

Abstract
Chemical cross-linking combined with mass spectrometry is a rapidly developing technique for structural proteomics. Cross-linked proteins are usually digested with trypsin to generate cross-linked peptides, which are then analyzed by mass spectrometry. The most informative cross-links, the interpeptide cross-links, are often large in size, because they consist of two peptides that are connected by a cross-linker. In addition, trypsin targets the same residues as amino-reactive cross-linkers, and cleavage will not occur at these cross-linker-modified residues. This produces high molecular weight cross-linked peptides, which complicates their mass spectrometric analysis and identification. In this paper, we examine a nonspecific protease, proteinase K, as an alternative to trypsin for cross-linking studies. Initial tests on a model peptide that was digested by proteinase K resulted in a “family” of related cross-linked peptides, all of which contained the same cross-linking sites, thus providing additional verification of the cross-linking results, as was previously noted for other post-translational modification studies. The procedure was next applied to the native (PrPC) and oligomeric form of prion protein (PrPβ). Using proteinase K, the affinity-purifiable CID-cleavable and isotopically coded cross-linker cyanurbiotindipropionylsuccinimide and MALDI-MS cross-links were found for all of the possible cross-linking sites. After digestion with proteinase K, we obtained a mass distribution of the cross-linked peptides that is very suitable for MALDI-MS analysis. Using this new method, we were able to detect over 60 interpeptide cross-links in the native PrPC and PrPβ prion protein. The set of cross-links for the native form was used as distance constraints in developing a model of the native prion protein structure, which includes the 90–124-amino acid N-terminal portion of the protein. Several cross-links were unique to each form of the prion protein, including a Lys185–Lys220 cross-link, which is unique to the PrPβ and thus may be indicative of the conformational change involved in the formation of prion protein oligomers.
Structural proteomics, which combines protein chemistry methods with modern mass spectrometry techniques for protein and peptides, is an emerging technology in structural biology. One of the tools for modern structural proteomics is chemical cross-linking combined with mass spectrometry (1–5). Cross-linking analysis provides the distance between two cross-linked amino acid residues, whereas mass spectrometry provides information on which residues are connected. The basis of this method is a chemical reaction of the cross-linking reagent with functional groups on a protein, leading to the formation of a covalent bond between each end of the reagent and the protein. The distance between two cross-linked sites is determined by the length of the spacer in the cross-linking reagent. Thus, identification of the cross-linked sites on a protein or a protein complex provides spatial information and distance constraints for the two amino acid residues that are connected by the cross-linker.
The use of chemical cross-linking combined with mass spectrometry is a rapidly developing technique for structural proteomics (4, 6–8). In recent years, we and others have been developing an array of cross-linking and modification reagents, software, and methods specifically designed for protein cross-linking experiments combined with MS analysis (9–18). For mass spectrometric determination of the cross-linking sites, the cross-linked protein is typically digested with proteolytic enzymes, and the peptides are examined by mass spectrometry.
Challenges associated with the cross-linking technique include finding the cross-linked peptides in the complex mixture that often results from this digestion. For this reason, isotopically coded cross-linkers are often used to produce a distinct “signature” for the cross-links in the mass spectra (19).
A cross-linker that can be cleaved with collision-induced dissociation is also highly beneficial because it facilitates identification of the linked peptides and determination of the cross-linking sites by MS/MS sequencing, after a peak corresponding to a cross-link (a cross-linked pair of peptides) has been found (20). To simplify the mass spectra and enhance the detectablility of the cross-linked peptides, affinity enrichment can be used (21).
Affinity purification is also important for enriching the sample in cross-linker-containing peptides. Cross-linkers with a biotin moiety incorporated in them, for example, can be enriched on avidin beads. Even these methods, however, will enrich the sample in all types of cross-links: dead-end and intrapeptide, as well as the more desirable interpeptide cross-links.
Another challenge in cross-linking studies is the lack of suitable cleavage sites. Although trypsin is the most widely used enzyme for proteomics studies, the existence of only a few tryptic cleavage sites in some proteins, including prions, limit the power of this traditional proteolytic enzyme, especially when used with amino-reactive cross-linkers, which target the same amino acids as trypsin and prevent cleavage at the modified sites. This often results in large interpeptide cross-links that are above the optimum range for successful detection by MALDI-MS and MALDI-MS/MS fragmentation. Double digestion with two different high specificity enzymes may somewhat reduce the overall size of the cross-links, but in many cases the interpeptide cross-links still remain fairly large.
In this paper, we examine the use of cyanurbiotindipropionylsuccinimide (CBDPS),1 an isotopically labeled affinity-purifiable CID-cleavable cross-linker, in combination with a nonspecific enzyme, proteinase K, to overcome all of these limitations. Proteinase K, a nonspecific protease, has already been successfully used for the mass spectrometric characterization of difficult-to-digest proteins (22, 23), and for the localization of disulfide bonds (24). Proteinase K digestion of a CBDPS-linked model peptide helped reveal the cleavage characteristics of this enzyme/cross-linker combination. We then applied the CBDPS/proteinase K method to two different conformations of the prion protein: the native noninfectious form (PrPC) and the β-oligomeric form (PrPβ).
MATERIALS AND METHODS
All materials were from Sigma-Aldrich unless otherwise noted.
Model Cross-linked Peptide
The model peptide Ac-TRTESTDIKRASSREADYLINKER (Creative Molecules Inc., Victoria, Canada) was cross-linked with an equimolar amount of CBDPS-H8/D8 reagent (cyanurbiotindipropionylsuccinimide; Creative Molecules Inc.), as previously described (10). The pH of the mixture was adjusted to 8.0–8.5 by the addition of 0.2 m Na2HPO4. The reaction mixture was incubated for 30 min at 25 °C and quenched with 50 mm ammonium bicarbonate. The cross-linked peptide was then digested with proteinase K (Invitrogen) for 1 h at 37 °C at a 1:1 (w/w) enzyme:substrate ratio.
Prion PrPC Protein
All of the prion constructs were obtained from Dr. Wishart's laboratory at the University of Alberta via the Prion Protein and Plasmid Production Platform Facility (PrP5) (www.prp5.ca) of PrioNet Canada (http://www.prionetcanada.ca/). Specifically, a synthetic gene corresponding to the Syrian hamster prion protein sequence 90–232 (shPrP90–232) with a 23-residue N-terminal fusion tag containing His6 and a thrombin cleavage site (MGSSHHHHHHSSGLVPRGSHMLE) was synthesized by DNA 2.0. The gene was cloned into a pET15b expression vector between XhoI and EcoRI restriction sites and heat shock transformed into Escherichia coli strain BL21 (DE3). For expression, the transformed cells were grown in 100 ml of LB plus 100 μg/ml ampicillin overnight to generate a starter culture. Between 1.5 and 2% of this starter culture was then used to inoculate 1 liter of LB medium (giving a starting A600 of 0.1). The cells were allowed to reach an A600 between 0.8 and 1.0 before induction with 1 mm isopropyl β-d-thiogalactopyranoside. Twelve to eighteen hours later, the cells were harvested by centrifugation at 5000 rpm for 30 min at 4 °C. The inclusion of the His6 tag afforded a standardized nickel affinity purification strategy previously described by Zahn et al. (27). PrPβ was generated by exposing the native PrPC protein to low pH (1.0) for 1 h and then dialyzing it back to pH 5.5. The details of the purification and PrPC to PrPβ conversion protocol have been described elsewhere (28).
