

Abstract
A protein's surface influences its role in protein-protein interactions and protein-ligand binding. Mass spectrometry can be used to give low resolution structural information about protein surfaces and conformations when used in combination with derivatization methods that target surface accessible amino acid residues. However, pinpointing the resulting modified peptides upon enzymatic digestion of the surface-modified protein is challenging because of the complexity of the peptide mixture and low abundance of modified peptides. Here a novel hydrazone reagent (NN) is presented that allows facile identification of all modified surface residues through a preferential cleavage upon activation by electron transfer dissociation coupled with a collision activation scan to pinpoint the modified residue in the peptide sequence. Using this approach, the correlation between percent reactivity and surface accessibility is demonstrated for two biologically active proteins, wheat eIF4E and PARP-1 Domain C.
In the past decade the number of protein sequences without solved structures in the Protein Data Bank library has increased dramatically (1). Determination of the correlation between protein structure and function remains a primary objective of biological research, thus motivating the development of advanced analytical tools for unraveling the three-dimensional structures of proteins. The most common techniques used to determine higher order structure of a protein are nuclear magnetic resonance (NMR)1 and x-ray crystallography; however these techniques are not universal because of practical limitations related to protein size, the inability to crystallize certain proteins, or limited sample amounts. Because of these restrictions, the development of mass spectrometry-based strategies for providing protein structural analysis has gained traction because of their speed and sensitivity.
A number of protein labeling techniques have been used in combination with tandem MS analysis to provide low resolution structural information including hydrogen-deuterium exchange, crosslinking, and covalent chemical modifications prior to proteolytic digestion (2). Hydrogen-deuterium exchange provides the most detailed information about a structure as it probes the entire protein backbone; however, spatial resolution can be greatly limited because of back-exchange of the deuterium to hydrogen before analysis (3) or hydrogen/deuterium scrambling during tandem MS (4). Electron-based dissociation methods have been able to overcome much of the error due to scrambling, but the limitations because of back-exchange still remain great (5, 6). Chemical modifications are used to covalently label specific or nonspecific amino acid side chains, thus eliminating the problem of back-exchange and scrambling (2). The foundation of this method is based on the premise that amino acids that are exposed to solvent and are therefore accessible to a chemical labeling agent will be modified, whereas those that are buried will be modified slowly or not at all. This type of labeling provides information about the identities of the amino acids accessible to the reagent in solution, resulting in a low resolution map of the protein's surface. In particular, the protein's surface and accessibility of amino acid side chains reflects their potential participation in protein-protein or protein-ligand interactions. The chemical modification methods are frequently combined with a bottom-up approach in which the modified proteins are subsequently enzymatically digested to facilitate MS/MS analysis. Crosslinking, a specific form of chemical modification, provides information about structure by creating new intramolecular or intermolecular bonds between specific amino acids with distance constraints on the location of the two amino acids linked based on the size of the linker.(7) Because new bonds are formed between contact areas of the protein, sequencing these sites through tandem MS can prove challenging. Identification of the crosslinked peptides provides distance parameters that can be used to reconstruct the contact points and conformation of the protein(s). With single covalent modifications of amino acids (not formation of crosslinks), the analysis is often more straightforward as the modification can be treated as a post-translational modification, thus allowing most searching algorithms to be used for the identification of the digested modified peptides. In this case, the identification of the modified residues reveals the exposed regions of the protein relative to the inaccessible regions, therefore reflecting conformational information.
Lysine is one of the most targeted amino acids for chemical labeling because of its intrinsically high reactivity, thus making it amenable to efficient modification. Moreover, the positively charged, polar side chain of lysine under physiological pH conditions means that lysines are more often located on the hydrophilic surface of proteins and consequently more often involved in protein-protein or protein-ligand interactions (2). These interactions can be studied through this covalent modification strategy based on monitoring the differential reactivity of selected residues in the presence/absence of the interacting protein or ligand, indicating their involvement in a binding interaction. Several different Lys-specific chemical modification methods have been reported such as aminoacetylation (8–13), amidination (14, 15), and many using biotin labeled reagents (16–20). These modifications, coupled with mass spectrometric analysis, have been used to characterize the topology of multiple proteins, protein-ligand complexes and protein-protein interactions (8–20).
