopy.10a
Scheme 24.

Intermolecular borylation using BCl3/AlCl3 and 2,6-lutidine or Me2NTol10b
Upon reaction with electron rich heteroarenes Ar-H at 25 °C, 311a afforded the corresponding borylation products Ar-BCl2 (312) on a timescale of hours (Scheme 24). Further treatment with pinacol in the presence of excess amine base produced the corresponding pinacol boronate esters 313a–c in high yield. On the other hand, 311a was not sufficiently reactive for the preparation of 313d–f. For these borylations, the reagent generated in situ from BCl3/AlCl3 and Me2NTol proved superior, and was used to prepare 313d–f under similar room temperature conditions. The overall sequence worked well on multigram scale. However, a borenium intermediate 311b could not be isolated and characteristic 11B signals for tricoordinate boron were not detected, apparantly due to a dynamic equilibrium process involving other Me2NTol complexes. If 311b is the reactive borylating agent present in equilibrium, then its reactivity appears to be considerably improved relative to that of 311a, but not sufficiently improved for the borylation of less activated substrates such as toluene. The authors did observe borylation of m-xylene at 140 °C using 311b, but the product mixture reflected competing isomerization of the xylene substrate. Overall, the Ingelson procedure has considerable potential for the borylation of electron rich arenes. The authors noted that borenium intermediates 311a or 311b may be the electrophiles that attack an aromatic ring carbon to form a C–B bond, but did not rule out other possibilities.
9. Extension of borenium chemistry to neighboring fields and unusual environments
9.1 N-Heterocyclic carbenes as stabilizing ligands for borenium salts
The recent focus on nucleophilic carbenes as stabilizing ligands for reactive species in both main group element and transition metal chemistry has stimulated logical extensions to borenium ion chemistry. To our knowledge, the first example of an N-heterocyclic carbene (NHC) ligand in a borenium environment appears in a 1997 report by Weber et al.121 Following a typical B-X bond heterolysis approach, 2-bromo-2,3-dihydro-1H-1,3,2-diazaborole 314 was reacted with the corresponding free carbenes 315a or 315b in benzene at RT to prepare the NHC-stabilized borenium salts 316 (Scheme 25). The cationic products were characterized by multinuclear NMR spectroscopy (316a δ 11B = 15.3 ppm; 316b δ 11B = 15.0 ppm) and X-ray crystallography in the case of 316a. The 11B chemical shifts are observed at unusually high field for tricoordinate boron species, but the same can be said for the neutral bromoborane 314 (δ 11B = 16.2 ppm). The unusual heteroaromatic environment appears to be primarily responsible for these boron chemical shift values. According to the 11B criterion, the tricoordinate borenium cations 316 resist formation of tetracoordinate species by addition of the bromide anion to boron. This degree of borenium stabilization probably reflects a cumulative effect of heteroaromatic delocalization as well as the substantial steric hindrance near the boron atom.
Scheme 25.

A more typical NHC-derived borenium salt 50 was mentioned in Section 3.2, Scheme 2 (Gabbaï et al.) to illustrate methods of borenium salt generation. The tricoordinate boron structure of 50 was established by X-ray crystallography,36 and was further supported by the 11B NMR chemical shift (δ = 66 ppm). In a related case (Lindsay et al.), the 9-BBN-derived borenium salt 318 was prepared using two independent pathways, (1) B-H hydride abstraction from 317 by TfOH, and (2) nucleophilic addition; heterolysis from 9-BBN-OTf (319) and carbene 320, and the structure 318 was established by X-ray crystallography and by δ 11B = 81.4 ppm.122 The chemical shifts of 50 and 318 are similar to values reported for analogous amine-ligated borenium salts such as 48 (δ 11B = 66 ppm, Section 3.2, Scheme 2) or the 9-BBN-containing 93 and 97 (δ 11B = 66.5 and 85.1 ppm, respectively, Section 3.5, Scheme 5), suggesting that replacement of the amine ligands in 48, 93 or 97 by the NHC ligand does not cause major electronic changes at boron. Importantly, the degree of ion pairing for 318 in CD2Cl2 was studied by diffusion ordered NMR spectroscopy (DOSY), a method that confirms the ion pair character of 318 and provides a welcome complement to the 11B chemical shift as evidence for the presence of tricoordinate boron in solution.
While exploring the applicability of NHCs to stabilization of a wide range of highly reactive main group molecules, Robinson et al. prepared borenium salt 322 by nucleophilic addition; heterolysis from reaction of (i-Pr)2NBCl2 with the carbene 321 (Scheme 25).123 The tricoordinate borenium chloride structure 322 was established by X-ray crystallography and by δ 11B = 32.2 ppm in solution. Apparently, the chloride anion resists coordination at the boron center of 322 due to the extreme steric congestion as well as some degree of cation stabilization by nitrogen lone pair delocalization involving the diisopropylamido substituent. Robinson et al. also reported an unusual reaction involving the reduction of 322 with KC8. This afforded a C-H insertion product 323, presumably formed via a divalent borylene intermediate.
Other classes of N-heterocyclic carbenes have begun to attract attention. So far, one example has been described in a non-aromatic heterocyclic environment as shown for the conversion from 324 to 325 (δ 11B = 51.4 ppm) by halide abstraction.124 The analogous pyridine derivative 326 (δ 11B = 42 ppm) as well as a related catechol boronate ester (δ 11B = 28 ppm) were also prepared, and X-ray data as well as computational evaluations suggested a stabilizing electrostatic interaction between tricoordinate boron and the π-system of the flanking aryl groups.
All of the NHC-containing borenium salts mentioned so far contain two substituents at boron in addition to the carbene ligand. In an attempt to generate an unsubstituted NHC-BH2+ salt, Alcarazo et al. treated the hindered NHC borane 327 with B(C6F5)3 as the electrophile for hydride abstraction (Scheme 25).125 However, the only product observed was the hydride-bridged dimer 328 according to X-ray and 11B NMR evidence. The authors concluded that δ-donation from the NHC substituent was not sufficient to prevent a 3c2e interaction between the incipient borenium cation with a B–H bond in the starting 327. On the other hand, when the carbodiphosphorane-derived borane 329 was reacted with B(C6F5)3, the primary borenium salt 330 was indeed formed and could be isolated as the crystalline solid. The structure of 330 is firmly established by X-ray evidence, by the 11B NMR signal at δ = 56.6 ppm (triplet, JBH = 29 Hz), and by the formation of a tetracoordinate boronium salt upon treatment with p-dimethylaminopyridine. The improved stability of 330 was attributed to π- as well as σ-donation from the carbodiphosphorane ligand to boron, and to steric protection by the flanking triphenylphosphonium substituents. By the criterion of borenium stability, the carbodiphosphorane subunit (Ph3P=C=PPh3) in 330 is a better electron donor than the NHC subunit in 327 or 328. Structure 330 is the only primary borenium salt known to date.