Cross-linking Analysis of Prion Proteins
A 10-μl aliquot of a 1 mg/ml solution of PrPC and PrPβ in PBS was mixed with 1 μl of a 0.5 mm CBDPS-H8/D8 solution in water, prepared from a 50 mm stock solution of the cross-linker in DMSO. The final concentration of the cross-linker reagent was chosen based on the preliminary titration of the reaction mixture where the intensity of the cross-linked dimer band of the β-oligomeric form of the prion protein (PrPβ) on SDS-PAGE was maximized, without the appearance of any nonspecific higher oligomeric cross-linked products of the native noninfectious prion protein (PrPC). The pH of the mixture was adjusted to 8.0–8.5 by the addition of 0.2 m Na2HPO4. The reaction mixture was incubated for 30 min at 25 °C and quenched with 50 mm ammonium bicarbonate. The cross-linked proteins were then digested with proteinase K, for 2.5 h at 37 °C at a 1:1 (w/w) enzyme:substrate ratio. A protease inhibitor mixture (P8465; Sigma-Aldrich) was added to the resulting peptide mixture.
Affinity Enrichment of CBDPS Cross-links
Monomeric avidin beads (ThermoFisher, Thermo Scientific, Mississauga, Canada) were used for the affinity enrichment. The mixture was affinity-purified with 80 μl of monomeric avidin-agarose beads slurry. The beads were washed three times with 120 μl of 100 mM ammonium acetate and then with water, and the affinity-bound material was eluted with 100 μl of 0.1% TFA and 100 μl of 0.1%TFA, 50% acetonitrile. Aliquots from the loading, flow-through, wash, and elution fractions were desalted using Zip-Tips C18 (Millipore) and were analyzed by MALDI-MS.
Nano-ESI-MS Analysis
ESI mass spectrometric analyses were performed on the Orbitrap Velos (Thermo Scientific) mass spectrometer, in the positive ion mode. The PK digest was loaded onto a C18 Zip-tip (Millipore, Billerica, MA) and eluted with 60% methanol, 0.1% formic acid. Nano-ESI of the eluate was performed using Proxeon nanoES capillaries (Thermo Scientific) at 1.6-kV spray voltage. ESI-MS spectra were acquired manually over an m/z range of 400–2000, using FT detection profile mode with a resolution of 60,000 and an isolation width of 1 Da. ESI-MS/MS spectra were manually acquired using CID fragmentation in the FT profile mode, with an isolation width of 2 Da, a collision energy setting of 30%, and a resolution of 60,000. Thermo Scientific Excalibur software, version 2.1.0 QF03489 build 1140, was used for the data acquisition. Thermo Proteome Discoverer 1.3, version 1.3.0.339 was used to generate the .mgf files.
LC-MALDI Analysis
The eluted fractions were combined, concentrated by lyophilization, and separated by nano-flow reversed phase HPLC on an Ultimate nano-LC system (Dionex/LC Packings, Sunnyvale, CA) equipped with an LC Packings (Sunnyvale, CA) 0.3 × 5 mm C18 PepMap trapping column (5-μm particle size, 100 Å pore size), and a 75-μm × 15-cm capillary column packed in-house with Magic C18 Aq (Michrom Bioresources Inc., Auburn, CA) particles (5 μm, 100 Å). This capillary LC system was operated at a flow rate of 300 nl/min, using a 55-min gradient from 5 to 60% acetonitrile (0.1% TFA). The column effluent was spotted at 1-min intervals (300 nl/spot) onto a stainless steel MALDI target using a Shimadzu (Kyoto, Japan) spotter. The spots were dried, overlaid with 4 mg/ml α-cyano-4-hydroxycinnamic acid matrix solution in 0.1% TFA, 50% acetonitrile, and analyzed by MALDI-MS and MS/MS, using a 4800 MALDI-TOF/TOF (AB Sciex, Concord, Canada). MS/MS spectra were acquired using “CID off,” 50-FHWM gate width, and a 1-kV MSMS method. In cases where additional fragmentation was desirable for the unambiguous cross-link assignments, spectra were reacquired using “CID on” and a 2-kV MSMS method.
The mass spectra were analyzed using the ICC-CLASS software (11) and the ICCLMSMS program (9) (version date: August 1, 2011; available free at www.creativemolecules.com/CM_Software.htm). Mass spectra peaklists were generated using Data Explorer Version 4.0 (AB Sciex). The mass spectra from each chromatographic fraction were searched for the undeuterated/deuterated CBDPS-cross-linked (D0/D8) doublets using the DX program of ICC-CLASS, with peak intensity cutoff of 50 and a mass tolerance setting of 0.01 Da for the 8.05824 Da theoretical mass difference between the masses of the light and heavy isotopes. The D0/D8 mass list obtained was used as inclusion list for automatic MS/MS spectra acquisition. The acquired MS/MS spectra were searched for isotopic signatures characteristic of CID cleavage of cross-links 401 Da apart, using the ICCLMSMS program. Interpeptide cross-links were recognized based on their CID cleavage patterns and were identified by the DXMSMS module of the ICC-CLASS software package based on the cross-link mass, the masses of the CID cleavage products, and the masses of the fragments of the cross-link, using the following settings: 100-ppm mass tolerance for the precursor mass, 300-ppm mass tolerance for MS/MS fragment ions, all possible cleavage sites, and KYST as allowed cross-linking sites.
Molecular Modeling
The NMR structure of hamster prion protein (Protein Data Bank code 1B10) (29) was used as the initial template, which covers only residues 125–231. The missing residues from the flexible N terminus (Gly68–Gly124) were modeled in an extended conformation and were connected to the initial template via superpositioning, followed by energy minimization of these residues using XPLOR-NIH (30).
MS cross-linking data was converted into distance constraints between the side chain nitrogen atoms of lysine residues, with an upper limit of 14 Å. For Gly68, the N-terminal residue of the N-terminal fusion tag, the position of the N-terminal nitrogen was used for the distance calculations. An ensemble of 200 models was generated by molecular dynamics in Cartesian space using XPLOR-NIH (30). Specifically, the positions of the N-terminal region (Gly68–Gly124) with respect to the initial template (Leu125–Gly228) was optimized with a simulated annealing protocol that included 6000 steps at 1000 K and 3000 cooling steps from 1000 K to 0 K, followed by 1000 steps of Powell minimization (31). In this initial model, the positions of the backbone atoms of the initial template were kept fixed. The structures were visualized using the molecular modeling program MOLMOL (32).