The development and application of new crosslinking and surface accessibility strategies has been impeded by the difficulty of detecting the low abundance crosslinked or modified peptides amid a large array of more abundant unmodified peptides produced upon enzymatic digestion of the proteins or protein complexes. This detection problem has been addressed by efforts to selectively enrich the modified peptides (21–27), incorporation of isotopic labels in the reagents to give the modified peptides distinctive isotopic signatures (28–32), and development of selectively cleavable reagents that yield a traceable neutral loss or reporter ions upon MS/MS analysis (33–40). In the context of surface accessibility studies, Reid et al. has addressed this issue using a “fixed charge” sulfonium ion with specificity for methionine (41), cysteine (42), and lysine (43) targets where upon collision induced dissociation (CID), the exclusive loss of a dialkylsulfide (e.g. a neutral loss of 62 Da) is indicative of a modified species. Our group has previously found that a N-N hydrazine bond can be selectively cleaved upon electron transfer dissociation (38). Bis-arylhydrazone crosslinked peptides were created by reacting succinimidyl 4-formylbenzoate modified peptides with succinimidyl 4-hydrazinonicotinate acetone hydrazone modified peptides (38). These crosslinked peptides were then readily identified through the production of two partially modified peptides, one even-electron product ion and one odd-electron product ion, following cleavage of the N-N hydrazine bond (38). Here we describe a new surface accessibility reagent, NN (see Fig. 1), that modifies primary amines (the side-chain of Lys and/or the N terminus) by incorporating an N-N hydrazine bond. Upon ETD, the preferential cleavage of the N-N bond leads to a dominant loss of 93 Da from all charge-reduced species that allows confident differentiation of modified peptides from unmodified ones. The N-N bond cleavage is both highly efficient and very selective, an outcome attributed in part because of the unique traits of electron-mediated activation. CID and/or ETD are used to pinpoint the exact locations of the modifications and to identify the modified peptides. This information is used in tandem with molecular modeling to map the surfaces of two biologically active proteins of considerable recent interest, wheat eIF4E and PARP-1 Domain C.
Fig. 1.
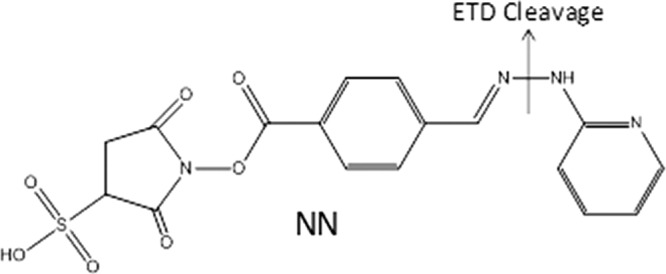
Structure of NN reagent; preferential ETD cleavage is marked indicating the loss of 93 Da from all charge reduced species.
EXPERIMENTAL PROCEDURES
Materials and Reagents
Ubiquitin, proteomics grade trypsin, endoproteinase Arg C, and Glu C were purchased from Sigma Aldrich (St. Louis, MO). PARP 1 Domain C protein was provided by Dr. Hung-wen Liu (Department of Pharmacy, University of Texas at Austin) and eIF4E was provided by Dr. Karen Browning (Department of Chemistry and Biochemistry, University of Texas at Austin). All other chemicals and solvents were purchased from Fisher Scientific (Fairlawn, NJ). The surface accessibility reagent, NN, was synthesized in house as summarized in the supplemental Material (see supplemental Fig. S1).