9.2 Miscellaneous related topics involving BH species
Borenium equivalents could be relevant to several areas beyond the mainstream organic chemistry context of this review. In addition to the definitive gas phase studies at the borinium-borenium interface mention in Section 3.3,3,43 other areas have emerged where a role for borenium equivalents is not assured, but may deserve consideration. These topics will be mentioned briefly in this paragraph to provide leading references. For example, hydrodefluorination has received some attention, and has been demonstrated using cationic alane- or silane-derived reagents, generated by the familiar trityl salt activation method.126 By analogy, hydrodefluorination might have some potential using activated cationic boranes. Indeed, one unusual example has already been encountered involving a borenium salt intermediate, although this was not by design (see partial dehydrofluorination of 71a, Scheme 3).47a A more extensively studied area is the activation of B–H bonds in amine or phosphine boranes using transition metal reagents or catalysts as well as Lewis acids. Possible applications of this chemistry include C–H insertion methodology, dehydrocoupling and dehydropolymerization, and reversible hydrogen storage.127,128 The key event in the latter examples is the conversion of a borane complex RR′ZH·BH3 (Z = N or P) to (RR′ZBH2)n and hydrogen. If the first step in this process is B–H activation, then a transient borenium equivalent may be involved.
Unusual pathways leading to observable or transient borenium salts have been reported in specialized systems. Thus, Piers et al. have developed an interesting variation of the hydride abstraction method where the substrate reacts with trityl cation at the allylic C–H bond of a B–C=C–CH segment to generate borenium salt 53 in the BODIPY series (Scheme 3).38b A more typical halide abstraction approach to the related cation 52 was described in Section 3.2.38a In a different application, Piers et al. have also reported an unusual variation of electrophilic activation by protonation of neutral borabenzene pyridine complexes. In this case, a transient borenium ion is generated by the protonation of the borabenzene ring.129 Protonation at carbon has also been used to form borenium salts from dienamines.41c
In two other recent examples, there is some evidence suggesting the generation of borenium salts130 or borenium equivalents131 by pathways that may not fit into the typical categories considered earlier. In the former example, the substrate can be represented by the formula iPr2NB(F)N(Ar)SiMe3 while the crystalline borenium byproduct isolated after treatment with AlCl3 contains the cation iPr2NB(C4H9)NH(Ar)+.130 The source of the butyl group in this structure, established by X-ray crystallography, is not known. The second example (Scheme 26)131 involves the conversion from 331 to the cyclic boronium salt 334 using excess diborane. The authors favored a pathway via 332 and 333, and discounted a role for other hydride-bridged 3c2e intermediates because treatment of the model substrate 335 with diborane gave no evidence for generation of an observable borenium salt 336 nor related boronium species. However, the high electrophilicity of an NHC-derived primary borenium ion as indicated by the conversion of 327 into 328 (Scheme 25) suggests that the role of potential 3c2e borenium equivalents in the cyclization of 332 may need to be re-evaluated. Furthermore, the known reductive quenching of other borenium equivalents by borohydride (Scheme 3) provides some evidence that 336 or derived hydride-bridged 3c2e cationic structures having borohydride counterions would not be observable because the equilibrium would strongly favor 335. Finally, an unusual triborane-derived borenium intermediate has been suggested to explain conversion from a B–Cl subunit to the corresponding B–OH using AgB[C6F5]4 in ether.132 The authors reported the GC detection of ethylene as supporting evidence, presumably resulting from the elimination of a hypothetical transient B–OEt2+ intermediate.
Scheme 26.

NHC borane-amine cyclization131
10. Summary
According to the discussion presented above, borenium ions are strongly electrophilic and Lewis acidic, and they resemble the isoelectronic carbenium ions in their reactions with nucleophiles. Even some of the weakest nucleophiles are sufficient to interact with non-stabilized cationic, tricoordinate boron species if the reaction conditions are carefully chosen to avoid solvent interference and the presence of potentially nucleophilic contaminants. Much of the borenium reactivity profile follows simple organic intuition based on the carbenium analogy, but some features stand in striking contrast and deserve a deeper analysis. For example, not a single example of a skeletal rearrangement involving borenium equivalents has been found in the literature, not even for potential sources of the non-stabilized, branched borenium cation 25 (Table 1) where the neopentyl analogy might imply that leaving group displacement would be very difficult. In fact, SN2 substitution of the corresponding primary iodide is quite easy (Section 2.4, eq. 8).22,23 Partly, this is due to the relatively long B–N bond, a factor that decreases steric crowding for SN2 attack. Another important factor is the formally positive heteroatom adjacent to boron, the same factor that so strongly enhances electrophilicity at boron. Skeletal rearrangement by one of the adjacent N–C bonds in 25 would have to form sextet cationic nitrogen that is even higher in energy than the borenium salt due to the greater electronegativity of nitrogen vs. boron. With no realistic option for skeletal rearrangement, the borenium precursors and borenium equivalents can often be used at elevated temperatures with surprising ease, provided that other undesired intramolecular events such as nucleophilic attack or fragmentation have higher activation energies.
Another contrast with carbenium chemistry results from the highly interactive nature of activated amine, phosphine, or NHC boranes. Their tendency to form relatively stable hydride-bridged dimers such as 78 and 84 (Scheme 4), 256 (eq. 30), 264 (Scheme 18), 299 (Scheme 22), or 328 (Scheme 25) raises an interesting question: what would be the reactivity of a non-stabilized borenium cation that is incapable of hydride bridging? Similar questions arise in connection with heteroatom-substituted borenium species that may form stabilized heteroatom-bridged “dimers” or various solvent adducts; how reactive would they be if such covalent stabilization is prevented or minimized by careful choice of substituents? Possible answers to such questions are only now beginning to emerge (Schemes 20, 21).
Given the huge range of borenium electrophilicities (Table 1), and the almost unlimited potential for fine-tuning structure and reactivity, it is safe to predict that the exploration of synthetic uses of borenium equivalents has barely begun, despite the multi-decades long history of borenium chemistry presented in this review. Fascinating applications that exploit chiral borenium equivalents in stoichiometric or catalytic applications are already well-established. Further developments in enantioselective catalysis can be anticipated from the potential of borenium electrophiles to interact with a broad range of nucleophiles, even including C–H σ-bonds.
The first indications of C–H insertion chemistry have recently been communicated.133 The example summarized in Scheme 27 proceeds at room temperature using stoichiometric trityl cation as the hydride abstracting agent, but the reaction can also be carried out catalytically with activation by 5% Tf2NH at 160 °C. The key activation event from 336 + Tr[B(C6F5)4] to the H-bridged dimer 337 and the borylation step to give a cyclic, nonstabilized borenium salt 339 followed by reductive quenching to give the amine borane 340 resemble the nitrogen-directed aromatic borylations of Scheme 18, but they do raise questions. In particular, the role of the “extra 40%” of Tr[B(C6F5)4] remains unclear. Is the black box intermediate simply the primary borenium salt 338? If so, does the extra activation step require hydride transfer from a cation (337) to another cation (Tr+) to effect the conversion to 338? These are typical of the many mechanistic questions that remain to be answered as we begin the ninth decade of research in borenium salt chemistry.
Scheme 27.