RESULTS
Digestion of the Cross-linked Model Peptide
To characterize the types of peptides produced by proteinase K digestion of the interpeptide cross-links, we analyzed a digest of the model peptide, Ac-TRTESTDIKRASSREADYLINKER, cross-linked with the CBDPS reagent (Fig. 1). CBDPS is homo-bifunctional NHS-based amine-reactive cross-linker, possessing additional features useful for mass spectomeric analyses, such as isotopic coding, CID cleavage, and affinity purification (10). Previous studies have shown that intrapeptide cross-links are generated by cross-linking this peptide with CBDPS. Digestion of this cross-linked peptide with proteinase K, a nonspecific enzyme, resulted in sets of short, related, interpeptide CBDPS cross-links, all of which contain the same cross-linked pair of lysine residues. As can be seen from Fig. 1A, proteinase K cleaves the peptides at or near the cross-linked residue, leaving two to four residues of each peptide still attached to the cross-linker. This usually results in a set of related isotopically labeled cross-links for each cross-linked pair of amino acids. In the example shown in Fig. 1A, there are multiple cross-links containing the same pair of cross-linked residues; for example, the cross-linked Lys-Lys pair (Lys9–Lys22) is found six times in the spectrum. Multiple cross-links often differed by one amino acid residue, creating a “sequence ladder” around the cross-linking site, which can be used as an additional verification tool for the cross-link assignment.
Fig. 1.


MALDI-MS and MS/MS analysis of the test peptide (Ac-TRTESDKIRASSREADYLINKER) cross-linked with CBDPS-H8/D8, followed by proteinase K digestion. A, MALDI-MS spectrum of the proteinase K digest. Inset, cross-links appear in the spectrum as doublets of signals 8.05 Da apart (theoretical), because of the isotopic coding of the cross-linking reagent. B, MS/MS spectrum of the m/z 1200 interpeptide IK-KER cross-link. (Note: masses of the isotopically coded peptide and fragment ion pairs are denoted as vertically aligned pairs of numbers corresponding to light and heavy isotopic forms; fragment ions are labeled with subscripts α or β, denoting the peptide from which they originated; fragments containing portions of the cross-linker and counterpart peptide are labeled with the subscript cl.) C, ESI-MS spectrum of proteinase K digest. Inset, expanded region of the doubly charged molecular ion of the cross-link, showing the isotopic labeling. D, ESI-MS/MS spectrum of the proteinase K digest. In positive ion ESI MS/MS, the fragmentation depends on the charge state of the precursor ion. The ESI-MS/MS spectrum of the +2 charge state of the precursor ion is shown.
Because of the molecular weight of the CBDPS cross-linker (∼500 Da), the interpeptide cross-links fall within the optimum mass range (1100–1500 Da), for MALDI-MS detection and MS/MS sequencing. An example of an MS/MS spectrum is shown in Fig. 1B. Use of the CID-cleavable cross-linker CBDPS allowed determination of the two component peptides forming the cross-link by MS/MS. The relatively short sequences from each cross-linked peptide simplify the MS/MS spectra and facilitate the sequence assignment of the cross-links. It should be noted that the proteinase K cleavage does not proceed completely to give only crosslinked dipeptides or K-CBDP-K crosslinks, possibly due to the experimental conditions, or steric hindrance from the crosslinker or from the connected peptide. Interestingly, in a paper on membrane proteins (done at higher pH) this “laddering” was also observed (22). Likewise, in a paper on localization of disulfides, peptides of varying lengths were also observed and cleavage sometimes occurred adjacent to the maleimido-butyryl-biocytin (MBB)-modified cysteines, giving peptides as small as C*G or GC* and SC* or SC* (C* = cysteine plus MBB) (24). In our case, had the reaction proceeded to give only crosslinked dipeptides, the sites would have more difficult to localize, and had the reaction proceeded all the way to the crosslinked residues, the crosslinking sites could not have been localized.
Notably, there are no ion signals from “free” (non-cross-linked) peptides present in the spectrum. Because of the digestion with proteinase K, which cleaves to within a few residues of the cross-links, and the +1 charge on the cross-links in MALDI MS, the masses of the CBDPS dead-end cross-links fall in the low mass region of the spectrum, below the mass range of the interpeptide cross-links (as can be seen in Fig. 1A), whereas non-cross-linked peptides are cleaved down to very small peptides, diamino acids (22, 23, 33) or possibly even to individual amino acids (34). In effect, this digestion “cleans up” the spectrum and leads to large regions of the spectrum, which predominantly contain signals from interpeptide cross-links. The MALDI MS/MS spectrum of a CBDPS cross-link is shown in Fig. 1B. Although the PK digestion is the same, in ESI, the spectra are more complex because of the presence of multiply charged ions (Fig. 1C), but the presence of the isotopic coding assists in the recognition of the cross-links. The presence and intensities of the structurally important fragment ions in the ESI MS/MS spectra are charge- and peptide-dependent, but ESI-MS/MS still gave easily interpretable results for the +2 charge state (Fig. 1D). Thus, this analysis can be performed in both ESI and MALDI, although if both ionization modes are available, we prefer to use MALDI because the simplicity of the spectra facilitates the interpretation.
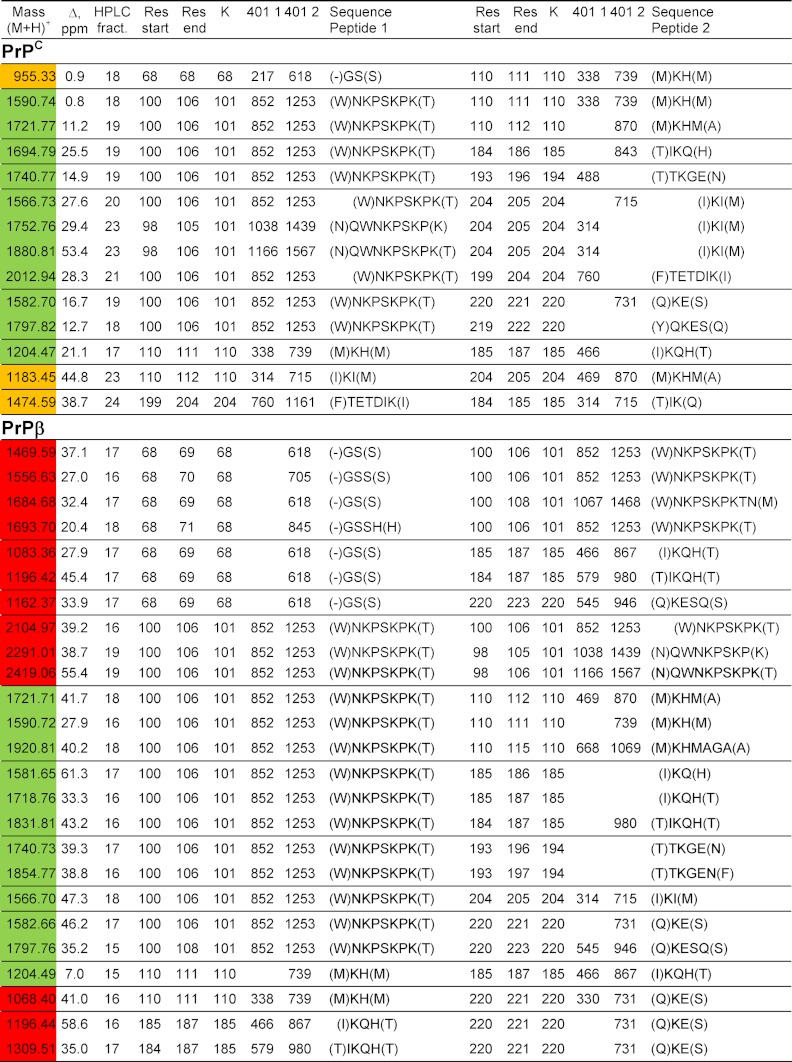
Table I shows the interpeptide cross-links and isotopically coded CID cleavage fragments from the CBDPS-cross-linked test peptide in MALDI. A similar pattern of digestion was observed with other cross-linkers, including di(N-succinimidyl) glutarate (DSG), disuccinimidyl suberate (DSS), photo-induced crosslinking of unmodified proteins (PICUP) (35), and ethylene glycol bis-[succinimidylsuccinate] (EGS) (Table II). For CBDPS, CID cleavage of the cross-linker bridge of the interpeptide cross-links produces short linear peptides from each cross-link (the columns labeled “401 1” and “401 2”), which are easy to assign.