Mass Spectrometry and Liquid Chromatography
All experiments were undertaken on a ThermoFisher LTQ XL linear ion trap mass spectrometer (San Jose, CA) equipped with an ETD unit. Direct infusion analysis on the LTQ XL was performed using an online nanoESI setup as previously described using a flow rate of 3 μl/min at a concentration of 10 μm in 49.5:49.5:1 MeOH:H2O:Acetic acid (44). An ESI voltage of 2 kV and a heated capillary temperature of 180 °C were used for experiments on the LTQ XL. Liquid chromatography was performed using a RLSC Dionex UltiMate 3000 system (Sunnyvale, CA). An Agilent ZORBAX 300Extend-C18 column (Santa Clara, CA) (150 × 0.3 mm, 3.5 μm particle size) was used for all separations. Eluent A consisted of 0.1% formic acid in water and eluent B 0.1% formic acid in acetonitrile. A linear gradient from 5% eluent B to 40% eluent B over 65 min at 0.3 μl/min was used. Injections of approximately one picomole were used for each digested sample. For all liquid chromatography-tandem MS (LC-MS/MS) runs, the first event was the full mass scan (m/z range of 400–2000) followed by five sets of consecutive MS/MS events on the five most abundant ions from the full mass scan. The first MS/MS event in each set was acquisition of an ETD spectrum using an electron transfer reaction time of 100 ms. Following ETD, CID was performed using a q-value of 0.25, a normalized collisional energy of 35%, and a collision activation of 30 ms. The maximum injection time for all events was set to 100 ms, and each mass spectrum and tandem mass spectrum was the average of five microscans.
Derivatization and Sample Preparation
Each protein (1 mm, 25 μl) was mixed with NN at a variety of molar ratios relative to NN (20 mm) in PBS buffer at pH 7.2–7.4. The protein:NN ratios were varied from 1:2 to 1:10. Higher protein/NN ratios were used to enhance the modification and subsequent detection of some of the less accessible sites that might otherwise be nondetectable under more limiting protein:NN reaction conditions. Upon covalent modification of any protein, slight changes in structure are possible following derivatization. Thus to preserve the structure of the protein as much as possible, the lowest possible protein/NN molar ratios are typically used to limit the number of modifications per protein. All reactions were carried out at room temperature for 30 min and cleaned up using 10 kDa MWCO filters. Derivatized samples were then split into three aliquots for digestion. For tryptic digestion, the derivatized protein (8 nmol) was diluted with 100 mm NH4HCO3 and digested with 1 mg/ml trypsin in 1 mm HCl in a 1:100 w/w ratio of protein to trypsin overnight at 37 °C. For GluC digestion, the derivatized protein (8 nmol) was diluted with the GluC Reaction Buffer from BioLabs, consisting of 50 mm Tris-HCl and 0.5 mm Glu-Glu buffer at pH 8.0, and digested with 1 mm GluC in water in a 1:20 w/w ratio overnight at 37 °C. For ArgC digestion, the derivatized protein (8 nmol) was diluted in 50 mm NH4HCO3 at a pH of 8.0 with a small addition of 20 mm CaCH3COOH to enhance digestion, and then digested using a 1:16 w/w molar ratio of protein to ArgC at 37 °C overnight. The digested samples were diluted to 10 μm before ESI analysis with 49.5/49.5/1 H2O/MeOH/Acetic Acid. Denatured samples were prepared by diluting the protein in 49.5/49.5/1 H2O/MeOH/acetic acid prior to derivatization.
Determination of Surface Accessibility
Upon identification as a modified peptide from the ETD spectrum based on the characteristic loss of 93 Da, peptides were sequenced manually using the subsequent CID fragmentation pattern of the charge-reduced species. The percent reactivity was calculated based on the sum of the peak areas of all peptides containing a modified residue based on the total ion chromatographic profiles and integrated using QualBrowser, divided by the sum of the area of all peptides containing the unmodified and modified residue as shown in equation 1.
 |
For proteins where a known structure was not available, ITASSER was used to predict a structure based on the primary sequence of the protein (1). Predicted surface accessibilities for all Lys side chains and the N terminus were calculated using GetArea software online (45) with all parameters set as the default values based on known or ITASSER model structures as indicated. All predicted accessibility values for the lysine residues are based on the entire lysine side-chain. The GetArea algorithm classifies residues as solvent accessible if the surface accessibility ratio is calculated to exceed 50%, whereas residues are categorized as buried if the calculated value falls below 20% (45, 46). Correlation between the percent reactivities associated with the reactions of NN with the proteins and the surface accessibilities calculated from modeling programs such as GetArea is not expected to be quantitative. For our experimental strategy, only residues that reside on the surface of a protein will readily react with the NN reagent, thus establishing the general correlation between the surface accessibility of the residue with its percent reactivity. However, as the GetArea computations consider the entire side chain and not only the reactive amine group, there is not an exact parallel between the two (i.e. surface accessibility from GetArea versus percent reactivities from our experimental method). Moreover, our reported strategy uses a bulkier reagent and monitors its ability to interact with accessible primary amines, whereas the computed surface accessibilities are derived from contact of small solvent molecules within van der Waal's distances when rolled along the protein.