Aliphatic C-H insertion
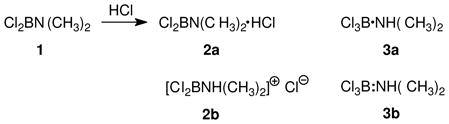 |
(1) |
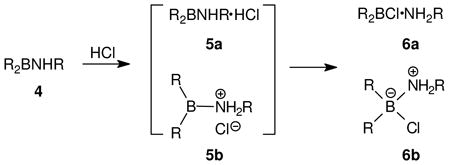 |
(2) |
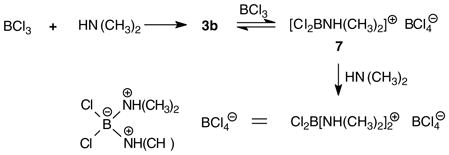 |
(3) |
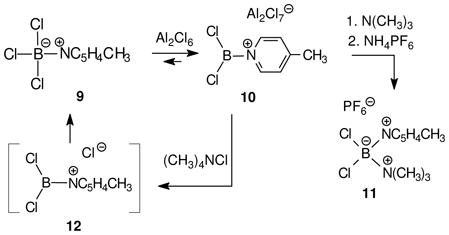 |
(4) |
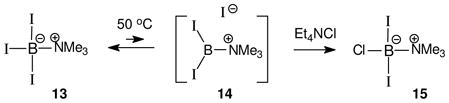 |
(5) |
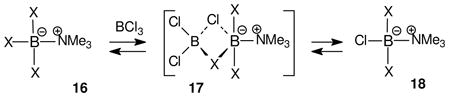 |
(6) |
 |
(7) |
 |
(8) |
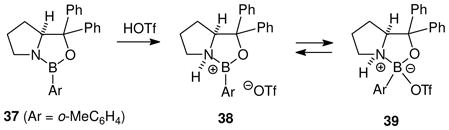 |
(9) |
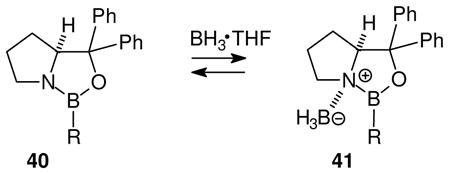 |
(10) |
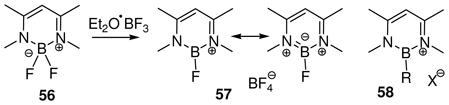 |
(11) |
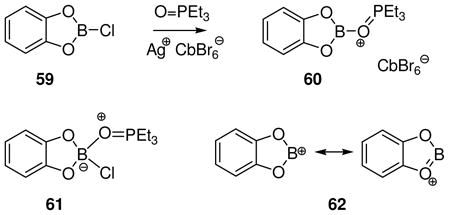 |
(12) |
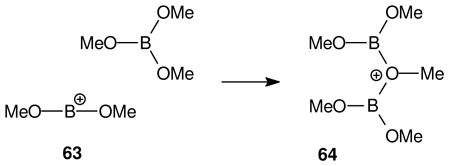 |
(13) |
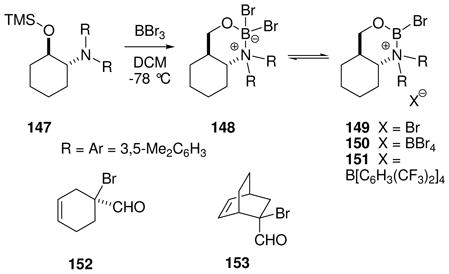 |
(14) |
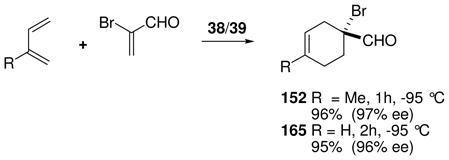 |
(15) |
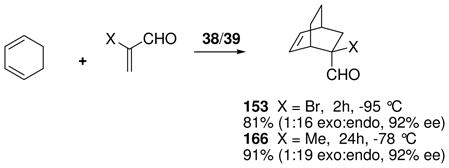 |
(16) |
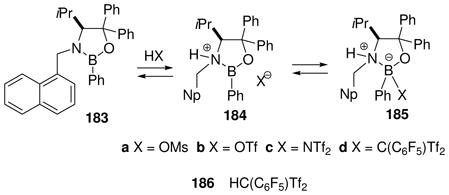 |
(17) |
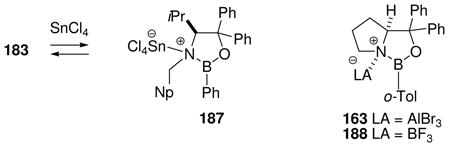 |
(18) |
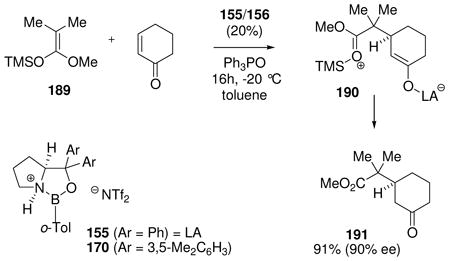 |
(19) |
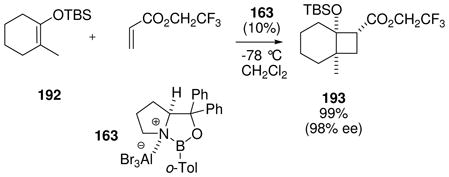 |
(20) |
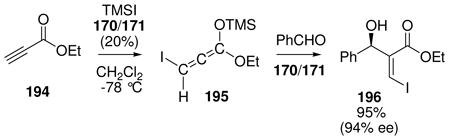 |
(21) |
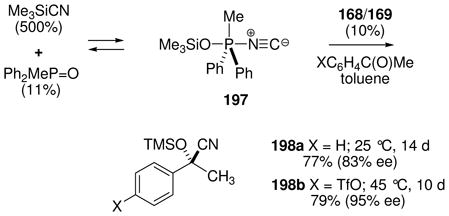 |
(22) |
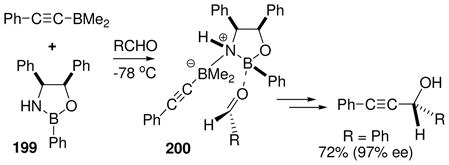 |
(23) |
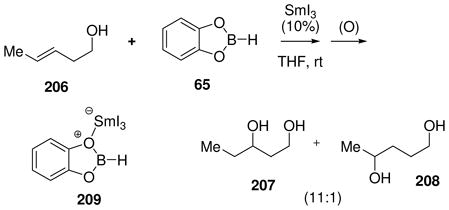 |
(24) |
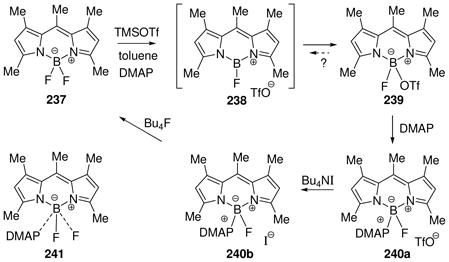 |
(25) |
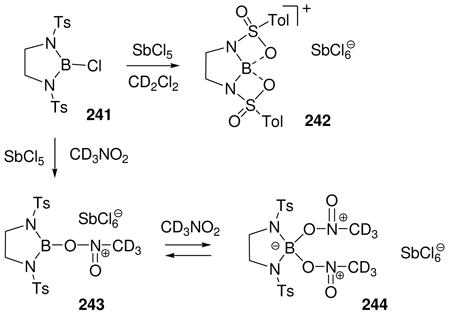 |
(26) |
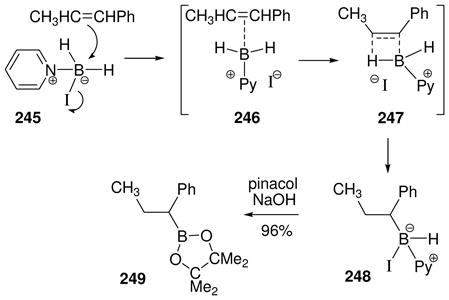 |
(27) |
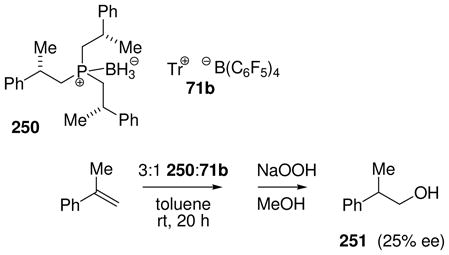 |
(28) |
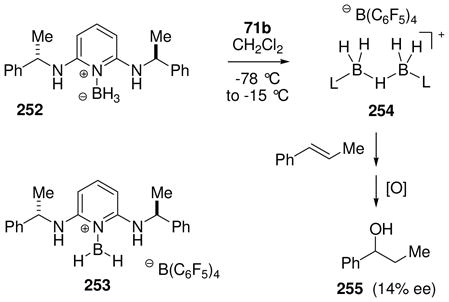 |
(29) |
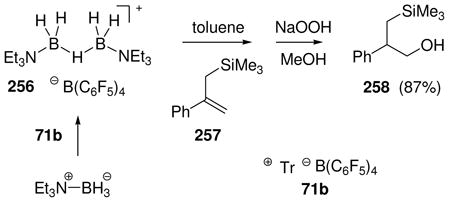 |
(30) |
 |
(31) |
 |
(32) |
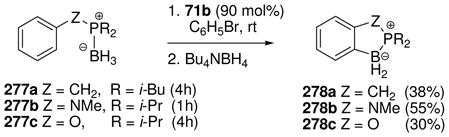 |
(33) |
Acknowledgments
The authors are grateful to Prof. J. Harvey and Prof. F. Weinhold for stimulating conversations and many useful suggestions regarding boron electrophiles and related matters of a fundamental nature. Special thanks are also due to Drs. S. F. Fields, M. R. Schrimpf, R. W. Chapman, S. R. Hitchcock, S. Lin, M. Scheideman, G. Wang, R-A. Rarig, and C. Cazorla as well as to my co-authors, whose combined experimental efforts kept this chemistry alive over many years. Financial support for the senior author’s current investigation of organoboron chemistry was generously supported by the taxpayers via the Institute of General Medical Sciences, NIH (GM067146)
Biographies
 Timothy De Vries was born in Highland, IN. He received his B.S. degree in Chemistry at Calvin College in Grand Rapids, MI, conducting research under Professor Ronald Blankespoor. He then moved to the University of Michigan, where he studied Lewis base complexes of borane as hydride sources, and C–B bond forming reactions of cationic, electrophilic boron intermediates under the direction of Professor Edwin Vedejs. He completed his Ph.D. in Organic Chemistry in 2007. After postdoctoral research investigating organolithium structures and kinetics under the supervision of Professor David Collum at Cornell University, Tim joined the Core R&D group within The Dow Chemical Company in 2010.
Timothy De Vries was born in Highland, IN. He received his B.S. degree in Chemistry at Calvin College in Grand Rapids, MI, conducting research under Professor Ronald Blankespoor. He then moved to the University of Michigan, where he studied Lewis base complexes of borane as hydride sources, and C–B bond forming reactions of cationic, electrophilic boron intermediates under the direction of Professor Edwin Vedejs. He completed his Ph.D. in Organic Chemistry in 2007. After postdoctoral research investigating organolithium structures and kinetics under the supervision of Professor David Collum at Cornell University, Tim joined the Core R&D group within The Dow Chemical Company in 2010.
