

Abstract
Currently, the reliable identification of peptides and proteins is only feasible when thoroughly annotated sequence databases are available. Although sequencing capacities continue to grow, many organisms remain without reliable, fully annotated reference genomes required for proteomic analyses. Standard database search algorithms fail to identify peptides that are not exactly contained in a protein database. De novo searches are generally hindered by their restricted reliability, and current error-tolerant search strategies are limited by global, heuristic tradeoffs between database and spectral information. We propose a Bayesian information criterion-driven error-tolerant peptide search (BICEPS) and offer an open source implementation based on this statistical criterion to automatically balance the information of each single spectrum and the database, while limiting the run time. We show that BICEPS performs as well as current database search algorithms when such algorithms are applied to sequenced organisms, whereas BICEPS only uses a remotely related organism database. For instance, we use a chicken instead of a human database corresponding to an evolutionary distance of more than 300 million years (International Chicken Genome Sequencing Consortium (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716). We demonstrate the successful application to cross-species proteomics with a 33% increase in the number of identified proteins for a filarial nematode sample of Litomosoides sigmodontis.
The identification of proteins from mass spectra is a key step for understanding cellular mechanisms, most of which occur on the protein level. Proteomic analysis steps such as the classification of proteomic patterns, the identification of biomarkers, or quantitative analyses only deliver additional understanding if their results can be linked to correctly identified proteins (1, 2). The success rate of protein identification depends on the proteome coverage of the available protein sequence databases, which are suboptimal even for important model organisms such as Chinese hamster or the African clawfrog Xenopus laevis (3), but also for economically relevant crops (4), let alone extinct species including dinosaurs (5–8). However, even in such cases, related species exist whose genome has been sequenced and can thus facilitate the identification of proteins, e.g. X. tropicalis for X. laevis. The existing information from the available reference genome could be used to make protein information feasible for currently unsequenced and unannotated genomes. Still, problems arise because the locations of substitutions and the nature of modifications are unknown. Even the number of substitutions cannot be estimated a priori. It can vary strongly between different proteins in the sample and even between different peptides in a protein. A similar problem of error-tolerant searches arises in the detection of single-nucleotide polymorphisms (SNPs), where departures from the reference database are of particular interest (9, 10) or with regard to the sequencing of antibodies. Next generation sequencing can increase the number of available genomes, but challenges persist in creating reliable and fully annotated reference genomes (11).
Three classes of approaches can be distinguished for the identification of proteins from mass spectra: spectral library searches, de novo sequencing, and database searches (see Refs. 1 and 12 for reviews). Spectral library approaches, which compare spectra with libraries of already identified peptides, are limited to exclusively find previously identified peptides. De novo approaches infer the sequence directly from the mass differences of the fragment ions in the spectra. Although they are error-tolerant by definition, they do not show sufficient reliability in low quality regions of spectra (13–15) and thus cannot currently be considered a full solution to the identification problem (2).
Database search procedures compare how well an observed spectrum fits to a theoretical spectrum obtained from sequences in a database. Popular methods include Mascot (16), Sequest (17), X!Tandem (18), PepSplice (19), and ProteinPilot (20). Although highly successful in identifying proteins present in a database, these approaches generally fail in their standard mode when the sequence of interest is not exactly contained. However, several error-tolerant extensions have been implemented.
The naïve solution of expanding the database (21) becomes infeasible when proteins containing substitutions and large databases are considered. For an average tryptic peptide consisting of 11 amino acids, considering limitations on the tryptic ends and the fact that isoleucine and leucine cannot be distinguished by mass spectrometry, the search space is expanded by a factor of 191 for a single amino acid substitution and by 16,452 for two amino acid substitutions within one peptide. Not only do run times and memory requirements of currently available approaches become excessive, but the risk of false positives is also strongly increased because of the enormous size of the search space. High mass accuracy can simplify the search problem by filtering out sequences with differing precursor mass, but this can only happen after full enumeration of all possible sequences.
Iterative search procedures have been proposed for identifying proteins with amino acid substitutions (22–24). These approaches rely on the assumption that every protein present in the sample is identifiable based on at least one peptide without any substitutions. Consequently, they run a database search on the unmodified database, and the full enumeration of changes to the protein sequence is only conducted for the proteins identified in the first run.
Tag-based methods first generate characteristic tags of 3–5 amino acids from a spectrum and filter those sequences from the database containing the tag (9, 25, 26). Multitag (27), LookUp Peaks (28), and ProteinPilot (20) use several shorter, de novo generated tags, whereas UStags (29, 30) and Gapped peptides (13) recover longer tags. Because only one part of the sequence (either the tag itself or one of the flanking masses) is allowed to depart from the original sequence, there is an upper limit on the number of modifications allowed (15) and thus on the error tolerance.
Extending the idea of a de novo search, several approaches combine initial de novo results with information of a database of a related species. DeNovoID (31) identifies those peptides with the most similar chemical composition from a database, whereas other approaches (32–34) apply FASTA-like algorithms to match de novo results to an unmodified database and use thresholds to decide whether a de novo error or a mutation is the reason for a mismatch. For individual proteins and multiple proteolytic enzymes, this approach has been expanded and gives very promising results (35, 36) because overlaps of several spectra can help to mitigate the de novo error, which is usually not possible for standard protocols.
Conversely, two-step approaches have been proposed which combine an initial database search with a de novo-BLAST step. There, all spectra are submitted to a database search with the unmodified protein database. Either all spectra below a prespecified cutoff threshold (37, 38) or those meeting certain quality filtering criteria (4, 39–41) are then subjected to a de novo interpretation with a subsequent BLAST search. The choice of the cutoff threshold is critical, and the results for modified peptides strongly depend on the accuracy of the de novo search.
A further idea relies on the hypothesis that even if a mutation cannot be found in a single related organism, the mutated sequence might be contained in the database derived by compiling a database of numerous related organisms. Although only applicable to cases when multiple related proteomes are available, this approach may identify a sufficient number of proteins for some analyses and can be combined with all database-driven strategies. However, it is only applicable and helpful in cases when multiple phylogenetically related species exist, which are not within the same line of heritage. Then, additional information may be gathered from including the several species as references. If two reference species are within the same line of heritage (with regard to the species of interest), all mutations should be included in the closest relative; adding another relative should not give any additional mutation that is helpful (but rather provide a basis for false positive identifications).
One difficulty shared by all approaches lies in the question of how the search space can be expanded to allow substitutions and modifications without allowing random hits or incurring absurd run times. It is imperative not to limit the number of allowed substitutions or modifications prematurely. Because individual spectra have varying amounts of substitutions as well as spectral reliability, arbitrary thresholds need to be avoided that combine information from the spectra with information from the database. Any such step may strongly reduce the amount of available information. We introduce a hybrid approach that builds on tags but adjusts the number of allowed modifications adaptively using an appropriate statistical regularization. This regularization does not require any thresholds, but trades the attainable increase in the goodness of fit of a sequence to a spectrum with the corresponding inflation of the size of the search space. For unmodified peptide fragment ion spectra, this allows to restrict of the search to a quick selection within a small search space. Conversely, at the same time a more extensive search is performed when necessary to allow multiple substitutions or modifications per peptide by increasing the search space accordingly. To avoid random hits, all modifications to the original sequence incur a penalty and are only admitted if the resulting increase in the goodness of fit to the spectrum outweighs this penalty.