RESULTS
Design of NN Surface Accessibility Reagent
NN (Fig. 1) was designed to selectively react with primary amines, such as the ε-amino groups on lysine side chains and the N terminus which have proven consistently to be among the most reactive sites of proteins, via conventional N-hydroxysuccinimide (NHS) coupling. NN contains a N-N hydrazine bond that has previously been shown to preferentially cleave upon ETD (38), thus specifically facilitating the identification of modified peptides based on an easily monitored MS/MS reaction. For NN, the characteristic fragmentation upon ETD results in the neutral loss of 93 Da from the charge-reduced precursor, and the number of consecutive neutral losses is indicative of the number of modified residues. This pathway is the dominant fragmentation pathway of NN-modified peptides upon ETD, thus providing a facile way to differentiate NN-modified peptides from unmodified peptides and allow confident sequencing by ETD or CID (Figs. 2 and 3). The characteristic N-N bond cleavage and neutral loss of 93 Da may also be used to implement a data-dependent scan mode in which the acquisition of a CID spectrum can be triggered by this neutral loss to identify the peptide and locate the modifications.
Fig. 2.

Preferential ETD cleavage of NN-modified peptides. ETD of triply charged MQIFVKTLTGK from ubiquitin with a single NN modification (A) and double NN modification (B). The sites of modification are indicated by bold underlined font. Preferential ETD cleavage is indicated by the arrows. Φ indicates the precursor ion in each spectrum and Δ indicates the NN modification maintained on fragment ion.
Fig. 3.

Identification of NN-modified peptides from PARP-1 Domain C. ETD (A) and CID (B) of doubly charged LLIFNKQQVPSGE. An ETD reaction time of 100 msec and a collision energy of 35% was used. The sites of modification are indicated by bold underlined font. Φ indicates the precursor ion in each spectrum, * depicts neutral losses, and Δ indicates the NN modification maintained on the fragment ion.
The use of ETD as the primary activation method for analysis of the peptides produced from the modified protein also offers a strategic advantage. Because the site of modification (side-chain of lysines) is also the cleavage site for trypsin, the most frequently used protease, missed cleavages at the modified lysine sites are common. Therefore, GluC and ArgC digests were used in combination with trypsin to gain a more detailed picture of each protein's structure. ETD proves to be remarkably efficient for activation of the larger, more highly charged peptides generated from the GluC and ArgC digests, thus making ETD a natural fit for this strategy. For the reactions of NN with each protein, the protein/NN ratios were varied over a range of values in order to optimize reaction efficiencies while maintaining the native tertiary protein structure. At lower molar ratios, only the most accessible sites are modified and the structural integrity is most readily maintained. With higher molar ratios, the reaction efficiencies of less accessible modification sites are increased, thus enhancing detection of those sites.
Method Verification
Ubiquitin contains eight primary amine sites including seven Lys and the N terminus. The reaction of ubiquitin with the NN reagent is very efficient, as evidenced by the ESI-mass spectrum of the protein after modification (see supplemental Figs. S2A and S2B). For ubiquitin, a 1:2 protein/NN ratio produced predominantly singly modified proteins with minor contributions of doubly and triply modified proteins. Subsequent tryptic digestion and LC-MS/MS analysis resulted in identification of nine unmodified peptides plus 14 modified peptides, the latter based on the characteristic N-N bond cleavage upon ETD followed by CID. The modified peptides were easily distinguished from unmodified peptides using ETD through the preferential cleavage of the NN bond (Fig. 2). This cleavage leads to a loss of 93 from all charge reduced species. In addition, sequential losses of 93 Da can be used to identify the number of modifications. For example, the N-terminal peptide for ubiquitin, MQIFVKTLTGK, has two possible modification sites, the N-terminal M1 and K6. Upon ETD of the singly modified peptide a single loss of 93 Da from both the singly and doubly charged reduced species reflects the incorporation of a single NN modification (Fig. 2A), whereas the doubly modified peptide shows two sequential losses of 93 Da, thus indicating two modifications (Fig. 2B). In this example, the sites of modification can be pinpointed directly from the ETD spectra. For the singly modified peptide, the unmodified c2, c3, z3, and z4 ions and the modified z8 and c7 ions localize the first modification on K6; for the doubly modified the absence of the unmodified c2, c3, z3, z4, z5, singly modified z8, and the doubly modified c7 ions indicates the second modification is located at M1. However, in some cases the location of modification cannot be pinpointed from the ETD spectrum alone because of insufficient fragment ions. Thus, a subsequent CID step was included to target the peptide (see supplemental Fig. S3 for an example from ubiquitin), thus ensuring the ability to sequence each modified peptide. A direct MS3 strategy to sequence the peptides is also possible in which CID is undertaken on the product formed upon the diagnostic neutral loss (–93 Da) in the subsequent ETD, making the strategy amenable to more elegant data-dependent work-flows.