Table I. Interpeptide cross-links identified by cross-linking test peptide (Ac-TRTESTDIKRASSREADYLINKER) with CBDPS followed by proteinase K digestion.
| Mass (M + H)+ | Δ (ppm) | Start residue | End residue | Lys | 401 1a | 401 2 | Sequence peptide 1 | Start residue | End residue | Lys | 401 1 | 401 2 | Sequence peptide 2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1200.51 | 24.4 | 8 | 9 | 9 | 314.16 | 715.17 | (D)IK(R) | 22 | 24 | 22 | 486.19 | 887.16 | (N)KER(−) |
| 1271.52 | 39.8 | 8 | 9 | 9 | 314.17 | 715.18 | (D)IK(R) | 20 | 23 | 22 | 557.25 | 958.22 | (L)INKE(R) |
| 1315.52 | 35.5 | 7 | 9 | 9 | 429.17 | 830.16 | (T)DIK(R) | 22 | 24 | 22 | 486.20 | 887.19 | (N)KER(−) |
| 1386.56 | 26.2 | 7 | 9 | 9 | 429.17 | 830.15 | (T)DIK(R) | 20 | 23 | 22 | 557.25 | 958.22 | (L)INKE(R) |
| 1416.58 | 24.3 | 6 | 9 | 9 | 530.27 | (S)TDIK(R) | 22 | 24 | 22 | 486.20 | 887.20 | (N)KER(−) | |
| 1503.60 | 34.1 | 5 | 9 | 9 | 617.18 | (E)STDIK(R) | 22 | 24 | 22 | 486.20 | 887.21 | (N)KER(−) |
a The central portion of the cross-linker weighs 401 Da. Because of the symmetrical nature of the cross-linker, cleavage on either side of this central portion leads to two pairs of peaks separated by 401 Da, corresponding to each peptide connected to a portion of the cross-linker. Because all of these cleaved fragments still contain the isotopically labeled propionic acid moiety of the CBDPS, they appear in the spectrum as doublets of peaks (10).
Table II. Interpeptide cross-links identified by cross-linking test peptide (Ac-TRTESTDIKRASSREADYLINKER) with DSG, DSS, EGS, and PICUP followed by proteinase K digestion.

Proteinase K Digestion of Cross-linked Prions
As a first application of this new method, we then applied the proteinase K digestion method to the cross-linking analysis of native monomeric PrPC and oligomerized PrPβ prion proteins. Prion proteins are characterized by template-induced conformational changes that lead to the conversion of the native structure and formation of the pathological protein aggregates (36–39). The molecular details of this process are currently unknown.
We investigated this structural problem using cross-linking combined with mass spectrometry. In CBDPS cross-linking experiments on PrPC and PrPβ, the experimental conditions were optimized so that interprotein cross-linked species were observed only for the β-oligomer (Fig. 2). We then combined CBDPS cross-linking with proteinase K digestion and compared the results with CBDPS and standard tryptic digestion. Employing tryptic digestion did not result in the detection of any interpeptide cross-links. Although the CBDPS-cross-linked protein produces a very complex mixture of peptides, amino acids, and cross-links with proteinase K, we were able to isolate numerous interpeptide cross-links (Fig. 3). This strategy produced a broad coverage of the cross-linked sites; as can be seen in Fig. 4, every lysine in the protein is represented in multiple cross-links. In addition, the masses of the interpeptide cross-links produced by proteinase K digestion of the cross-linked protein were appropriate for MALDI-MS analysis. Using proteinase K and all of the distinguished features of the CBDPS cross-linker (CID cleavage, isotopic coding, and affinity purification), over 60 interpeptide cross-links were found for the native and the β-forms of the prion protein. Several of these cross-links were unique to the each form of the prion protein (Table III).
Fig. 2.
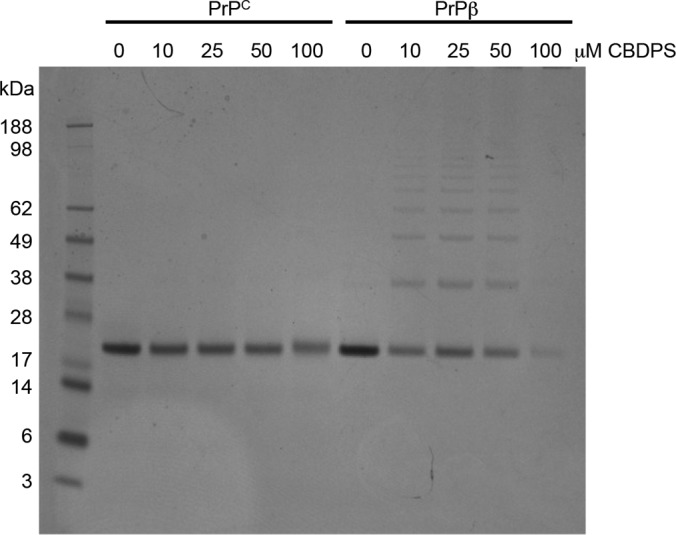
CBDPS cross-linking of PrPC and PrPβ. PrPC and PrPβ samples were cross-linked with increasing concentrations of CBDPS and separated by SDS-PAGE. Interprotein cross-linked species were observed only for the β-oligomer.
Fig. 3.