MATERIALS AND METHODS
The overall workflow of the Bayesian information criterion-driven error-tolerant peptide search (BICEPS)1 is detailed in Fig. 1. In the first steps (Fig. 1, A–C), error-tolerant sequence tags are generated by de novo procedures and mapped to candidate sequences from the database. We introduce reasonable choices for the goodness of fit function (fit) as well as the distance function (distmodify) and apply the idea of the Bayesian information criterion to find λ, the parameter that quantifies the tradeoff between database and spectral information (Fig. 1, D and E). Then we derive an upper bound that allows early termination (Fig. 1F). It requires an exhaustive search through the entire search space only when appropriate (Fig. 1G). We motivate a strategy to determine a confidence level estimate (Fig. 1H) and aggregate the results to identify proteins (Fig. 1I).
Fig. 1.

Flow chart of the proposed workflow from tag generation through scoring and complexity estimation to confidence level estimation. After de novo tag generation (A), error-tolerant sequence candidates are determined (B), and peptide sequences of appropriate length are extracted (C). The resulting matches to the spectrum are scored (D) and weighted against the size of the search space using the BIC score (E). An early termination criterion allows abortion of the search without requiring the full search space (F). However, when better suited candidates may still be available, a deeper search with more tags or increased error tolerance is started (G). We apply a mixture model strategy to determine a confidence level estimate (H) and aggregate the results to identify proteins (I).
Mathematical Formulation of the Error-tolerant Search Problem
In general, an error-tolerant database search can be understood as a statistical regularization problem. We formalize the description in mathematical terms to motivate our approach. Given a spectrum S, a candidate sequence M, and a set of amino acid positions in this sequence A, we define modify: (M,A) ↛ M to be a modification function that changes the sequence (e.g. by a substitution of an amino acid or a post-translational modification) at the positions A. distmodify: (M,M) ↛ [0,∞) is a modification distance function that measures the difference between the modified and the original sequence. Further, we define fit: (M,S) ↛ [0,1] to be a function that measures how well a sequence fits to the given spectrum. Finally, we define a tradeoff parameter, λ ∈ [0,∞), between the goodness of fit and the current modification distance. The problem of finding the best matching modified sequence can be rewritten as a regularization problem.
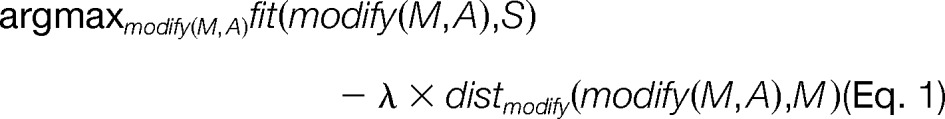 |
The first part of the equation quantifies the goodness of fit, whereas the second part quantifies the increase in the search space weighted by λ. The goal is to find that modification to a sequence which best combines these two aspects. For λ → ∞, this corresponds to a database search without any modifications because any departure from the original sequence carries an infinite penalty; setting λ = 0 corresponds to a de novo approach because there is no penalty for departing from the sequence M. In this case, the original sequence M is irrelevant for the scoring and could be replaced by any other sequence without further penalty. Any remaining value of λ provides a weighted tradeoff between database and spectral information. A major focus of this contribution lies in the strategy for finding λ.
Tag Generation
Tags are generated using DirecTag (42) and Pepnovo (43). Up to 20 tags of length 5 are computed for each spectrum with each de novo approach, and the database is screened for protein sequences containing at least one of the tags. The departure of a single amino acid from the derived tag is allowed to increase error-tolerance. In combination with the large number of tags, this increases the sensitivity while maintaining specificity. Because the following steps depend on the success of the tag generation, a thorough analysis is given in the supplemental material.
Database Filtering
Once all detectable tags are collected, all protein sequences from the database containing a specific sequence tag are identified. Including the complete sequence of a protein for a search would comprise many peptides not containing the tag itself and would thus lead to potential random hits. However, restricting the candidate sequence to the peptide containing the tag alone might decrease the sensitivity because miscleaved peptides or nontryptic ends might be missed. To balance these two ideas, we apply a cutout procedure to the protein sequence.
A subsequence comprising the tag as well as adjacent amino acids is excised from the protein sequence so that the subsequence matches the measured precursor mass. Then, we add two further amino acids at both ends of the subsequence to ensure that even in the case of substitutions from heavier to lighter amino acids the full peptide sequence is included (Fig. 1C).
Database Search and Goodness of Fit Function
For scoring how well a certain sequence fits to a spectrum, we use the fast scoring scheme of PepSplice (19) and extend it to allow the scoring of spectra of triply charged precursor ions. PepSplice applies a hypergeometric scoring scheme (44), yielding a probabilistic interpretation of the goodness of fit function (fit()).
Search Space Size as a Measure of the Cost of a Modification
PepSplice measures the distance of a modified candidate sequence to the original sequence by the resulting logarithmic increase of the search space size (19). This is necessary because the increase in search space goes hand in hand with an increased risk of random hits. Still, this statistical consideration does not incorporate prior knowledge because the likelihood of substitutions between different amino acids varies significantly. To do justice to both ideas, we apply a combined procedure. Rather than penalizing all substitutions with the same penalty, we rank all substitutions by their biochemical likelihood of occurrence based on a PAM 1 substitution matrix (45) and assign penalties based on these ranks. The kth ranked substitution thus obtains a penalty pen(k) = k/n × p, where n is the overall number of substitutions, and p is the penalty that is derived in PepSplice (19) for all substitutions. Hence, biochemically more likely substitutions are preferred to other substitutions, whereas the overall cost of a modification class remains unchanged.
The Bayesian Information Criterion as a Tradeoff Procedure between Goodness of Fit and Modification Cost
A critical step is balancing the goodness of fit and the cost of a modification and thereby defining the tradeoff parameter λ. We need to evaluate whether an increase in the search space, i.e., in the number of allowed departures from the original sequences, is warranted by the increase in the quality of the match. Following the idea of the Bayesian information criterion (BIC) (46) and its motivation from the Bayes factor (47), we compare the goodness of fit of a spectrum S and one modified version of a sequence modifyi(M,A) to the fit of S to competing modified versions. For any sequence modification function modifyi and all possible corresponding amino acid position sets A○, the logarithmic probability of the spectrum being explained by a modification, log(P(S|modifyi(M,A○))), can be approximated by the following,
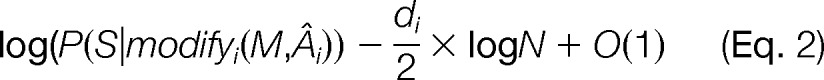 |
using a Laplace approximation to the integral (47), where Âi is the position of the modification with the maximum likelihood among all possible positions given the search space as defined by modifyi(M), di is the size of the corresponding search space, and N is the number of measured fragment ions in the spectrum that could result in a match to a theoretical fragment ion. Similar to the BIC itself, we disregard the O(1) remainder and define the BIC score B for a spectrum to a modified sequence with a slightly rearranged order of multiplication for future ease of notation:
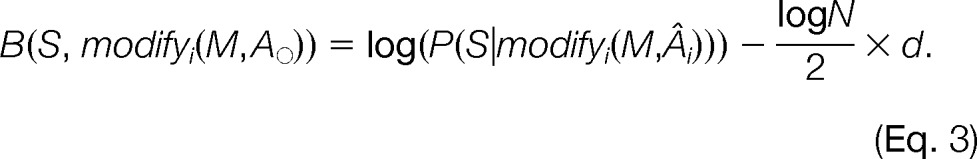 |
Now, we can see that the derived BIC score B corresponds to the solution of the regularization in Equation 1. We define our goodness of fit function fit(M,Âi) = log(P(S|modifyi(M,Âi))) and use the logarithm of the hypergeometric score from PepSplice to estimate this best possible score for a given class of modification allowed for a modification M. As a distance function, distmodify, we use the size of the search space, di, and approximate it by the penalty, pen, introduced above. Finally, we set the tradeoff parameter λ = (log N)/2. Thereby, it depends on the number of measured fragment ions, N, which is a measure of the spectral information content.