 Aleksandrs Prokofjevs was born in 1985 and obtained his Bachelor’s degree from the University of Latvia, Riga, in 2006. While still in high school,. Aleksandrs began research work at the Latvian Institute of Organic Synthesis, Riga, under the supervision of Dr. Chem. Peteris Trapencieris, and continued this project throughout his undergraduate years. In 2006 he moved to the University of Michigan, Ann Arbor, where he is currently completing his Ph.D. degree with Prof. Edwin Vedejs. His current research is centered on the experimental and computational investigations of highly electrophilic boron cations and their use for synthetically valuable transformations, including borylations, C-H bond functionalizations, and hydroborations.
Aleksandrs Prokofjevs was born in 1985 and obtained his Bachelor’s degree from the University of Latvia, Riga, in 2006. While still in high school,. Aleksandrs began research work at the Latvian Institute of Organic Synthesis, Riga, under the supervision of Dr. Chem. Peteris Trapencieris, and continued this project throughout his undergraduate years. In 2006 he moved to the University of Michigan, Ann Arbor, where he is currently completing his Ph.D. degree with Prof. Edwin Vedejs. His current research is centered on the experimental and computational investigations of highly electrophilic boron cations and their use for synthetically valuable transformations, including borylations, C-H bond functionalizations, and hydroborations.
 Edwin Vedejs was born in 1941 in Riga, Latvia. His family left in 1944 because of the impending Soviet takeover and emigrated to the United States in 1950. After completion of his BS (University of Michigan, 1962) and PhD (University of Wisconsin, 1966), he spent a postdoctoral year with E. J. Corey at Harvard. Prof. Vedejs then joined the chemistry faculty at the University of Wisconsin (1967). In 1999, he moved to the University of Michigan as the Moses Gomberg Professor of Chemistry. He served as Associate Editor for Synthesis and Natural Products Chemistry, J. Am. Chem. Soc., 1994–1999, and as Chair of the Organic Division of ACS, 2003. His recent awards include the H. C. Brown Award for Creative Research in Synthetic Methods, 2004, and the Order of Three Stars, Latvia, 2006.
Edwin Vedejs was born in 1941 in Riga, Latvia. His family left in 1944 because of the impending Soviet takeover and emigrated to the United States in 1950. After completion of his BS (University of Michigan, 1962) and PhD (University of Wisconsin, 1966), he spent a postdoctoral year with E. J. Corey at Harvard. Prof. Vedejs then joined the chemistry faculty at the University of Wisconsin (1967). In 1999, he moved to the University of Michigan as the Moses Gomberg Professor of Chemistry. He served as Associate Editor for Synthesis and Natural Products Chemistry, J. Am. Chem. Soc., 1994–1999, and as Chair of the Organic Division of ACS, 2003. His recent awards include the H. C. Brown Award for Creative Research in Synthetic Methods, 2004, and the Order of Three Stars, Latvia, 2006.