Cross-linking analysis of the prion proteins. A, MALDI MS spectrum of an HPLC fraction of proteinase K digest of the β-oligomeric sample cross-linked with CBDPS-H8/D8. The proteins were cross-linked with CBDPS-H8/D8 and digested, and the cross-links were affinity-purified with immobilized avidin, separated by HPLC, and analyzed by MALDI MS and MS/MS. Inset, differential cross-link, present only in the β-oligomer sample. B, MS/MS spectrum of the 1309 m/z cross-link identified as a Lys185–Lys220 interpeptide cross-link.
Fig. 4.

Locations of the interpeptide cross-links found for both PrPC (green) and PrPβ (red). Sequences for the peptide components of the Lys-Lys interpeptide cross-links are highlighted.
Table III. Interlysine CBDPS cross-links of prion proteins after proteinase K digestion.
Proteins were cross-linked with isotopically coded CBDPS-H8/D8 reagent and digested with proteinase K. Cross-linked peptides were affinity-enriched using immobilized monomeric avidin beads, and the resulting peptide cross-links were separated by HPLC and analyzed by MALDI MS and MS/MS. Cross-links that were unique to the native form of prion are highlighted in yellow; those unique to the β-oligomeric form are highlighted in red; and those found in both samples are in green. 401 1 and 401 2 indicate the masses of CID cleavage products of the interpeptide cross-links, which carry a specific isotopic signature: two 4.03-Da doublets of signals 401 Da apart.
Molecular Modeling
Initial analysis of PrP models indicated that multiple cross-link-based restraints to Lys101 could not be satisfied by a single conformation. Therefore, cross-link-based distance constraints were split into two sets to separate Lys101 cross-linking partners that are located on the opposite sides of the protein: set A included cross-links Lys101-Lys194 and Lys101-Lys220; set B included cross-links Lys101–Lys185 and Lys101–Lys204. This yielded 123 and 200 models, respectively, with no violations of the distance constraints. These models are shown in Fig. 5. This model contains the 90–124-amino acid portion of the N-terminal sequence that could not be determined by NMR spectroscopy. Even the most recent NMR-based model of prions, published on-line in July 2011, did not obtain sufficient constraints to determine a structure for this flexible portion of the protein (40). Our new model is based on mass spectrometrically derived cross-linking restraints and includes the N-terminal flexible region (amino acids 90–123) plus a 23-residue extension to facilitate purification. This model indicates that there is an interaction of the N-terminal part of PrP with the C-terminal part of helix 2 and helix 2-helix 3 loop.
Fig. 5.

Conformations of the native form of the PrP 90–232-amino acid protein, modeled using the interlysine cross-link distance constraints. The dashed lines indicate constraints that were applied to the cross-linked residues. A and B show cross-linker distances for constraint sets A and B, respectively. C and D illustrate “open” and “closed” conformational bundles of the flexible N-terminal region (blue lines) that are identified by constraint sets A and B, respectively. B1, B2, H1, H2, and H3 are β-strands 1 and 2, and helices 1, 2, and 3, respectively. Both conformational bundles are consistent with transient interactions of the N-terminal flexible region with the C-terminal part of helix 2 and the loop between helices 2 and 3.
DISCUSSION
The ionization efficiency of MALDI-MS drops as the molecular weight of peptides increases, so there is always a concern in mass spectrometry-based cross-linking studies as to whether all of the cross-links present in the digest have been detected. This is even more of a concern for protein sequences that lack cleavage sites for high specificity proteolytic enzymes and that may lose these cleavage sites because of cross-linking. This can lead both to reduced peptide sensitivity because of the size of the peptide (41–44), thereby leading to increased susceptibility to suppression effects, particularly in complex mixtures, as well as reduced fragmentation efficiencies (44, 45). To address this issue, we explored use of the nonspecific proteolytic enzyme proteinase K for the digestion of the cross-linked proteins. Low specificity enzymes are not often used in proteomics studies because they usually divide the peptide signals among several possible digestion peptide products and because they complicate the identification of the peptides. Knowledge of an enzyme's specificity (i.e., where it cleaves a protein) is used by most database search engines to reduce the number of theoretical peptides against which the data is searched.
Proteinase K has been used for the study of prions since the early 1980s. In fact, proteinase K was used in initial experiments that proved that the infectious agent in scrapies was, in fact, a protein and not a virus or viroid (46–48). Although in these studies digestion with proteinase K reduced the infectivity of the samples, it was also noted that resistance to proteinase K correlated with the titer of the infectious agent (49, 50). This led to a patented method for the determination of PrPC and PrPSc in biological samples, such as blood (51).
To overcome the partial resistance of PrPβ to proteinase K digestion, fairly high enzyme:substrate ratios were used in our study. In our cross-linking experiments, we found that proteinase K digestion appears to stop within 2–6 residues of cross-linking sites. Even with very high enzyme:substrate ratios, K-CL-K cross-links are not observed. An important additional benefit of this method is that proteinase K digestion effectively eliminates the noninformative free (non-cross-linked) peptides and dead-end cross-links from the mass region of the spectra that contains the interpeptide cross-links. Dead-end cross-links, which are the largest group of cross-links, will be efficiently digested by proteinase K and are therefore below the usual mass range of MALDI-MS. In fact, if the minimum sequence length requirement for the proteinase K reaction is 4–6 residues, for CBDPS this would bring non-cross-linked peptide products into a mass range below 600 Da and the dead-ends into the range below ∼1000 Da, whereas most of interpeptide cross-links would be observed in the mass range 1200–1800 Da, the ideal mass range for MALDI-MS/MS fragmentation. This is a major advantage of this method and is a significant advantage when it comes to the detection of interpeptide cross-links. In fact, when proteinase K digestion was used for cross-linking studies, we found that any drawbacks caused by the lack of specificity of the enzyme were more than outweighed by the reduced size of the cross-linked peptides and the simplicity of the interpeptide cross-links produced.
Although the short cross-linked peptides produced could potentially lead to ambiguous cross-linking site identification in large or complex systems, the fact that these cross-links are observed will at least provide a starting point for subsequent strategies for their targeted determination, such as the use of alternative enzymes. Removing the digestion restrictions also increases the computational challenges for the cross-linking data analysis software, especially for complex protein systems. As the systems increase in size, this will require further upgrades to search algorithms and automated data processing procedures. This work is currently in progress in our laboratory.
Representation of the same cross-linked sites by several interpeptide cross-links will certainly dilute the cross-link signal. However, as has been noted for other studies using PK on post-translationally modified peptides (22), the presence of these “families” of cross-links provides additional confirmation of the cross-linking sites. These “families” also provide an additional tool for the search and assignment of the interpeptide cross-links.