As a consequence, we have a statistically derived criterion to balance the database information against the spectral information, which changes according to spectral complexity and does not need to be predefined beforehand.
Upper Bound on the Bayesian Information Criterion
Using the BIC score B, we can compute the optimal solution for a class of modifications modifyi, but we still need to compare all possible changes to the sequences. However, both the search space and the run times grow exponentially when considering all possible modifications. To address this problem, we introduce an upper bound on the BIC score B based on the idea that the maximum hypergeometric score from PepSplice, being a probability, cannot exceed 1. We incrementally increase the search space starting from the unmodified sequences. After the kth search, the BIC score Bstep k is computed, and the current maximal BIC score, Bstep kmax = max(Bstep 1 … Bstep k) among all prior searches is compared with the theoretically highest possible score at step (k + 1), which is the BIC score bound BSBstep k+1. We obtain it by adjusting Equation 3. We set the likelihood to 1 and insert the search space size in the (k + 1)th step, dk+1:
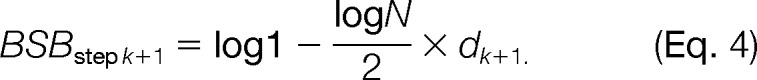 |
If the currently best score Bstep kmax > BSBstep k+1, then an increase in the search space can never result in a higher score and is thus never worthwhile. Thus, we can stop increasing the search space because we already found a better solution than we can obtain by searching any further. In the vast majority of cases, this allows an early termination after only a few steps.
Steps of the Iterative Search Space Expansion
In a first step, the seven best tags for each spectrum are selected without allowing an error within the tag, and the procedure is iterated with an increasing search space to allow more and more substitutions in the peptide sequence, but not the tag itself. If the BSB bound is not yet reached, this is followed by a search on the best 20 tags, and eventually an error is allowed within the tag itself, and the search is iterated again with an increasing number of substitutions in the peptide sequence (see the supplemental material).
False Discovery Rate Estimation
Any peptide identification should be supported by a false discovery rate (FDR) statement. Here, we rely on a mixture modeling approach (48). One of the key features of this approach, which is of particular importance for the problem at hand, is the elimination of the need for decoy databases. Although the standard procedure for obtaining a FDR is through decoy database searches, these searches require that the decoy database does not contain any peptides occurring in the forward database. When searching for peptides with possible substitutions, there is no complete forward database available, because the exact sequence of the proteins from the forward database is unknown. Consequently, it is also impossible to filter the decoy database against the forward database, making the computation of a standard FDR unfeasible. However, our mixture modeling approach does not rely on a decoy database and is thus not affected by this restriction.
Protein Identification and FASTA Output
BICEPS focuses on identifying peptides. However, it is possible to aggregate BICEPS peptide level results to the protein level using standard methods; an overview is given in Ref. 12. To allow easy interfacing to standard database search algorithms, BICEPS outputs a new FASTA database in which all the substitutions of the identified peptides have been included. The novel FASTA database contains a single entry for each protein that includes all detected substitutions for this protein. This database can then be used for database searching in a non-error-tolerant way. For the experiments described in this contribution, we used ProteinPilot for this step.
Implementation
BICEPS is implemented in C++ and available from http://hci.iwr.uni-heidelberg.de/MIP/Software/ and http://software.steenlab.org. Computation time for the test sample (8457 spectra) searched against the IPI.chicken database as described below was ∼8 h on a dual core desktop computer, corresponding to an average computation time of 4 s per spectrum for the error-tolerant search. This search time can be easily reduced when parallelizing the searching onto several cores/CPUs.
Experimental Setup
BICEPS results were evaluated on the peptide level and compared with results of existing error-tolerant approaches. In addition, we also compared its reliability in error-tolerant settings to the reliability of standard search approaches in regular, not error-tolerant settings. As an application of BICEPS, we also show two experiments in which BICEPS was used in a protein identification setting. However, BICEPS focuses on the peptide level, whereas on protein level multiple further factors could confound the identification results. Comparative results shown are examples, but not necessarily representative for performance results. BICEPS does not provide its own protein inference but relies on another search engine, ProteinPilot in this case. Thus, the comparison was limited to ProteinPilot to restrict the number of confounding factors. Experimental details, data set availability as well as search parameters are described in the supplemental material.
Peptide Level Experiment: Human Sample Searched against a Chicken Database
On the peptide level, we base the analysis on a previously described and publically available human HeLa cell data set (48, 49), which was analyzed on an LTQ-Orbitrap classic (Thermo Scientific) resulting in 8457 tandem mass spectra. We establish a de facto ground truth for comparison by searching these spectra against the IPI human database using Mascot, Sequest, and ProteinPilot in their standard mode and a peptide FDR of 5%. Further, we also artificially create an error-tolerant search problem by assuming that the human database was not available and searching these spectra of a human sample against the IPI chicken database. The evolutionary distance of chicken to human is estimated to be more than 300 million years (50). Overlaps of peptides based on this artificially difficult search with the de facto ground truth can be regarded as correct identifications and allow us to compare BICEPS, ProteinPilot in its tag-driven substitution scheme, and Mascot in its iterative error-tolerant mode. It should be noted that this mode of Mascot is designed only for detecting single-nucleotide substitutions and allows either the detection of such a substitution, a variable modification, or a departure from cleavage specification, but no combination thereof. Mascot is thus used here outside of its designed settings. Further, we also run Pepnovo and PEAKS as popular de novo sequencing tools on these spectra. FDR cutoffs at the 5% level were computed based on the mixture model described in Ref. 48. We only analyze the best scored peptide spectrum match (PSM) for each search engine. BICEPS only provides the best PSM in its output because its early-stopping criterion is focused on identifying the best PSM and second best PSMs may be missed because of the stopping of the search procedure.
Protein Level Experiment: CHO Sample Searched against a Rat Database
Chinese hamster ovary (CHO) cells are widely used in the biotech and biopharma industries because they have proven to be an excellent mammalian expression system for recombinant protein therapeutics. However, CHO cells are not amenable to mass spectrometry-based proteomics experiments because of the lack of a comprehensive protein sequence database for Cricetulus griseus. As of August 2011, there are 232 UniProtKB/SWISSPROT entries and 341 UniProtKB/TREMBL entries for C. griseus, i.e., far too few for any kind of comprehensive protein discovery experiment.
For this proteomics experiment, whole CHO cell lysate was fractionated by SDS-PAGE. The gel lane was cut into 10 bands of similar size prior to in gel digestion. The extracted peptides were analyzed on an LTQ-Orbitrap classic resulting in 83,769 spectra overall.