References
- 1.Shitov OP, Ioffe SL, Tartakovskii VA, Novikov SS. Russ Chem Rev. 1970;39:905. [Google Scholar]
- 2.Kölle P, Nöth H. Chem Rev. 1985;85:399. [Google Scholar]
- 3.Piers WE, Bourke SC, Conroy KD. Angew Chem Int Ed. 2005;44:5016. doi: 10.1002/anie.200500402. [DOI] [PubMed] [Google Scholar]
- 4.(a) Wiberg E, Schuster K. Z Anorg Allg Chem. 1933;213:77. [Google Scholar]; (b) Wiberg E, Schuster K. Z Anorg Allg Chem. 1933;213:89. [Google Scholar]
- 5.Wiberg E, Hertwig K. Z Anorg Chem. 1947;255:141. [Google Scholar]
- 6.Brown CA, Osthoff RC. J Am Chem Soc. 1952;74:2340. [Google Scholar]
- 7.Goubeau J, Rahtz M, Becher HJ. Z Anorg Allg Chem. 1954;275:161. [Google Scholar]
- 8.Gerrard W, Hudson HR, Mooney EF. J Chem Soc. 1960:5168. [Google Scholar]
- 9.Ryschkewitsch GE, Myers WH. Synth React Inorg Metalorg Chem. 1975;5:123. [Google Scholar]
- 10.Ryschkewitsch GE, Wiggins JW. J Am Chem Soc. 1970;92:1790.Although 10 was not isolated, the closely related 2,6-lutidine derivative was recently prepared and was fully characterized, including the nearly identical 11B chemical shift (δ = 46.9 ppm) and an X-ray structure determination as the tetrachloroaluminate salt: Del Grosso A, Helm MD, Solomon SA, Caras-Quintero D, Ingleson MJ. Chem Commun. 2011;47:12459. doi: 10.1039/c1cc14226g.
- 11.Nöth H, Schweizer P, Ziegelgänsberger F. Chem Ber. 1966;99:1089. [Google Scholar]
- 12.Heaton GS, Riley PNK. J Chem Soc A. 1966:952. [Google Scholar]
- 13.Lowe JR, Uppal SS, Weidig C, Kelly HC. Inorg Chem. 1970;9:1423. [Google Scholar]
- 14.Krishnamurthy SS, Lappert MF. Inorg Nucl Chem Lett. 1971;7:919. [Google Scholar]
- 15.Benton BW, Miller JM. Can J Chem. 1974;52:2866. [Google Scholar]
- 16.Farquharson MJ, Hartman JS. Can J Chem. 1989;67:1711. [Google Scholar]
- 17.Farquharson MJ, Hartman JS. Can J Chem. 1996;74:1309. [Google Scholar]
- 18.Hartman JS, Yuan Z, Fox A, Nguyen A. Can J Chem. 1996;74:2131. [Google Scholar]
- 19.Hartman JS, Ilnicki EI, Shoemaker JAW, Szerminski WR, Yuan Z. Can J Chem. 1998;76:1317. [Google Scholar]
- 20.Shoemaker JAW, Hartman JS. Can J Chem. 1999;77:1856. [Google Scholar]
- 21.Hartman JS, Shoemaker JAW. Can J Chem. 2001;79:426. [Google Scholar]
- 22.Nainan KC, Ryschkewitsch GE. J Am Chem Soc. 1969;91:330. [Google Scholar]
- 23.Mathur MA, Ryschkewitsch GE. Inorg Chem. 1980;19:887. [Google Scholar]
- 24.Bratt PJ, Brown MP, Seddon KR. J Chem Soc, Dalton Trans. 1974:2161. [Google Scholar]
- 25.Bratt PJ, Brown MP, Seddon KR. J Chem Soc, Dalton Trans. 1976:353. [Google Scholar]
- 26.Denniston ML, Chuisano M, Brown J, Martin DR. J Inorg Nucl Chem. 1976;38:379. [Google Scholar]
- 27.Ref. 25 describes a puzzling solvolysis from 23 to a product said to be BH2(NH3)2I (fits elemental analysis; no NMR data) after reaction in liquid ammonia (44 h), followed by evaporation and heating to 120 °C under vacuum to remove unreacted 23. Conversion from 23 is said to be accelerated by addition of NaBr due to a special salt effect, and the authors use this evidence to argue that a species drawn as [Me3N·BH2]I is an intermediate. Presumably, an ion pair corresponding to 25 was intended. No follow-up work has appeared to confirm this suggestion as far as we are aware, and the earlier studies described in references 23,24 were not cited.
- 28.Nöth H, Fritz P. Z Anorg Allg Chem. 1963;322:297. [Google Scholar]
- 29.Narula CK, Nöth H. Inorg Chem. 1984;23:4147. [Google Scholar]
- 30.Both the Nöth and the Piers reviews classify several methods for boron cation formation, and Piers lists Methods 1–6 in the same order as Nöth’s text headings IIA–IIF. However, Nöth’s text headings A–D are not the same as Methods A–D defined below his Table I. Our discussion presents methods specific to borenium ions according to mechanistic categories and makes comparisons with the Nöth/Piers text headings in the simpler cases.
- 31.Corey EJ, Shibata T, Lee TW. J Am Chem Soc. 2002;124:3808. doi: 10.1021/ja025848x. [DOI] [PubMed] [Google Scholar]
- 32.For a review, see: Corey EJ, Helal CJ. Angew Chem Int Ed. 1998;37:1986. doi: 10.1002/(SICI)1521-3773(19980817)37:15<1986::AID-ANIE1986>3.0.CO;2-Z.
- 33.Uddin MK, Nagano Y, Fujiyama R, Kiyooka S, Fujio M, Tsuno Y. Tetrahedron Lett. 2005;46:627. [Google Scholar]
- 34.Kiyooka S, Fujiyama R, Kawai T, Fujimoto H, Goh K. Tetrahedron Lett. 2001;42:4151. [Google Scholar]
- 35.Chiu CW, Gabbaï FP. Organometallics. 2008;27:1657. [Google Scholar]
- 36.Matsumoto T, Gabbaï FP. Organometallics. 2009;28:4252. [Google Scholar]
- 37.Wood TK, Piers WE, Keay BA, Parvez M. Chem Commun. 2009:5147. doi: 10.1039/b911982e. [DOI] [PubMed] [Google Scholar]
- 38.(a) Bonnier C, Piers WE, Parvez M, Sorensen TS. Chem Commun. 2008:4593. doi: 10.1039/b808739c. [DOI] [PubMed] [Google Scholar]; (b) Bonnier C, Piers WE, Parvez M. Organometallics. 2011;30:1067. [Google Scholar]
- 39.Kato T, Tham FS, Boyd PDW, Reed CA. Heteroatom Chem. 2006;17:209.Similar subporphyrin borenium salts have also been prepared by abstraction of a methoxy group in place of chloride: Tsurumaki E, Hayashi S, Tham FS, Reed CA, Osuka A. J Am Chem Soc. 2011;133:11956. doi: 10.1021/ja2056566.For a review on the chemistry of subphthalocyanines, see: Claessens CG, González-Rodríguez D, Torres T. Chem Rev. 2002;102:835. doi: 10.1021/cr0101454.
- 40.Kuhn N, Kuhn A, Lewandowski J, Speis M. Chem Ber. 1991;124:2197. [Google Scholar]
- 41.Qian B, Baek SW, Smith MR., III Polyhedron. 1999;18:2405.Cowley AH, Lu Z, Jones JN, Moore JA. J Organomet Chem. 2004;689:2562.Similar heteroaromatic borenium cations have been prepared by the protonation of a conjugated cyclic dienamine with TfOH: Someya CI, Inoue S, Präsang C, Irran E, Driess M. Chem Commun. 2011;47:6599. doi: 10.1039/c1cc11789k.Heteroaromatic borenium salts can be generated by the reaction of 1-ethyl-2-trifluorosulfonyloxy-1,2-azaborine with nucleophiles following an addition-heterolysis sequence (see Scheme 5 for mechanistic analogies): Marwitz AJV, Jenkins JT, Zakharov LN, Liu S-Y. Angew Chem Int Ed. 2010;49:7444. doi: 10.1002/anie.201004084.Marwitz AJV, Jenkins JT, Zakharov LN, Liu S-Y. Organometallics. 2011;30:52. doi: 10.1021/om101083p.