The abundances of the unmodified peptides and NN modified counterparts were quantified through manual integration of peak areas in the TIC profile using QualBrowser software, and the results are summarized in supplemental Table S1 in terms of percent reactivities along with the surface accessibilities estimated from the GetArea algorithm. The surface accessibilities of the sites predicted by the GetArea algorithm based on the previously determined tertiary structure of ubiquitin ranged from 11 to 77% (supplemental Table S1). Ubiquitin was reacted with NN using four protein/NN molar ratios from 1:2 to 1:10, yielding up to three modifications for the low molar ratio of 1:2 and up to five modifications for the higher molar ratios of reagent to intact protein. These results are in general agreement with the expected surface accessibilities of the amine sites given that four Lys residues have surface accessibilities greater than 50% and one Lys is slightly lower at 47% based on the GetArea predictions. These five Lys residues are situated on the surface of the protein and therefore are more accessible to modification whereas the other three primary amines reside on the interior of the tertiary structure, making their side-chains largely inaccessible and unreactive. Three proteases, trypsin, GluC, and ArgC, were used to digest the protein prior to LCMS/MS analysis, thus facilitating identification of the modified peptides in a bottom-up approach. Although trypsin is the most popular protease for conventional proteomics strategies, it proves to be problematic for proteins modified at lysine sites, such as the NN-modified proteins in the present study, in which the modification disrupts proteolytic cleavage after the lysines. However, trypsin does give the most comprehensive sequence coverage of all three digests and thus provided information about some of the more inaccessible residues. Note that any differences in ionization efficiencies of the peptides, whether NN-modified or not and regardless of the sizes, charge states, or elution times, were not compensated nor corrected because the percent reactivity values reflect ratios of abundances of modified to sums of modified and unmodified peptides. Thus, the percent reactivities show relative differences in reactivities of various accessible (or inaccessible) amino acids, not absolute values.
For ubiquitin, K27 and K29 were shown to be the least accessible sites as they displayed no reactivity based on the GluC and ArgC results and very little reactivity for the set of trypsin results as shown in supplemental Table S1. These sites are heavily involved in hydrogen bonding on the interior of the protein. Our general reactivity trend for all three proteases was Lys6 ≈ Lys63 ≈ Lys 48 > Met 1 > Lys 33 > Lys 11 > Lys 27, Lys 29; in which the only discrepancy from previously reported results was for the N-terminal methionine which was found to be slightly less reactive than Lys 6, Lys 63, and Lys 48. This trend agrees well with the surface accessibility predictions for the tertiary structure of ubiquitin from GetArea as the N terminus had only a 14% surface accessibility whereas Lys 6, Lys 63, and Lys 48 all averaged about 60% reactivities.