We searched the sample against the 573 proteins available for C. griseus using Mascot in its standard mode to illustrate the need for a large database. Then we searched against all rat sequences in the IPI database (39879 proteins) using ProteinPilot and BICEPS in their error-tolerant settings. For the BICEPS search, all identified substitutions were included in an updated fasta file as described under “Materials and Methods,” and then this file was searched using ProteinPilot in its standard mode (without allowing substitutions) to obtain protein identifications.
Protein Level Experiment: Litomosoides sigmodontis Samples Searched against a Brugia malayi Database
L. sigmodontis is a nematode that is frequently used as a model organism for filarial research (51). However, there is only a single UniProtKB/SWISSPROT entry and only 60 UniProtKB/TREMBL entries for L. sigmodontis (August 2011), making protein identification infeasible. However, the recent sequencing of the genome of a related nematode, B. malayi, resulted in 11,742 B. malayi protein sequences in the RefSeq database version 36 (52), which we can use as sequence input for error-tolerant searches. B. malayi and L. sigmodontis are both spirurida and thus should share far more substitution sites among each other than with any other class of nematodes (compare (53) for a phylogenetic analysis).
A total of 27,597 spectra were acquired on an LTQ-Orbitrap classic instrument from 11 in-gel digestions of cell lysates from L. sigmodontis. ProteinPilot and BICEPS were run in their error-tolerant mode. For the BICEPS search, all identified substitutions were included in an updated fasta file as described under “Materials and Methods,” and then this file was searched using ProteinPilot in its standard mode (without allowing substitutions) to obtain protein identifications.
RESULTS
The experiments were designed to allow a comparison of BICEPS with existing peptide identification strategies. For evaluation, we took spectra from a human sample, but assumed that human protein sequences are not available; instead we sought to identify peptides in this sample using a chicken database. To evaluate our BICEPS approach, we used the tandem mass spectra from a HeLa cell lysate (see above) and searched them against the chicken IPI protein sequence database, pretending that the human protein sequences are not available. Peptide identifications based on the chicken database were considered to be confirmed when they coincided with identifications from a database search against the human database. The application of BICEPS to protein identifications was based on Chinese hamster ovary and L. sigmodontis samples.
Peptide Level Results for the Human Dataset Searched against a Chicken Database
BICEPS Performance for a Human Sample against a Chicken Database Is Competitive to a Standard Database Search against a Human Database
Fig. 2 and Table I show the number of peptides recovered (i) from the standard database searches with Mascot, ProteinPilot, and Sequest against a human database, (ii) by the various error-tolerant search procedures using the chicken database, and (iii) by the de novo approaches. Despite the large phylogenetic difference between human and chicken, BICEPS shows a high number of identified peptides with a significant ratio of identifications confirmed by the searches on the human database. Surprisingly, the overlap of BICEPS and the standard database searches is similar to the overlap among different standard database search algorithms. This is particularly noteworthy as BICEPS searched the human sample-derived data against the chicken database, whereas the standard database search algorithm searched the human data against the appropriate, i.e., human database. The overlap between the various standard database search algorithm is smaller than expected and may be explained by the comparatively high confidence limit of 5%, which was chosen to avoid the possibility that correctly identified substitutions were suppressed by restrictive filtering.
Fig. 2.

Four-way Venn diagram overlap of peptides identified in a human sample. The numbers in the various overlaps of the ellipses show the numbers of coinciding identifications at 5% error rate for each tool. Mascot, ProteinPilot, and Sequest use the human database, whereas our approach BICEPS uses the chicken database. The overlap of BICEPS with the other approaches is similar to the overlap between the remaining approaches. It also demonstrates that no single search engine can reliably identify all spectra (confer Table I).
Table I. Overlap of peptides identified in a human sample.
Table of the overlap of peptides identified in a human sample at 5% error rate for various tools when using a human or a chicken database. The overlap of BICEPS using a chicken database with the other approaches is similar to the overlap between the remaining approaches using the human database. It also demonstrates that no single search engine can reliably identify all spectra.
| Approach | Database | Overlap with Mascot on IPI.hum | Overlap with ProteinPilot on IPI.hum | Overlap with Sequest on IPI.hum | Overlap with BICEPS on IPI.hum |
|---|---|---|---|---|---|
| Mascot | IPI.chicken | 300 | 215 | 198 | 262 |
| ProteinPilot | IPI.chicken | 411 | 501 | 390 | 517 |
| BICEPS | IPI.chicken | 646 | 704 | 554 | 904 |
| Pepnovo | – | 146 | 133 | 67 | 140 |
| PEAKS | – | 48 | 29 | 40 | 39 |
| Mascot | IPI.hum | (1877) | 659 | 841 | 974 |
| ProteinPilot | IPI.hum | 659 | (3194) | 509 | 751 |
| Sequest | IPI.hum | 841 | 509 | (1653) | 820 |
| BICEPS | IPI.hum | 974 | 751 | 820 | (2193) |
Overall, the database driven error-tolerant approaches resulted in 4–30 times more confirmed identifications than the de novo approaches, which again demonstrates that it is appropriate to take advantage of the available information from a database of a related species. Among the error-tolerant database search procedures, BICEPS shows more confirmed identifications than Mascot or ProteinPilot. This is independent of which database search algorithm was used for the confirmation on the human database.
Peptide length can be an important factor for the performance of database search engines. When evaluating the influence of peptide length in error-tolerant settings, we observe that Mascot shows better performance for shorter peptides and ProteinPilot requires longer peptides, BICEPS shows good results across peptides of different length (see supplemental material).
BICEPS Identifies the Highest Number of Confirmed Substitutions
One plausible explanation for the good results of the error-tolerant searches is that they may predominantly identify peptides without substitutions which could indicate that actually no error tolerance is required. Thus, we additionally investigate the number of peptides with substitutions among those peptides that have been confirmed on the human database searches. We consider a substitution to be confirmed if the peptide sequence is in our de facto ground truth, based on the searches on the human database, but not in the chicken database.
More than 20% of the confirmed identified peptides of BICEPS contained at least one substitution. This is well above the rate for the other database-driven error-tolerant approaches. Table II and Fig. 3 show that twice as many confirmed substitutions were found by BICEPS than by any other approach, including the de novo approaches. Mascot in its error-tolerant mode does not identify any peptides with substitutions above the 5% peptide FDR threshold and identifies only six over all. Pepnovo, as a de novo approach, has a ratio of 43% confirmed peptides with substitutions but finds much fewer peptides and substitutions in absolute numbers.
Table II. Number of confirmed peptides with substitutions on the human sample at a 5% peptide FDR.
Among the identified peptides which were confirmed by the standard database search with Mascot, ProteinPilot or Sequest on the human database, we evaluated for each of the error-tolerant approaches how many peptides contained substitutions. BICEPS showed more than three times as many confirmed substitutions as any other approach. A graphical representation of the data is shown in Figure 3.
| Approach | Substitutions found at 5% confidence | Substitutions confirmed by Mascot on IPI.hum | Substitutions confirmed by ProteinPilot on IPI.hum | Substitutions confirmed by Sequest on IPI.hum | Substitutions confirmed by any approach on IPI.hum | Peptides confirmed by any approach on IPI.hum |
|---|---|---|---|---|---|---|
| Mascot | 0 | 0 | 0 | 0 | 0 | 739 |
| ProteinPilot | 488 | 62 | 87 | 71 | 136 | 937 |
| BICEPS | 793 | 139 | 153 | 144 | 260 | 1297 |
| Pepnovo | 305 | 67 | 47 | 36 | 84 | 206 |
| PEAKS | 1068 | 19 | 11 | 18 | 23 | 38 |
Fig. 3.