- 42.Del Grosso A, Pritchard RG, Muryn CA, Ingleson MJ. Organometallics. 2010;29:241. [Google Scholar]
- 43.Hettich RL, Cole T, Freiser BS. Int J Mass Spectrom. 1987;81:203.For other gas phase studies of the borinium/borenium interface, see: Forte L, Lien MH, Hopkinson AC, Bohme DK. Can J Chem. 1989;67:1576.Ranatunga TD, Kenttämaa HI. J Am Chem Soc. 1992;114:8600.DePuy CH, Gareyev R, Hankin J, Davico GE, Damrauer R. J Am Chem Soc. 1997;119:427.DePuy CH, Gareyev R, Hankin J, Davico GE, Krempp M, Damrauer R. J Am Chem Soc. 1998;120:5086.
- 44.Dureen MA, Lough A, Gilbert TM, Stephan DW. Chem Commun. 2008:4303. doi: 10.1039/b808348g. [DOI] [PubMed] [Google Scholar]
- 45.Lata CJ, Crudden CM. J Am Chem Soc. 2010;132:131. doi: 10.1021/ja904142m. [DOI] [PubMed] [Google Scholar]
- 46.Benjamin LE, Carvalho DA, Stafiej SF, Takacs EA. Inorg Chem. 1970;9:1844. [Google Scholar]
- 47.(a) Vedejs E, Nguyen T, Powell DR, Schrimpf MR. Chem Commun. 1996:2721. [Google Scholar]; (b) De Vries TS, Prokofjevs A, Harvey JN, Vedejs E. J Am Chem Soc. 2009;131:14679. doi: 10.1021/ja905369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Vries TS, Vedejs E. Organometallics. 2007;26:3079. doi: 10.1021/om070228w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stephens FH, Baker RT, Matus MH, Grant DJ, Dixon DA. Angew Chem Int Ed. 2007;46:746. doi: 10.1002/anie.200603285.Hydride bridged cationic phosphine boranes were considered as potential reaction intermediates, but spectroscopic data were not reported: Kameda M, Kodama G. Inorg Chem. 1997;36:4369. doi: 10.1021/ic9705195.
- 50.Prokofjevs A. Ph D Dissertation. University of Michigan; 2012. [Google Scholar]
- 51.Narula CK, Nöth H. Inorg Chem. 1985;24:2532.(b) The minor 11B chemical shift (δ = 59 ppm) is consistent with contamination by (9-BBN)2O.
- 52.Prokofjevs A, Kampf JW, Vedejs E. Angew Chem Int Ed. 2011;50:2098. doi: 10.1002/anie.201005663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider WF, Narula CK, Nöth H, Bursten BE. Inorg Chem. 1991;30:3919. [Google Scholar]
- 54.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215. [Google Scholar]
- 55.(a) Plumley JA, Evanseck JD. J Chem Theory Comput. 2008;4:1249. doi: 10.1021/ct800210e. [DOI] [PubMed] [Google Scholar]; Plumley JA, Evanseck JD. J Phys Chem A. 2009;113:5985. doi: 10.1021/jp811202c. [DOI] [PubMed] [Google Scholar]; Janesko BG. J Chem Theory Comput. 2010;6:1825. doi: 10.1021/ct1000846. [DOI] [PubMed] [Google Scholar]; (b) Brown HC, Bartholomay H, Taylor MD. J Am Chem Soc. 1944;66:435. [Google Scholar]
- 56.Ryschkewitsch GE, Garrett JM. J Am Chem Soc. 1968;90:7234. [Google Scholar]
- 57.Toyota S, Ito F, Nitta N, Hakamata T. Bull Chem Soc Jpn. 2004;77:2081. [Google Scholar]
- 58.(a) Vedejs E, Fields SC, Hayashi R, Hitchcock SR, Powell DR, Schrimpf MR. J Am Chem Soc. 1999;121:2460. [Google Scholar]; (b) Vedejs E, Fields SC, Lin S, Schrimpf MR. J Org Chem. 1995;60:3028. [Google Scholar]
- 59.Chapman RW. Ph D Dissertation. University of Wisconsin; 1994. [Google Scholar]
- 60.Vedejs E, Chapman RW, Lin S, Müller M, Powell DR. J Am Chem Soc. 2000;122:3047. [Google Scholar]
- 61.Kaiser PF, White JM, Hutton CA. J Am Chem Soc. 2008;130:16450. doi: 10.1021/ja8044629. [DOI] [PubMed] [Google Scholar]
- 62.Braun M, Schlecht S, Engelmann M, Frank W, Grimme S. Eur J Org Chem. 2008;2008:5221. [Google Scholar]
- 63.Heard PJ. Chem Soc Rev. 2007;36:551. doi: 10.1039/b514855n. [DOI] [PubMed] [Google Scholar]
- 64.(a) Itsuno S, Ito K, Hirao A, Nakahama S. Chem Commun. 1983:469. [Google Scholar]; (b) Itsuno S, Sakurai Y, Ito K, Hirao A, Nakahama S. Bull Chem Soc Jpn. 1987;60:395. [Google Scholar]
- 65.(a) Corey EJ, Bakshi RK, Shibata S. J Am Chem Soc. 1987;109:5551. [Google Scholar]; (b) Corey EJ, Bakshi RK, Shibata S, Chen C-P, Singh VK. J Am Chem Soc. 1987;109:7925. [Google Scholar]
- 66.Oxazaborolidines have also been demonstrated to catalyze the enantioselective reduction of oximes in ref 64b and in the following related reports: Sakito Y, Yoneyoshi Y, Suzukamo G. Tetrahedron Lett. 1988;29:223.Itsuno S, Sakurai Y, Shimizu K, Ito K. J Chem Soc, Perkin Trans. 1990;1:1859.
- 67.Corey EJ, Cimprich KA. J Am Chem Soc. 1994;116:3151. [Google Scholar]
- 68.For chiral borane catalysts, see Corey EJ. Angew Chem, Int Ed. 2002;41:1650. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b.For chiral borenium catalysts, see Corey EJ. Angew Chem, Int Ed. 2009;48:2100. doi: 10.1002/anie.200805374.
- 69.Hayashi Y, Rohde JJ, Corey EJ. J Am Chem Soc. 1996;118:5502. [Google Scholar]
- 70.(a) Beall H, Bushweller CH, Dewkett WJ, Grace M. J Am Chem Soc. 1970;92:3484. [Google Scholar]; (b) Bacon J, Gillespie RJ, Hartman JS, Rao URK. Mol Phys. 1970;18:561. [Google Scholar]
- 71.Moniz WB, Gutowsky HS. J Chem Phys. 1963;38:1155. [Google Scholar]
- 72.Ryu DH, Corey EJ. J Am Chem Soc. 2003;125:6388. doi: 10.1021/ja035393r. [DOI] [PubMed] [Google Scholar]
- 73.Ryu DH, Lee TW, Corey EJ. J Am Chem Soc. 2002;124:9992. doi: 10.1021/ja027468h. [DOI] [PubMed] [Google Scholar]
- 74.Canales E, Corey EJ. Org Lett. 2008;10:3271. doi: 10.1021/ol8011502. [DOI] [PubMed] [Google Scholar]
- 75.Liu D, Canales E, Corey EJ. J Am Chem Soc. 2007;129:1498. doi: 10.1021/ja068637r. [DOI] [PubMed] [Google Scholar]
- 76. [accessed 2/15/11]; Oxazaborolidine 37 is available from the Aldrich Chemical company: http://www.sigmaaldrich.com/catalog/ProductDetail.do?lang=en&N4=654302|ALDRICH&N5=SEARCH_CONCAT_PNO.