A set of 60 cross-links was found, including cross-links that were specific to each form of the protein. Nine PrPc interpeptide cross-links (Fig. 4) were used to model the orientation of the N-terminal region (Gly68–Gly124) with respect to the N-terminal domain of PrP, using 14 Å as the maximum span of the CBDPS cross-linker (Fig. 5). To our knowledge, these cross-links provide the first experimental evidence of an interaction of the flexible N-terminal region of PrP with the C-terminal part of helix 2 and helix 2-helix 3 loop (Fig. 5), the first regions to undergo conformational transition under destabilizing conditions such as low pH (28, 52–54). The propensity of these PrP low stability spots to interact with the extended polypeptide chain of the N-terminal region would be consistent with helix 2 and the H2-H3 loop being involved in interactions between PrPC and PrPSc during disease propagation.
The PrPC interpeptide cross-links and the PrPC model are consistent with changes in the NMR chemical shifts in helix 2 and the helix 2-helix 3 loop, which were induced by the presence of the flexible N-terminal region in human and hamster prion proteins (55, 56). This points to direct interactions between this flexible N-terminal region and these portions of the PrP C-terminal domain as a cause of the observed chemical shift changes. Interestingly, a Gly90 (N terminus) to Glu152 cross-link has been reported using 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC), trypsin, and mass spectrometric detection (57). In our current model, these residues are too far apart to be consistent with the length of an EDC cross-linker. However, the large distance between Gly90 and Glu152 can also be associated with a different buffer conditions or/and the different protein constructs that were used the two studies. It is important to stress that a reliable interpretation of the cross-linking results in the light of prion biology was not the primary focus of this study. This would require full-length PrPC construct (23–230) and removal of any purification tags attached to the protein. Efforts to reproduce the cross-linking results for PrP(23–230) are underway in our laboratory.
Using proteinase K, a CBDPS cross-link between Lys185 and Lys220 was also found, but only in the β-oligomeric form of the prion protein (PrPβ). This cross-link can originate either from rearrangements of the PrP structure upon conversion to PrPβ or from intermolecular interactions among subunits of the β-oligomer and is experimental evidence for the conformational differences between PrPc and PrPβ. In the native structure, the distance between Lys185 and Lys220 is greater than 26 Å, and these two residues are situated on opposite surfaces of the protein, separated by the 127–165-amino acid loop. A conformational change involving unfolding of the 127–165-amino acid loop would expose residues Lys185 and Lys220, which would then be able to be cross-linked. This hypothesis is currently being tested in our laboratory by other structural proteomics methods, such as limited proteolysis (58), surface modification (59), and hydrogen/deuterium exchange (60, 61).
Cross-linking combined with mass spectrometry holds great promise for providing constraint data for molecular modeling studies, especially for flexible portions of proteins that cannot be determined by NMR. EDC, a zero-length cross-linker, combined with tryptic digestion and mass spectrometry, was used to demonstrate cross-links in a PrP dimer (57), and a Gly90–Gly90 cross-link was recently found in PrPSc using bis(sulfosuccinimidyl) suberate (11.4 Å spacer length), thus providing the first distance constraint for PrPSc (62). The structures of PrPC, PrPβ, and PrPSc have also been studied using surface chemical modification with nitration (25, 26) or acetylation (25), combined with mass spectrometric detection. Based on our proof-of-principle experiments, proteinase K combined with cleavable cross-linkers will be useful for mass spectrometry-based structural proteomics studies of prions and other difficult-to-digest proteins. We are confident that the use of this new technique, combined with molecular modeling, will lead to the determination of the structures of these particularly challenging proteins.
Conclusion
This study illustrates the utility of the nonspecific enzyme proteinase K for cross-linking combined with mass spectrometry experiments. For the proteins studied thus far, the broad specificity and high activity of this enzyme leads to an extensive digestion of the cross-linked proteins, providing comprehensive representation of the cross-linked sites. Digestion apparently stops within few residues of the cross-linked sites, producing a very clean mass spectrum containing effectively only interpeptide cross-links. Cleavage with proteinase K produces interpeptide cross-links with molecular masses amenable to MALDI-MS detection and MS/MS analysis, which greatly facilitates identification of the cross-linked peptides and the cross-linked residues. The digestion produces a series of related interpeptide cross-link species differing by single amino acid residues, which provides additional confirmation of the cross-link assignments. These features of proteinase K digestion of cross-linked proteins make it an attractive alternative to high specificity enzymes for cross-linking applications using mass spectrometric analysis.
Footnotes
* This work was supported by Genome Canada, Genome British Columbia, and PrioNet Canada.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- CBDPS
- cyanurbiotindipropionylsuccinimide
- PrPC
- native (noninfectious) prion protein
- PrPβ
- β-oligomeric form of the prion protein
- EDC
- 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride].
REFERENCES
- 1. Sinz A. (2007) Chemical cross-linking and mass spectrometry for investigation of protein-protein interactions. Mass Spectrom. Protein Interact. 83–107 [Google Scholar]
- 2. Chakravarti B., Lewis S. J., Chakravarti D. N., Raval A. (2006) Three dimensional structures of proteins and protein complexes from chemical cross-linking and mass spectrometry: A biochemical and computational overview. Curr. Proteomics 3, 1–21 [Google Scholar]
- 3. Sinz A. (2003) Chemical crosslinking and mass spectrometry for mapping three-dimensional structures of proteins and protein complexes. J. Mass Spectrom. 38, 1225–1237 [DOI] [PubMed] [Google Scholar]
- 4. Petrotchenko E. V., Borchers C. H. (2010) Crosslinking combined with mass spectrometry for structural proteomics. Mass Spectrom. Rev. 29, 862–876 [DOI] [PubMed] [Google Scholar]