Total number of confirmed peptides at a 5% FDR cutoff on the chicken database and number of confirmed peptides containing at least a single substitution. All of the confirmations are based on independent searches on the human database. BICEPS shows the highest number of confirmed peptides, as well as the highest number of confirmed peptides containing substitutions. Mascot searches do not result in any peptides with confirmed substitutions.
BICEPS also Identifies Peptides with Two Confirmed Substitutions
Among the 260 confirmed peptides with substitutions for BICEPS, 41 show more than a single substitution per peptide. A full list of these sequences is given in supplemental Table 1. In contrast, the other database-driven error-tolerant approaches do not recover a single peptide with more than one substitution. To further evaluate these results, we manually analyzed the corresponding MS2 spectra and compared the performance of BICEPS on the spectra with two substitutions to the other approaches.
Two examples of manual validation are shown in Fig. 4, and additional examples are in the supplemental material. Although some substitutions are confirmed by clear signals for both fragment ion series, this is not the case for all substitutions. Still, we could not find any evidence against any of the identified substitutions in the examples studied. Of note, BICEPS worked very well even on MS2 spectra acquired in a linear quadrupole ion trap (here LTQ), which is normally characterized by missing fragment ions in the low m/z range. The challenges arising from these missing ions become evident when analyzing false positive identifications by BICEPS based on runs against the human database with full error tolerance (a more detailed discussion is given in the supplemental material).
Fig. 4.

Example of MS2 spectra for peptides with two substitutions identified by BICEPS. In the case of the upper spectrum, we see strong evidence supporting the first substitutions (Ser to Glu). Both fragment ion series support the identification as indicated by the upper and lower diagonal lines in the sequence representation. In contrast, there is only limited direct evidence supporting the Ala to Gly substitution with general few clear signals in the corresponding spectral region. However, the precursor mass and the observed fragment ions provide very strong support for the indicated substitution. In the case of the lower spectrum, we see clear evidence in the spectrum for both substitutions occurring at the end of the sequence with clear spacings in the y ion series as indicated by the bolts. Additional examples are given in the supplemental material.
For these sequences containing two substitutions, we compared again the overlap with standard database search algorithms on the human database. As shown in the supplemental material, the overlaps of BICEPS are again comparable with the overlaps among the standard database searches, even though BICEPS has to overcome the difference between the chicken database and the human sample.
BICEPS Rarely Requires a Large Search Space
Even though BICEPS identifies a substantial number of peptides with substitutions, BICEPS requires its full search depth only in less than 2% of all cases. In approximately half of all cases, the search could be directly aborted without any extension of the search space when the optimal solution was identified. The search space considered by BICEPS was less than 2 orders of magnitude larger than the size of the unmodified database in the majority of all cases, whereas a search space of more than 4 orders of magnitude larger was considered when necessary. This underscores the effectiveness of applying the BIC score to dynamically limiting the search space (see supplemental material).
Protein Level Results for the Chinese Hamster Ovary Sample
After validating BICEPS on the human dataset, searched against a chicken protein sequence database, we applied BICEPS to a whole cell lysate derived from CHO cells. Using Mascot in its standard mode to search against the Uniprot-derived C. griseus database with 569 sequences, 213 proteins were identified (at 1% global protein FDR). Given the complexity of the sample with more than 80,000 spectra, it was obvious that this small number of identified proteins did not do justice to the sample because of the incomplete nature of the C. griseus database. This notion was further underscored by the large number of high quality spectra that could not be assigned by Mascot because of the lack of appropriate database entries.
We then ran ProteinPilot and BICEPS in their error-tolerant mode and opted for rat as a related organism with complete genome information available. A Venn diagram of the number of identifed proteins is shown in Fig. 5. The ProteinPilot search resulted in 2,295 proteins. Applying BICEPS, this number could be further increased to 2,504 proteins (at 1% global protein FDR). Investigating the overlapping and unique protein identifications, 2,194 proteins were identified by both ProteinPilot and BICEPS, i.e., approximately 94% and 86%, respectively, of the individually identified proteins. These significant overlaps corroborate the validity of the error-tolerant searches.
Fig. 5.
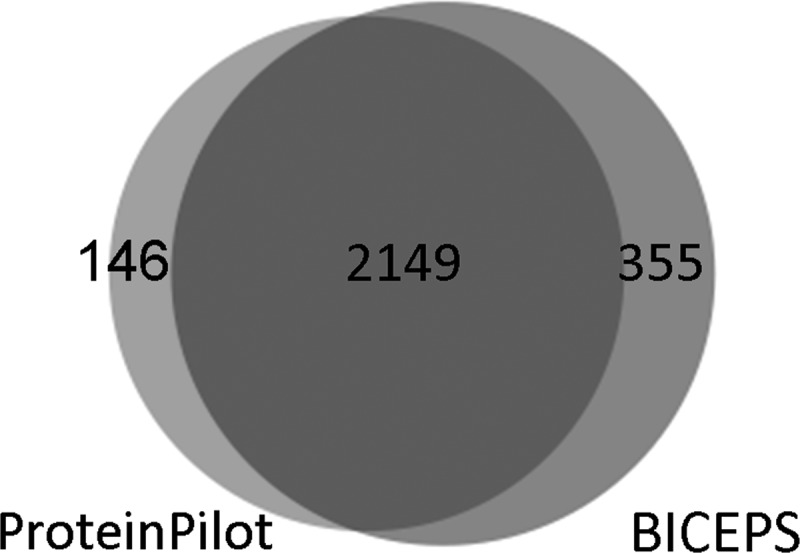
Chinese hamster protein identification using a rat database. The Venn diagram shows the overlap of identified proteins at a 1% protein FDR level between ProteinPilot and BICEPS for the Chinese hamster ovary sample searched against the rat database. ProteinPilot (2,295 proteins) and BICEPS (2,504) show a large overlap with BICEPS identifying more proteins.
Protein Level Results for the L. sigmodontis Sample
The L. sigmodontis sample was searched against the database of a related nematode species, B. malayi. In its error-tolerant setting, ProteinPilot identified 27 proteins at a 1% protein false discovery rate. BICEPS identified as many as 36 proteins as indicated in Fig. 6, increasing the number of identified proteins by ∼33%.
Fig. 6.