Our NN-based accessibility results are in general agreement with other surface accessibility studies that have been conducted for ubiquitin (47, 48). Upon amidination of the intact protein, the Reilly group found that the only inaccessible Lys was K27 whereas all other possible modification sites were fully amidinated (47). This Lys residue resides in the bottom of a hydrophobic pocket of ubiquitin's tertiary structure, thus leaving it completely shielded from the solvent. A top-down approach that utilized N-hydroxysuccinimidyl acetate to acetylate primary amines in ubiquitin found the reactivity trend to be Met1 ≈ Lys6 ≈ Lys48 ≈ Lys 63 > Lys 33 > Lys 11> Lys 27, Lys 29 (48). The Met1, Lys6, Lys48, and Lys63 sites were found to be the most reactive as they were involved in only weak hydrogen bonds involving other backbone carbonyl groups, whereas Lys 11, Lys 27, and Lys 29 were the least accessible because of their involvement in strong hydrogen bonds to carboxylic acids on other amino acid side chains.
Upon denaturing, the extent of NN reaction significantly changed (supplemental Figs. S2 and S4). These changes in reactivity confirm that the accessibility and thus reactivity of NN is dependent on the conformation of the protein, as desirable for a chemical probe of tertiary structure. Circular dichroism was also used to monitor any potential structural changes that occurred upon NN binding. No notable differences were seen for the NN-modified and native proteins, as exemplified by the CD comparison for native PARP to NN-bound PARP in supplemental Fig. S4. The CD results support that the NN reactions do not significantly disrupt the secondary structures of the proteins.
Surface Accessibility of Wheat eIF4E and PARP-1 Domain C
Upon successful demonstration of ETD-selective cleavage of the NN-modified peptides from ubiquitin, the surface maps of two other proteins, eukaryotic translation initiation factor-4E (eIF4E) and Poly(ADP-ribose) polymerase-1 (PARP-1) domain C, were evaluated using a similar strategy. The structure of eIF4E has been extensively studied in mammalian and yeast cells but only recently in wheat (49). Upon crystallization, a dimeric structure has been observed because of the artifact formation of a disulfide bond. The only known monomeric structure was determined for a mutant bound to 7-methyl-GDP that cannot form the disulfide bond (49). Wheat eIF4E has 15 Lys and the N terminus as possible modification sites. At the low molar protein:NN ratios utilized for ubiquitin, minimal modifications were observed for eIF4E; however, at higher protein/NN molar ratios of 1:15 to 1:20, the dominant species was the singly modified protein (Fig. 4A). The relatively high protein/NN ratio required for this protein, as well as the rather low modification rate, suggests that there are fewer highly accessible Lys residues and the native structure is only minimally disrupted after the first modification. The NN-modified protein was then subjected to enzymatic digestion using three proteases (trypsin, GluC, ArgC), followed by LCMS/MS analysis. In each case, spectral acquisition involved collection of full ESI mass spectra, followed by ETD and CID spectra of the five most abundant ions. All peptides were manually identified. The characteristic loss of 93 Da upon ETD was used to pinpoint the NN-modified peptides, and then CID was subsequently used to identify the sequence (see supplemental Fig. S5 for an example from eIF4E). Unmodified peptides were identified using the ETD spectra for those not exhibiting the loss of 93 Da. Percent reactivities were calculated based on Equation 1 in which the peak area for each modified residue was integrated and compared with the peak areas of all peptides containing the residue using QualBrowser software.
Fig. 4.

ESI-mass spectra of the NN-modified proteins. A, Wheat eIF4E at a 1:15 protein:NN molar ratio, B, PARP-1 Domain C at a 1:5 protein:NN molar ratio; Δ = NN adduct.