- 5. Young M. M., Tang N., Hempel J. C., Oshiro C. M., Taylor E. W., Kuntz I. D., Gibson B. W., Dollinger G. (2000) High throughput protein fold identification by using experimental constraints derived from intramolecular cross-links and mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 97, 5802–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Back J. W., de Jong L., Muijsers A. O., de Koster C. G. (2003) Chemical cross-linking and mass spectrometry for protein structural modeling. J. Mol. Biol. 331, 303–313 [DOI] [PubMed] [Google Scholar]
- 7. Sinz A. (2006) Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom. Rev. 25, 663–682 [DOI] [PubMed] [Google Scholar]
- 8. Lee Y. J. (2008) Mass spectrometric analysis of cross-linking sites for the structure of proteins and protein complexes. Mol. BioSystems 4, 816–823 [DOI] [PubMed] [Google Scholar]
- 9. Petrotchenko E. V., Serpa J. J., Borchers C. H. (2010) Use of a combination of isotopically coded cross-linkers and isotopically coded N-terminal modification reagents for selective identification of inter-peptide crosslinks. Anal. Chem. 82, 817–823 [DOI] [PubMed] [Google Scholar]
- 10. Petrotchenko E. V., Serpa J. J., Borchers C. H. (2011) An isotopically-coded CID-cleavable biotinylated crosslinker for structural proteomics. Mol. Cell. Proteomics 10.1074/mcp.M110.001420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Petrotchenko E. V., Borchers C. H. (2010) ICC-CLASS: Isotopically-coded cleavable crosslinking analysis suite. BMC Bioinformatics 11, 64, doi: 10.1186/1471-2105-11-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petrotchenko E. V., Xiao K., Cable J., Chen Y., Dokholyan N. V., Borchers C. H. (2009) BiPS, a photocleavable, isotopically coded, fluorescent cross-linker for structural proteomics. Mol. Cell. Proteomics 8, 273–286 [DOI] [PubMed] [Google Scholar]
- 13. Petrotchenko E. V., Thomas J. M., Borchers C. H. (2008) A collection of novel isotopically-coded crosslinkers for structural proteomics. Presented at the 56th American Society for Mass Spectrometry Conference on Mass Spectrometry and Allied Topics, Denver, CO, June 1–5, 2008 [Google Scholar]
- 14. Petrotchenko E. V., Borchers C. H. (2008) Cross-linking as a tool to examine protein complexes: Examples of cross-linking strategies and computational modeling. In Mass Spectrometry Analysis for Protein-Protein Interactions and Dynamics (Chance M., ed.) Wiley and Sons, Hoboken, NJ [Google Scholar]
- 15. Petrotchenko E. V., Olkhovik V. K., Borchers C. H. (2005) Isotopically-coded cleavable crosslinker for studying protein-protein interaction and protein complexes. Mol. Cell. Proteomics 4, 1167–1179 [DOI] [PubMed] [Google Scholar]
- 16. Leitner A., Walzthoeni T., Kahraman A., Herzog F., Rinner O., Beck M., Aebersold R. (2010) Probing native protein structures by chemical cross-linking, mass spectrometry and bioinformatics. Mol. Cell. Proteomics 9, 1634–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rappsilber J., Mann M. (2002) What does it mean to identify a protein in proteomics? Trends Biochem. Sci. 27, 74–78 [DOI] [PubMed] [Google Scholar]
- 18. Singh P., Panchaud A., Goodlett D. R. (2010) Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal. Chem. 82, 2636–2642 [DOI] [PubMed] [Google Scholar]
- 19. Müller D. R., Schindler P., Towbin H., Wirth U., Voshol H., Hoving S., Steinmetz M. O. (2001) Isotope-tagged cross-linking reagents: A new tool in mass spectrometric protein interaction analysis. Anal. Chem. 73, 1927–1934 [DOI] [PubMed] [Google Scholar]
- 20. Soderblom E. J., Goshe M. B. (2006) Collision-induced dissociative chemical cross-linking reagents and methodology: Applications to protein structural characterization using tandem mass spectrometry analysis. Anal. Chem. 78, 8059–8068 [DOI] [PubMed] [Google Scholar]
- 21. Hurst G. B., Lankford T. K., Kennel S. J. (2004) Mass spectrometric detection of affinity purified crosslinked peptides. J. Am. Soc. Mass Spectrom. 15, 832–839 [DOI] [PubMed] [Google Scholar]
- 22. Wu C. C., MacCoss M. J., Howell K. E., Yates J. R., 3rd (2003) A method for the comprehensive proteomic analysis of membrane proteins. Nat. Biotechnol. 21, 532–538 [DOI] [PubMed] [Google Scholar]
- 23. Papasotiriou D. G., Jaskolla T. W., Markoutsa S., Baeumlisberger D., Karas M., Meyer B. (2010) Peptide mass fingerprinting after less specific in-gel proteolysis using MALDI-LTQ-Orbitrap and 4-chloro-α-cyanocinnamic acid. J. Proteome Res. 9, 2619–2629 [DOI] [PubMed] [Google Scholar]
- 24. Hwa J., Klein-Seetharaman J., Khorana H. G. (2001) Structure and function in rhodopsin: Mass spectrometric identification of the abnormal intradiscal disulfide bond in misfolded retinitis pigmentosa mutants. Proc. Natl. Acad. Sci. U.S.A. 98, 4872–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gong B., Ramos A., Vázquez-Fernández E., Silva C. J., Alonso J., Liu Z., Requena J. R. (2011) Probing structural differences between PrPC and PrPSc by surface nitration and acetylation: Evidence of conformational change in the C-terminus. Biochemistry 50, 4963–4972 [DOI] [PubMed] [Google Scholar]
- 26. Lennon C. W., Cox H. D., Hennelly S. P., Chelmo S. J., McGuirl M. A. (2007) Probing structural differences in prion protein isoforms by tyrosine nitration. Biochemistry 46, 4850–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zahn R., von Schroetter C., Wüthrich K. (1997) Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 417, 400–404 [DOI] [PubMed] [Google Scholar]
- 28. Bjorndahl T. C., Zhou G. P., Liu X., Perez-Pineiro R., Semenchenko V., Saleem F., Acharya S., Bujold A., Sobsey C. A., Wishart D. S. (2011) Detailed biophysical characterization of the acid-induced PrPc to PrPb conversion process. Biochemistry 50, 1162–1173 [DOI] [PubMed] [Google Scholar]
- 29. James T. L., Liu H., Ulyanov N. B., Farr-Jones S., Zhang H., Donne D. G., Kaneko K., Groth D., Mehlhorn I., Prusiner S. B., Cohen F. E. (1997) PDB ID 1B10: Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc. Natl. Acad. Sci. U.S.A. 94, 10086–10091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M. (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 [DOI] [PubMed] [Google Scholar]
- 31. Powell M. J. (1973) On search directions for minimization algorithms. Math. Programming 4, 193–201 [Google Scholar]
- 32. Koradi R., Billeter M., Wüthrich K. (1996) MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics Modelling 14, 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- 33. Ebeling W., Hennrich N., Klockow M., Metz H., Orth H. D., Lang H. (1974) Proteinase K from Tritirachium album Limber. Eur. J. Biochem. 47, 91–97 [DOI] [PubMed] [Google Scholar]
- 34. Roche Diagnostics GmbH (2006) Proteinase K, recombinant, PCR grade. https://e-labdoc.roche.com/LFR_PublicDocs/ras/03115879001_en_06.pdf