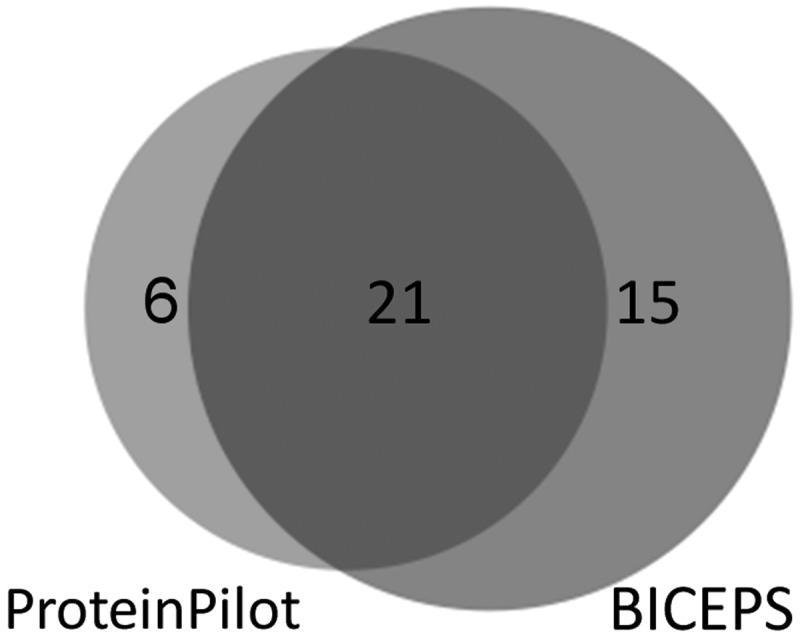
L. sigmodontis protein identification using a B. malayi database. The Venn diagram shows the overlap of identified proteins at a 1% protein FDR level between ProteinPilot in its error-tolerant mode and BICEPS. While showing a strong overlap with ProteinPilot (27 proteins), BICEPS identifies 33% more proteins (36 proteins).
With the single exception of one protein (p27), all proteins found by BICEPS showed at least a single substitution at the peptide level. The maximum number of substitutions added up to 20 substitutions within 11 peptides, which were identified on the 95% confidence level for histone H4. Fig. 7 shows the number of substitutions found by BICEPS for all proteins.
Fig. 7.
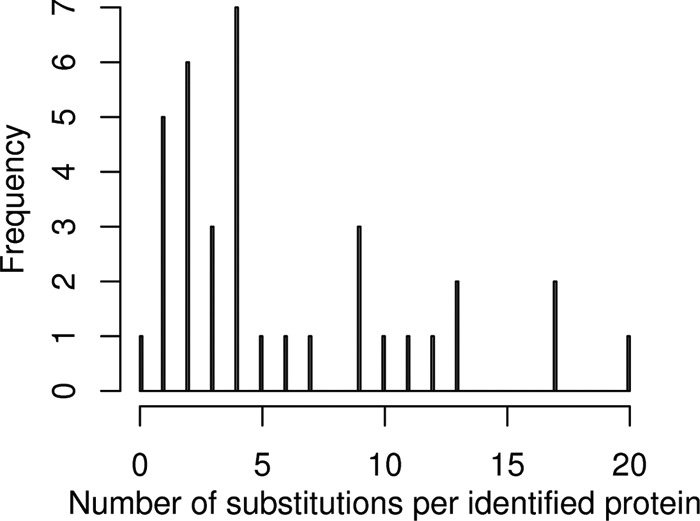
Number of substitutions per protein identified by BICEPS on the L. sigmodontis data set. We observed an average of 6.1 substitutions per protein, which was identified in an error-tolerant search against the B. malayi database with an overall range of 0–20 substitutions.
DISCUSSION
BICEPS Performs Error-tolerant Searches without a Decrease in Accuracy
As shown in the peptide level analysis, the error-tolerant performance of BICEPS is excellent. In a contrived experiment (designed to offer ground truth), we find that the performance of BICEPS compares well to standard database search algorithms such as Mascot, ProteinPilot, or Sequest even in cases when BICEPS has to overcome a large phylogenetic distance (here from chicken to human), whereas the standard approaches have the database of the species of interest available. The overlap between BICEPS results using a database of a distantly related species, and the results of standard database search approaches against the appropriate target are similar to the overlap between the results from the various standard database search approaches. This means that there is no price to pay with regard to the quality of the peptide identification results for an error-tolerant search with BICEPS. This even holds when BICEPS only has a database of a distantly related species compared with the appropriate target database, which may not be available. The results indicate that BICEPS also shows excellent performance for strongly modified sequences with two substitutions.
BICEPS increases the search time by approximately 1 order of magnitude in comparison with traditional database search algorithms, which do not allow substitutions. Still, this increase is small when considering that the theoretical increase in the search space is already at 4 orders of magnitude for sequences with only two substitutions.
BICEPS Provides a Significant Improvement over Current Error-tolerant Approaches
In comparison with Mascot or ProteinPilot in their error-tolerant mode, BICEPS identifies more sequences at the same confidence level, shows larger overlap with the ground truth, and finds more confirmed substitutions. Considering that the ground truth itself was created using Mascot and ProteinPilot in their traditional mode, the results are actually biased in favor of Mascot and ProteinPilot and are thus conservative with regard to BICEPS. The small number of identified substitutions for Mascot was expected because the error-tolerant mode is designed for more limited search problems. Its iterative search approach favors small distances but shows clear limitations as the number of substitutions increases.
Protein Level Results Indicate That BICEPS Is Especially Powerful for Overcoming Large Differences
Also on the protein level, BICEPS shows additional identifications when compared with searches with ProteinPilot in its error-tolerant mode at the same false discovery rate. However, it should be noted that the protein identification comparisons are influenced and confounded by multiple factors, e.g. biological factors such as the presence of orthologs and/or isoforms as well as computational factors. For instance, BICEPS does not provide its own protein inference tool, but we used ProteinPilot for this step; results may differ if another tool is applied. Still, the comparison of the performance of BICEPS between experiments is noteworthy. Although BICEPS resulted in more protein identifications than ProteinPilot, this increase is rather moderate for the CHO dataset. BICEPS identified 9% more proteins than ProteinPilot on the CHO data, whereas on the peptide level comparison of a human sample against a chicken database, we observed an increase of up to 33%. We saw a similarly large benefit of using BICEPS on the protein level when analyzing the L. sigmodontis sample against a B. malayi database. One possible explanation is the smaller phylogenetic distance between rat and Chinese hamster as compared with chicken and human or between the two nematodes. Rat and Chinese hamster are so similar in terms of phylogeny and protein sequences that many protein and peptide sequences are unchanged. Then very simple error-tolerant searches suffice for successful identifications; such infrequent, single substitution searches are rather well handled by ProteinPilot when used in its substitution mode. In contrast, human and chicken as well as L. sigmodontis and B. malayi are so distant in phylogenetic and protein sequence similarity terms such that single substitutions are often insufficient for comprehensive protein identification. Error-tolerant searches with more than one substitution are the strength of the BICEPS search strategy. The nematode sample comparison with an increase of 33% in the number of identified proteins demonstrates the need for these robust error-tolerant searches test.
BICEPS Provides a Statistically Motivated, Adaptive Criterion instead of Arbitrary Thresholds
BICEPS does not require an a priori limitation of the size of the search space but adaptively increases the search space and balances the increase in the number of identified peptides with the risk of random matches. Consequently, no arbitrary tradeoffs between sensitivity and specificity or a de novo error and a homology search error need to be specified. Instead they are automatically estimated from the data. This estimation is not performed on a global level but on the individual spectra level. Thus, BICEPS has an adaptive search complexity; it allows more substitutions and deeper searches in strongly modified regions of a protein, while only considering a small search space in well conserved regions. An efficient early stopping criterion limits the run time and helps to minimize the number of false positives. This approach allows us to search a de facto 10,000 times larger search space in 10 times the search time when compared with standard protein identification approaches. Here, we applied this criterion to the identification of peptides containing amino acid substitutions, but this principle can easily be applied to other error-tolerant search settings such as the detection of post-translational modifications.