- 77.Hu QY, Rege PD, Corey EJ. J Am Chem Soc. 2004;126:5984. doi: 10.1021/ja048808x. [DOI] [PubMed] [Google Scholar]
- 78.Payette JN, Yamamoto H. J Am Chem Soc. 2007;129:9536. doi: 10.1021/ja0735958. [DOI] [PubMed] [Google Scholar]
- 79.For comparisons of HC(C6F5)Tf2, TfOH, and Tf2NH, see Hasegawa A, Ishihara K, Yamamoto H. Angew Chem, Int Ed. 2003;42:5731. doi: 10.1002/anie.200352382.
- 80.Payette JN, Yamamoto H. Angew Chem, Int Ed. 2009;48:8060. doi: 10.1002/anie.200904339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Futatsugi K, Yamamoto H. Angew Chem, Int Ed. 2005;44:1484. doi: 10.1002/anie.200461319. [DOI] [PubMed] [Google Scholar]
- 82.Liu D, Hong S, Corey EJ. J Am Chem Soc. 2006;128:8160. doi: 10.1021/ja063332y. [DOI] [PubMed] [Google Scholar]
- 83.Canales E, Corey EJ. J Am Chem Soc. 2007;129:12686. doi: 10.1021/ja0765262. [DOI] [PubMed] [Google Scholar]
- 84.Senapati BK, Hwang GS, Lee S, Ryu DH. Angew Chem, Int Ed. 2009;48:4398. doi: 10.1002/anie.200900351. [DOI] [PubMed] [Google Scholar]
- 85.(a) Ryu DH, Corey EJ. J Am Chem Soc. 2004;126:8106. doi: 10.1021/ja0475959. [DOI] [PubMed] [Google Scholar]; (b) Ryu DH, Corey EJ. J Am Chem Soc. 2005;127:5384. doi: 10.1021/ja050543e. [DOI] [PubMed] [Google Scholar]; (c) Corey EJ, Cimprich KA. J Am Chem Soc. 1994;116:3151. [Google Scholar]
- 86.Wei P, Atwood DA. Inorg Chem. 1998;37:4934. doi: 10.1021/ic971595a. [DOI] [PubMed] [Google Scholar]
- 87.Evans DA, Muci AR, Stuermer R. J Org Chem. 1993;58:5307. [Google Scholar]
- 88.(a) Kennedy JWJ, Hall DG. J Am Chem Soc. 2002;124:11586. doi: 10.1021/ja027453j. [DOI] [PubMed] [Google Scholar]; (b) Rauniyar V, Hall DG. J Am Chem Soc. 2004;126:4518. doi: 10.1021/ja049446w. [DOI] [PubMed] [Google Scholar]
- 89.Ishiyama T, Ahiko T, Miyaura N. J Am Chem Soc. 2002;124:12414. doi: 10.1021/ja0210345.For a computational evaluation of AlCl3 activation supporting coordination to a B–O substituent see Sakata K, Fujimoto H. J Am Chem Soc. 2008;130:12519. doi: 10.1021/ja804168z.
- 90.Chen M, Roush WR. Org Lett. 2010;12:2706. doi: 10.1021/ol1007444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.(a) Yu SH, Ferguson MJ, McDonald R, Hall DG. J Am Chem Soc. 2005;127:12808. doi: 10.1021/ja054171l. [DOI] [PubMed] [Google Scholar]; (b) Elford TG, Arimura Y, Yu SH, Hall DG. J Org Chem. 2007;72:1276. doi: 10.1021/jo062151b. [DOI] [PubMed] [Google Scholar]
- 92.Rauniyar V, Hall DG. J Org Chem. 2009;74:4236. doi: 10.1021/jo900553f. and references therein. [DOI] [PubMed] [Google Scholar]
- 93.Ishihara K, Ohara S, Yamamoto H. J Org Chem. 1996;61:4196. doi: 10.1021/jo9606564.Latta R, Springsteen G, Wang B. Synthesis. 2001:1611.Maki T, Ishihara K, Yamamoto H. Org Lett. 2005;7:5047–5050. doi: 10.1021/ol052061d.For the related esterification of α-hydroxycarboxylic acids see Maki T, Ishihara K, Yamamoto H. Org Lett. 2005;7:5043. doi: 10.1021/ol052060l.
- 94.Agou T, Kobayashi J, Kawashima T. Inorg Chem. 2006;45:9137. doi: 10.1021/ic061055q. [DOI] [PubMed] [Google Scholar]
- 95.Welch GC, Cabrera L, Chase PA, Hollink E, Masuda JD, Wei P, Stephan DW. Dalton Trans. 2007:3407. doi: 10.1039/b704417h. [DOI] [PubMed] [Google Scholar]
- 96.Hudnall TW, Chiu C-W, Gabbaï FP. Acc Chem Res. 2009;42:388. doi: 10.1021/ar8001816. [DOI] [PubMed] [Google Scholar]
- 97.(a) Lee MH, Agou T, Kobayashi J, Kawashima T, Gabbaï FP. Chem Commun. 2007:1133. doi: 10.1039/b616814k. [DOI] [PubMed] [Google Scholar]; (b) Hudnall TW, Kim Y-M, Bebbington MWP, Bourissou D, Gabbaï FP. J Am Chem Soc. 2008;130:10890. doi: 10.1021/ja804492y. [DOI] [PubMed] [Google Scholar]; (c) Kim Y, Gabbaï FP. J Am Chem Soc. 2009;131:3363. doi: 10.1021/ja8091467. [DOI] [PubMed] [Google Scholar]
- 98.Hudnall TW, Gabbaï FP. J Am Chem Soc. 2007;129:11978. doi: 10.1021/ja073793z. [DOI] [PubMed] [Google Scholar]
- 99.(a) Kim Y, Zhao H, Gabbaï FP. Angew Chem, Int Ed. 2009;48:4957. doi: 10.1002/anie.200901275. [DOI] [PubMed] [Google Scholar]; (b) Hudnall TW, Gabbaï FP. Chem Commun. 2008:4596. doi: 10.1039/b808740g. [DOI] [PubMed] [Google Scholar]
- 100.(a) Chiu C-W, Gabbaï FP. Dalton Trans. 2008:814. doi: 10.1039/b715534d. [DOI] [PubMed] [Google Scholar]; (b) Xu Z, Kim SK, Han SJ, Lee C, Kociok-Kohn G, James TD, Yoon J. Eur J Org Chem. 2009:3058. [Google Scholar]
- 101.Review: Wade CR, Broomsgrove AEJ, Aldridge S, Gabbaï F. Chem Rev. 2010;110:3958. doi: 10.1021/cr900401a.Chiu C-W, Kim Y, Gabbaï FP. J Am Chem Soc. 2009;131:60. doi: 10.1021/ja808572t.