- 35. Fancy D. A., Kodadek T. (1999) Chemistry for the analysis of protein-protein interactions: Rapid and efficient cross-linking triggered by long wavelength light. Proc. Natl. Acad. Sci. U.S.A. 96, 6020–6024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Govaerts C., Wille H., Prusiner S. B., Cohen F. E. (2004) Evidence for assembly of prions with left-handed β-helices into trimers. Proc. Natl. Acad. Sci. U.S.A. 101, 8342–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wille H., Bian W., McDonald M., Kendall A., Colby D. W., Bloch L., Ollesch J., Borovinskiy A. L., Cohen F. E., Prusiner S. B., Stubbs G. (2009) Natural and synthetic prion structure from x-ray fiber diffraction. Proc. Natl. Acad. Sci. U.S.A. 106, 16990–16995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stöhr J., Weinmann N., Wille H., Kaimann T., Nagel-Steger L., Birkmann E., Panza G., Prusiner S. B., Eigen M., Riesner D. (2008) Mechanisms of prion protein assembly into amyloid. Proc. Natl. Acad. Sci. U.S.A. 105, 2409–2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baskakov I. V., Legname G., Baldwin M. A., Prusiner S. B., Cohen F. E. (2002) Pathway complexity of prion protein assembly into amyloid. J. Biol. Chem. 277, 21140–21148 [DOI] [PubMed] [Google Scholar]
- 40. Biljan I., Ilc G., Giachin G., Raspadori A., Zhukov I., Plavec J., Legname G. (2011) Toward the molecular basis of inherited prion diseases: NMR structure of the human prion protein with V210I mutation. J. Mol. Biol. 412, 660–673, 10.1016/j.jmb.2011.07.067 [DOI] [PubMed] [Google Scholar]
- 41. Tench R. J., Balooch M., Bernardez L., Allen M. J., Siekhaus W. J., Olander D. R., Wang W. (1991) Clusters formed in laser-induced ablation of silicon, silicon carbide, platinum, uranium dioxide and evaporation of UO2 observed by laser ionization time-of-flight mass spectrometry and scanning tunneling microscopy. J. Vacuum Sci. Technol. 9, 820–824 [Google Scholar]
- 42. Chen X., Westphall M. S., Smith L. M. (2003) Mass spectrometric analysis of DNA mixtures: Instrumental effects responsible for decreased sensitivity with increasing mass. Anal. Chem. 75, 5944–5952 [DOI] [PubMed] [Google Scholar]
- 43. Trauger S. A., Webb W., Siuzdak G. (2002) Peptide and protein analysis with mass spectrometry. Spectroscopy 16, 15–28 [Google Scholar]
- 44. Tran B. Q., Hernandez C., Waridel P., Potts A., Barblan J., Lisacek F., Quadroni M. (2011) Addressing trypsin bias in large scale (phospho)proteome analysis by size exclusion chromatography and secondary digestion of large post-trypsin peptides. J. Proteome Res. 10, 800–811 [DOI] [PubMed] [Google Scholar]
- 45. Laskin J., Futrell J. H. (2003) Collisional activation of peptide ions in FT-ICR mass spectrometry. Mass Spectrom. Rev. 22, 158–181 [DOI] [PubMed] [Google Scholar]
- 46. Prusiner S. B., Groth D. F., Cochran S. P., Masiarz F. R., McKinley M. P., Martinez H. M. (1980) Molecular properties, partial purification, and assay by incubation period measurements of the hamster scrapie agent. Biochemistry 19, 4883–4891 [DOI] [PubMed] [Google Scholar]
- 47. Prusiner S. B., McKinley M. P., Groth D. F., Bowman K. A., Mock N. I., Cochran S. P., Masiarz F. R. (1981) Scrapie agent contains a hydrophobic protein. Proc. Natl. Acad. Sci. U.S.A. 78, 6675–6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Diener T. O., McKinley M. P., Prusiner S. B. (1982) Viroids and prions. Proc. Natl. Acad. Sci. U.S.A. 79, 5220–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bolton D. C., McKinley M. P., Prusiner S. B. (1982) Identification of a protein that purifies with the scrapie prion. Science 218, 1309–1311 [DOI] [PubMed] [Google Scholar]
- 50. McKinley M. P., Bolton D. C., Prusiner S. B. (1983) A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57–62 [DOI] [PubMed] [Google Scholar]
- 51. Prusiner S. B., Safar J. (2000) Method for detecting prions. United States Patent and Trademark Office, pp. http://patft.uspto.gov/netacgi/nph-Parser?Sect2=PTO1&Sect2=HITOFF&p=1&u=%2Fnetahtml%2FPTO%2Fsearch-bool.html&r=1&f=G&l=50&d=PALL&RefSrch=yes&Query=PN%52F6620629, The Regents of the University of California [Google Scholar]
- 52. Calzolai L., Zahn R. (2003) Influence of pH on NMR structure and stability of the human prion protein globular domain. J. Biol. Chem. 278, 35592–35596 [DOI] [PubMed] [Google Scholar]
- 53. Bae S. H., Legname G., Serban A., Prusiner S. B., Wright P. E., Dyson H. J. (2009) Prion proteins with pathogenic and protective mutations show similar structure and dynamics. Biochemistry 48, 8120–8128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bjorndahl T. C., Zhou G. P., Liu X., Perez-Pineiro R., Semenchenko V., Saleem F., Acharya S., Bujold A., Sobsey C. A., Wishart D. S. (2011) Detailed biophysical characterization of the acid-induced PrPc to PrPβ conversion process. Biochemistry 50, 1162–1173 [DOI] [PubMed] [Google Scholar]
- 55. Zahn R., Liu A., Lührs T., Riek R., von Schroetter C., López García F., Billeter M., Calzolai L., Wider G., Wüthrich K. (2000) NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. U.S.A. 97, 145–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Donne D. G., Viles J. H., Groth D., Mehlhorn I., James T. L., Cohen F. E., Prusiner S. B., Wright P. E., Dyson H. J. (1997) Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. U.S.A. 94, 13452–13457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kaimann T., Metzger S., Kuhlmann K., Brandt B., Birkmann E., Höltje H. D., Riesner D. (2008) Molecular model of an α-helical prion protein dimer and its monomeric subunits as derived from chemical cross-linking and molecular modeling calculations. J. Mol. Biol. 376, 582–596 [DOI] [PubMed] [Google Scholar]
- 58. Hall M. C., Shcherbakova P. V., Fortune J. M., Borchers C. H., Dial J. M., Tomer K. B., Kunkel T. A. (2003) DNA binding by yeast Mlh1 and Pms1: Implications for DNA mismatch repair. Nucleic Acids Res. 31, 2025–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Glocker M. O., Borchers C., Fiedler W., Suckau D., Przybylski M. (1994) Molecular characterization of surface topology in protein tertiary structures by amino-acylation and mass spectrometric peptide mapping. Bioconjugate Chem. 5, 583–590 [DOI] [PubMed] [Google Scholar]
- 60. Pan J., Han J., Borchers C. H., Konermann L. (2009) Hydrogen/deuterium exchange mass spectrometry with top-down electron capture dissociation for characterizing structural transitions of a 17 kDa protein. J. Am. Chem. Soc. 131, 12801–12808 [DOI] [PubMed] [Google Scholar]
- 61. Pan J., Han J., Borchers C. H., Konermann L. (2010) Characterizing short-lived protein folding intermediates by top-down hydrogen exchange mass spectrometry. Anal. Chem. 82, 8591–8597 [DOI] [PubMed] [Google Scholar]
- 62. Onisko B., Fernández E. G., Freire M. L., Schwarz A., Baier M., Camiña F., García J. R., Rodríguez-Segade Villamarín S., Requena J. R. (2005) Probing PrPSc structure using chemical cross-linking and mass spectrometry: Evidence of the proximity of Gly90 N termini in the PrP 27–30 aggregate. Biochemistry 44, 10100–10109 [DOI] [PubMed] [Google Scholar]