Conclusion
In this paper, we introduced BICEPS, a search strategy that makes error-tolerant cross-species peptide identification as powerful as peptide identification in non-error-tolerant standard settings. We showed its competitive performance to current state-of-the-art database search algorithms such as Mascot, Sequest, or ProteinPilot in an example of a human sample searched by the state-of-the-art approaches against a human database and searched by BICEPS against a distant chicken database. Our strategy is based on a statistical regularization idea and automatically finds a tradeoff between the goodness of fit of a spectrum to a sequence and the number of modifications and substitutions. This allows for an adaptive and user-independent decision on the number of substitutions allowed on the peptide level. This strategy also avoids an uncontrolled increase of random hits, which would result from an increased search space containing all possible solutions. The idea of a regularization of the search space could also be generalized to similar settings in protein identification such as modified or cross-linked peptides. In this contribution, we applied the regularization scheme to the effective and cache-optimized hypergeometric scoring scheme of PepSplice, but the general idea is applicable to any scoring scheme with a probabilistic goodness-of-fit score. BICEPS is an open source tool and freely available from our webpages.
Acknowledgments
We thank Michael Hanselmann, Xinghua Lou, Anna Kreshuk, and Ullrich Köthe (Interdisciplinary Center for Scientific Computing, University of Heidelberg); Peter Bühlmann (Seminar for Statistics, ETH Zürich); and Flavio Monigatti (Departments of Pathology, Children's Hospital Boston and Harvard Medical School) for fruitful and inspiring discussions. We thank Hanns Soblik (Bernhard Nocht Institute, Hamburg) for L. sigmodontis sample processing and Sven Giese (NG4, Robert Koch Institute, Berlin) for critical evaluation.
Footnotes
* This work was supported by Deutsche Forschungsgemeinschaft Grant HA 4364/2-1 (to B. Y. R.), Alexander von Humboldt Foundation Grant DEU/1134241 (to M. K.), funds from the postdoctoral program of the German Academic Exchange Service (to D. W.), Marie Curie International Reintegration Grant PIRG-GA-2010-277062 (to A. T.), funds from the Israeli Centers of Research Excellence program at Center 41/11 (to A. T.), and National Institutes of Health Grant R01-GM094844-01 (to H. S.).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- BICEPS
- Bayesian information criterion-driven error-tolerant peptide search
- BIC
- Bayesian information criterion
- FDR
- false discovery rate
- PSM
- peptide spectrum match
- CHO
- Chinese hamster ovary.
REFERENCES
- 1. McHugh L., Arthur J. W. (2008) Computational methods for protein identification from mass spectrometry data. PLoS Comput. Biol. 4, e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wright J. C., Beynon R. J., Hubbard S. J. (2010) Cross species proteomics. Methods Mol. Biol. 604, 123–135 [DOI] [PubMed] [Google Scholar]
- 3. Liska A. J., Sunyaev S., Shilov I. N., Schaeffer D. A., Shevchenko A. (2005) Error-tolerant EST database searches by tandem mass spectrometry and MultiTag software. Proteomics 5, 4118–4122 [DOI] [PubMed] [Google Scholar]
- 4. Grossmann J., Fischer B., Baerenfaller K., Owiti J., Buhmann J. M., Gruissem W., Baginsky S. (2007) A worflow to increase the detection rate of proteins from unsequenced organisms in high-throughput proteomics experiments. Proteomics 7, 4245–4254 [DOI] [PubMed] [Google Scholar]
- 5. Asara J. M., Schweitzer M. H., Freimark L. M., Phillips M., Cantley L. C. (2007) Protein sequences from mastodon and Tyrannosaurus rex revealed by mass spectrometry. Science 316, 280–285 [DOI] [PubMed] [Google Scholar]
- 6. Buckley M., Walker A., Ho S. Y., Yang Y., Smith C., Ashton P., Oates J. T., Cappellini E., Koon H., Penkman K., Elsworth B., Ashford D., Solazzo C., Andrews P., Strahler J., Shapiro B., Ostrom P., Gandhi H., Miller W., Raney B., Zylber M. I., Gilbert M. T., Prigodich R. V., Ryan M., Rijsdijk K. F., Janoo A., Collins M. J. (2008) Comment on “Protein sequences from mastodon and Tyrannosaurus rex revealed by mass spectrometry.” Science 319, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pevzner P. A., Kim S., Ng J. (2008) Comment on “Protein sequences from mastodon and Tyrannosaurus rex revealed by mass spectrometry.” Science 321, 1040. [DOI] [PubMed] [Google Scholar]
- 8. Schweitzer M. H., Zheng W., Organ C. L., Avci R., Suo Z., Freimark L. M., Lebleu V. S., Duncan M. B., Vander Heiden M. G., Neveu J. M., Lane W. S., Cottrell J. S., Horner J. R., Cantley L. C., Kalluri R., Asara J. M. (2009) Biomolecular characterization and protein sequences of the Campanian hadrosaur B. canadensis. Science 324, 626–631 [DOI] [PubMed] [Google Scholar]
- 9. Dasari S., Chambers M. C., Slebos R. J., Zimmerman L. J., Ham A. J., Tabb D. L. (2010) TagRecon: High-throughput mutation identification through sequence tagging. J. Proteome Res. 9, 1716–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li J., Su Z., Ma Z. Q., Slebos R. J., Halvey P., Tabb D. L., Liebler D. C., Pao W., Zhang B. (2011) A bioinformatics workflow for variant peptide detection in shotgun proteomics. Mol. Cell. Proteomics 10.1074/mcp.M110.006536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Florea L., Souvorov A., Kalbfleisch T. S., Salzberg S. L. (2011) Genome assembly has a major impact on gene content: A comparison of annotation in two Bos taurus assemblies. PLoS ONE 6, e21400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nesvizhskii A. I., Vitek O., Aebersold R. (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat. Methods 4, 787–797 [DOI] [PubMed] [Google Scholar]
- 13. Kim S., Bandeira N., Pevzner P. A. (2009) Spectral profiles: A novel representation of tandem mass spectra and their applications for de novo peptide sequencing and identification. Mol. Cell. Proteomics 8, 1391–1400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu C., Yan B., Song Y., Xu Y., Cai L. (2006) Peptide sequence tag-based blind identification of post-translational modifications with point process model. Bioinformatics 22, E307–E313 [DOI] [PubMed] [Google Scholar]
- 15. Shevchenko A., Valcu C. M., Junqueira M. (2009) Tools for exploring the proteomosphere. J. Proteomics 72, 137–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 17. Eng J. K., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem massspectral data of peptides with amino-acid-sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 18. Craig R., Beavis R. C. (2004) TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 19. Roos F. F., Jacob R., Grossmann J., Fischer B., Buhmann J. M., Gruissem W., Baginsky S., Widmayer P. (2007) PepSplice: Cache-eficient search algorithms for comprehensive identification of tandem mass spectra. Bioinformatics 23, 3016–3023 [DOI] [PubMed] [Google Scholar]