- 102.Fe cations: Dusemund C, Samankumara Sandanayake KRA, Shinkai S. J Chem Soc, Chem Commun. 1995:333.Venkatasubbaiah K, Nowik I, Herber RH, Jäkle F. Chem Commun. 2007:2154. doi: 10.1039/b701807j.Day JK, Bresner C, Coombs ND, Fallis IA, Ooi L-L, Aldridge S. Inorg Chem. 2008;47:793. doi: 10.1021/ic701494p.Ir cations: Zhao Q, Li F, Liu S, Yu M, Liu Z, Yi T, Huang C. Inorg Chem. 2008;47:9256. doi: 10.1021/ic800500c.Broomsgrove AEJ, Addy DA, Di Paolo A, Morgan IR, Bresner C, Chislett V, Fallis IA, Thompson AL, Vidovic D, Aldridge S. Inorg Chem. 2010;49:157. doi: 10.1021/ic901673u.Pyridinium cations: Wade CR, Gabbaï FP. Dalton Trans. 2009:9169. doi: 10.1039/b913030f.Ru cations: Wade CR, Gabbaï FP. Inorg Chem. 2010;49:714. doi: 10.1021/ic9020349.
- 103.Reviews: Vidovic D, Pierce GA, Aldridge S. Chem Commun. 2009:1157. doi: 10.1039/b816042b.Braunschweig H, Dewhurst RD, Schneider A. Chem Rev. 2010;110:3924. doi: 10.1021/cr900333n.Braunschweig H, Chiu C-W, Radacki K, Brenner P. Chem Commun. 2010:916. doi: 10.1039/b923652j.Addy DA, Pierce GA, Vidovic D, Mallick D, Jemmis ED, Goicoechea JM, Aldridge S. J Am Chem Soc. 2010;132:4586. doi: 10.1021/ja101192e.Saleh LMA, Birjkumar KH, Protchenko AV, Scwartz AD, Aldridge S, Jones C, Kaltsoyannis N, Mountford P. J Am Chem Soc. 2011;133:3836. doi: 10.1021/ja2007092.
- 104.Kiyooka S, Fujiyama R, Uddin MK, Goh K, Nagano Y, Fujio M, Tsuno Y. Tetrahedron Lett. 2005;46:209. [Google Scholar]
- 105.Karatjas AG, Vedejs E. J Org Chem. 2008;73:9508. doi: 10.1021/jo8020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scheideman M, Wang G, Vedejs E. J Am Chem Soc. 2008;130:8669. doi: 10.1021/ja0774663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.De Vries TS. Ph D Dissertation. University of Michigan; 2007. [Google Scholar]
- 108.Clay JM. Ph D Dissertation. University of Michigan; 2007. [Google Scholar]
- 109.See ref. 52, footnote 39.
- 110.Genaev AM, Nagy SM, Salnikov GE, Shubin VG. Chem Commun. 2000:1587. [Google Scholar]
- 111.The originally reported toluene ortho-borylation product has been reassigned as the meta-regioisomer (personal communication from Prof. M. J. Ingleson).
- 112.Del Grosso A, Singleton PJ, Muryn CA, Ingleson MJ. Angew Chem, Int Ed. 2011;50:2102. doi: 10.1002/anie.201006196. [DOI] [PubMed] [Google Scholar]
- 113.Cho J-Y, Iverson CN, Smith MR., III J Am Chem Soc. 2000;122:12868.Paul S, Chotana GA, Holmes D, Reichle RC, Maleczka RE, Jr, Smith MR., III J Am Chem Soc. 2006;128:15552. doi: 10.1021/ja0631652.Murphy JM, Tzschucke CC, Hartwig JF. Orglett. 2007;9:757. doi: 10.1021/ol062903o.Review: Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890. doi: 10.1021/cr900206p.
- 114.Muetterties EL. J Am Chem Soc. 1959;81:2597. [Google Scholar]
- 115.Bujwid ZJ, Gerrard W, Lappert MF. Chem & Ind. 1959:1091. [Google Scholar]
- 116.Muetterties EL. J Am Chem Soc. 1960;82:4163. [Google Scholar]
- 117.Muetterties EL, Tebbe FN. Inorg Chem. 1968;7:2663. [Google Scholar]
- 118.Olah GA. Angew Chem, Int Ed. 1993;32:767. [Google Scholar]
- 119.Hurd DT. J Am Chem Soc. 1948;70:2053. [Google Scholar]
- 120.Ishida N, Moriya T, Goya T, Murakami M. J Org Chem. 2010;75:8709. doi: 10.1021/jo101920p. [DOI] [PubMed] [Google Scholar]
- 121.Weber L, Dobbert E, Stammler H-G, Neumann B, Boese R, Bläser D. Chem Ber. 1997;130:705. [Google Scholar]
- 122.McArthur D, Butts CP, Lindsay DM. Chem Commun. 2011;47:6650. doi: 10.1039/c1cc10767d. [DOI] [PubMed] [Google Scholar]
- 123.Wang Y, Robinson GH. Inorg Chem. 2011;50:12326. doi: 10.1021/ic200675u. [DOI] [PubMed] [Google Scholar]
- 124.Mansaray HB, Rowe ADL, Phillips N, Niemeyer J, Kelly M, Addy DA, Bates JI, Aldridge S. Chem Commun. 2011;47:12295. doi: 10.1039/c1cc15259a. [DOI] [PubMed] [Google Scholar]
- 125.Inés B, Patil M, Carreras J, Goddard R, Thiel W, Alcarazo M. Angew Chem Int Ed. 2011;50:8400. doi: 10.1002/anie.201103197. [DOI] [PubMed] [Google Scholar]
- 126.Meier G, Braun T. Angew Chem, Int Ed. 2009;48:1546. doi: 10.1002/anie.200805237. [DOI] [PubMed] [Google Scholar]
- 127.Piers WE. Angew Chem, Int Ed. 2000;39:1923. doi: 10.1002/1521-3773(20000602)39:11<1923::aid-anie1923>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 128.Short review: Staubitz A, Robertson APM, Sloan ME, Manners I. Chem Rev. 2010;110:4023. doi: 10.1021/cr100105a.
- 129.Ghesner I, Piers WE, Parvez M, McDonald R. Chem Commun. 2005:2480. doi: 10.1039/b502448j. [DOI] [PubMed] [Google Scholar]
- 130.Ott H, Matthes C, Ringe A, Magull J, Stalke D, Klingbeil U. Chem Eur J. 2009;15:4602. doi: 10.1002/chem.200802669. [DOI] [PubMed] [Google Scholar]
- 131.Tsai J-H, Lin S-T, Yang RB-G, Yap GPA, Ong T-G. Organometallics. 2010;29:4004. [Google Scholar]
- 132.Hayashi Y, Segawa Y, Yamashita M, Nozaki K. Chem Commun. 2011;47:5888. doi: 10.1039/c1cc11334h. [DOI] [PubMed] [Google Scholar]
- 133.Prokofjevs A, Kampf JW, Vedejs E. J Am Chem Soc. 2011;133:20056. doi: 10.1021/ja208093c. [DOI] [PMC free article] [PubMed] [Google Scholar]