- 20. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The Paragon algorithm: A next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 21. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 22. Craig R., Beavis R. C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17, 2310–2316 [DOI] [PubMed] [Google Scholar]
- 23. Creasy D. M., Cottrell J. S. (2002) Error tolerant searching of uninterpreted tandem mass spectrometry data. Proteomics 2, 1426–1434 [DOI] [PubMed] [Google Scholar]
- 24. Starkweather R., Barnes C. S., Wyckoff G. J., Keightley J. A. (2007) Virtual polymorphism: Finding divergent peptide matches in mass spectrometry data. Anal. Chem. 79, 5030–5039 [DOI] [PubMed] [Google Scholar]
- 25. Mann M., Wilm M. (1994) Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal. Chem. 66, 4390–4399 [DOI] [PubMed] [Google Scholar]
- 26. Tabb D. L., Saraf A., Yates J. R., 3rd (2003) GutenTag: High-throughput sequence tagging via an empirically derived fragmentation model. Anal. Chem. 75, 6415–6421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sunyaev S., Liska A. J., Golod A., Shevchenko A., Shevchenko A. (2003) MultiTag: Multiple error-tolerant sequence tag search for the sequence-similarity identification of proteins by mass spectrometry. Anal. Chem. 75, 1307–1315 [DOI] [PubMed] [Google Scholar]
- 28. Bern M., Cai Y., Goldberg D. (2007) Lookup peaks: A hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal. Chem. 79, 1393–1400 [DOI] [PubMed] [Google Scholar]
- 29. Shen Y., Tolić N., Hixson K. K., Purvine S. O., Anderson G. A., Smith R. D. (2008) De novo sequencing of unique sequence tags for discovery of post-translational modifications of proteins. Anal. Chem. 80, 7742–7754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shen Y., Tolić N., Hixson K. K., Purvine S. O., Pasa-Tolić L., Qian W. J., Adkins J. N., Moore R. J., Smith R. D. (2008) Proteome-wide identification of proteins and their modifications with decreased ambiguities and improved false discovery rates using unique sequence tags. Anal. Chem. 80, 1871–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Halligan B. D., Ruotti V., Twigger S. N., Greene A. S. (2005) DeNovoID: A web-based tool for identifying peptides from sequence and mass tags deduced from de novo peptide sequencing by mass spectroscopy. Nucleic Acids Res. 33, W376–W381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DiMaggio P. A., Jr., Floudas C. A., Lu B., Yates J. R., 3rd (2008) A hybrid method for peptide identification using integer linear optimization, local database search, and quadrupole time-of-flight or OrbiTrap tandem mass spectrometry. J. Proteome Res. 7, 1584–1593 [DOI] [PubMed] [Google Scholar]
- 33. Han Y., Ma B., Zhang K. (2005) SPIDER: Software for protein identification from sequence tags with de novo sequencing error. J. Bioinform. Comput. Biol. 3, 697–716 [DOI] [PubMed] [Google Scholar]
- 34. Searle B. C., Dasari S., Turner M., Reddy A. P., Choi D., Wilmarth P. A., McCormack A. L., David L. L., Nagalla S. R. (2004) High-throughput identification of proteins and unanticipated sequence modifications using a mass-based alignment algorithm for MS/MS de novo sequencing results. Anal. Chem. 76, 2220–2230 [DOI] [PubMed] [Google Scholar]
- 35. Bandeira N., Pham V., Pevzner P., Arnott D., Lill J. R. (2008) Automated de novo protein sequencing of monoclonal antibodies. Nat. Biotechnol. 26, 1336–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Han Y., Yuen D., Ma B. (2009) Automated protein (re)sequencing with MS/MS and a homologous database yields almost full coverage and accuracy. Bioinformatics 25, 2174–2180 [DOI] [PubMed] [Google Scholar]
- 37. Habermann B., Oegema J., Sunyaev S., Shevchenko A. (2004) The power and the limitations of cross-species protein identification by mass spectrometry-driven sequence similarity searches. Mol. Cell. Proteomics 3, 238–249 [DOI] [PubMed] [Google Scholar]
- 38. Shevchenko A., Sunyaev S., Loboda A., Shevchenko A., Bork P., Ens W., Standing K. G. (2001) Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal. Chem. 73, 1917–1926 [DOI] [PubMed] [Google Scholar]
- 39. Junqueira M., Spirin V., Balbuena T. S., Thomas H., Adzhubei I., Sunyaev S., Shevchenko A. (2008) Protein identification pipeline for the homology-driven proteomics. J. Proteomics 71, 346–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Waridel P., Frank A., Thomas H., Surendranath V., Sunyaev S., Pevzner P., Shevchenko A. (2007) Sequence similarity-driven proteomics in organisms with unknown genomes by LC-MS/MS and automated de novo sequencing. Proteomics 7, 2318–2329 [DOI] [PubMed] [Google Scholar]
- 41. Wielsch N., Thomas H., Surendranath V., Waridel P., Frank A., Pevzner P., Shevchenko A. (2006) Rapid validation of protein identifications with the borderline statistical confidence via de novo sequencing and MS BLAST searches. J. Proteome Res. 5, 2448–2456 [DOI] [PubMed] [Google Scholar]
- 42. Tabb D. L., Ma Z. Q., Martin D. B., Ham A. J., Chambers M. C. (2008) DirecTag: Accurate sequence tags from peptide MS/MS through statistical scoring. J. Proteome Res. 7, 3838–3846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frank A., Pevzner P. (2005) PepNovo: De novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 44. Sadygov R. G., Yates J. R., 3rd (2003) A hypergeometric probability model for protein identification and validation using tandem mass spectral data and protein sequence databases. Anal. Chem. 75, 3792–3798 [DOI] [PubMed] [Google Scholar]
- 45. Dayhofi M. O., Schwartz R. M., Orcutt B. C. (1978) A model of evolutionary change in proteins. Atlas Protein Sequence Struct. 5, 345–352 [Google Scholar]
- 46. Schwarz G. (1978) Estimating the dimension of a model. Ann. Statistics 6, 461–464 [Google Scholar]
- 47. Hastie T., Tibshirani R., Friedman J. (2001) The Elements of Statistical Learning: Data Mining, Inference, and Prediction, Springer Verlag, New York [Google Scholar]
- 48. Renard B. Y., Timm W., Kirchner M., Steen J. A., Hamprecht F. A., Steen H. (2010) Estimating the confidence of peptide identifications without decoy databases. Anal. Chem. 82, 4314–4318 [DOI] [PubMed] [Google Scholar]
- 49. Renard B. Y., Kirchner M., Monigatti F., Ivanov A. R., Rappsilber J., Winter D., Steen J. A., Hamprecht F. A., Steen H. (2009) When less can yield more: Computational preprocessing of MS/MS spectra for peptide identification. Proteomics 9, 4979–4984 [DOI] [PubMed] [Google Scholar]
- 50. International Chicken Genome Sequencing Consortium (2004) Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 [DOI] [PubMed] [Google Scholar]
- 51. Hoffmann W., Petit G., Schulz-Key H., Taylor D., Bain O., Le Goff L. (2000) Litomosoides sigmodontis in mice: Reappraisal of an old model for filarial research. Parasitol. Today 16, 387–389 [DOI] [PubMed] [Google Scholar]
- 52. Pruitt K. D., Tatusova T., Maglott D. R. (2007) NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Blaxter M. L., De Ley P., Garey J. R., Liu L. X., Scheldeman P., Vierstraete A., Vanfleteren J. R., Mackey L. Y., Dorris M., Frisse L. M., Vida J. T., Thomas W. K. (1998) A molecular evolutionary framework for the phylum Nematoda. Nature 392, 71–75 [DOI] [PubMed] [Google Scholar]