The artifact dimeric structure of wheat eIF4E previously determined by x-ray and NMR measurements (49) arises from disulfide bond formation and does not give a good representation of the monomer because of the extensive artificial protein-protein interface. A structure for a known mutant of wheat eIF4E has also been characterized previously as well, but this mutant is formed upon binding to 7-methyl-GDP which may cause considerable changes in tertiary structure, especially near the binding pocket. Therefore the tertiary structure of monomeric eIF4E was predicted using molecular modeling software, ITASSER (1), prior to calculation of the surface accessibility values for all amino acid side-chains via the GetArea program which were compared to those of the known mutant monomer and dimeric forms (Table I). The known structure for the dimer, the 7-methyl-GDP mutant, and the monomer structure predicted from ITASSER are shown in Fig. 5 with key Lys residues labeled. The surface accessibility values calculated by GetArea for each structure were compared with the experimental percent reactivities determined for each enzymatic digest based on the LC/ETD/CID strategy in Table I. Based on the surface accessibilities calculated for the ITASSER-predicted monomer, three to four Lys residues are expected to exhibit much higher accessibility (above 90%) than all other possible modification sites; K18, K65, K122, and K169. For the ESI-MS/MS results of the tryptic digest of the NN-modified protein, all four of these sites were identified as the primary sites of modification, thus indicating substantial NN reactivities. The N terminus (A1) exhibited low accessibility (2%). Upon increasing the molar ratio of protein/NN from 1:15 to 1:20, the reactivities for K65, K122, and K169 all increased to 100% whereas the reactivity of K18 showed little change and that of the N terminus increased to 26%. The latter change may signal a structural change for this part of the protein upon binding of NN to K18, thus opening up the structure so that the N terminus is more readily accessible. For the GluC digest, significant reactivity was observed for A1, K18, K122, and K169 as seen in the tryptic digest. However, based on the GluC digest, no reactivity values could be determined for K63, K65, K80, or K89 as no peptides were identified that contained these residues. We believe the absence of these peptides arises from the sizes of the peptides produced via GluC proteolysis and possibly spontaneous disulfide bonds forming following digestion. For the ArgC digest, again K18, K65, K122 and K169 proved to be the most reactive residues with A1 showing a small amount of reactivity at a 1:15 molar ratio. The lack of reactivity of other Lys sites indicates these Lys residues reside on the interior of the protein, restricting their exposure to the NN reagent.
Table I. Comparison of Surface Accessibility to Percent Reactivity for wheat eIF4E reacted with NN at a 1:15 protein/NN molar ratio. Surface accessibility was calculated using GetArea. Percent Reactivity was calculated using equation 1. “NA” indicates a peptide that was not found in either its modified or unmodified form. Standard deviations ranged from 10–20% for all percent reactivities.
| Residue | Solvent accessibility % |
% Reactivity |
||||
|---|---|---|---|---|---|---|
| Dimer | Mutant | Monomer | Tryptic | GluC | ArgC | |
| Ala 1 | 46 | 65 | 41 | 12 | 11 | 5 |
| Lys 18 | 43 | 34 | 96 | 19 | 5 | 25 |
| Lys 52 | 76 | 74 | 50 | 0 | 0 | 0 |
| Lys 63 | 5 | 9 | 20 | 0 | NA | 0 |
| Lys 65 | 70 | 100 | 94 | 32 | NA | 11 |
| Lys 69 | 100 | 20 | 48 | 0 | 0 | 0 |
| Lys 80 | 27 | 20 | 20 | 0 | NA | 0 |
| Lys 89 | 77 | 80 | 67 | NA | NA | NA |
| Lys 122 | 40 | 38 | 92 | 79 | 5 | 70 |
| Lys 131 | 31 | 26 | 31 | 0 | 0 | 0 |
| Lys 144 | 68 | 73 | 66 | 1 | 2 | 0 |
| Lys 147 | 22 | 15 | 13 | 0 | 0 | 0 |
| Lys 153 | 42 | 42 | 84 | 0 | 0 | 0 |
| Lys 165 | 80 | 43 | 50 | 0 | 0 | 0 |
| Lys 169 | 40 | 81 | 91 | 60 | 9 | 34 |
| Lys 172 | 28 | 94 | 72 | 0 | 0 | 0 |
Fig. 5.

Structures for wheat eIF4E. Dimer = PDB 2IDR, 7-Me-GDP mutant = PDB 2IDV. The monomer is a predicted model from ITASSER, and key Lys residues (dark gray) are labeled.
PARP-1 Domain C (50, 51) has recently been isolated and studied by NMR spectroscopy to determine its solution structure (52). The NMR characterization yielded the “shortened” structure shown in Fig. 6 because three amino acids at the N terminus and 15 amino acids at the C terminus were not detected. No other NMR or x-ray crystallographic structures have been reported for the true full monomeric form of the protein. In its full form, PARP-1 domain C contains 18 Lys residues plus the N terminus as possible modification sites. The “shortened” version determined by NMR maintains all 18 Lys residues but one potential surface accessible reactive site (the true N terminus amine) is excluded in the structural map because the three amino acids at the N terminus are invisible by NMR. Because the tertiary structure of the full sequence version of domain C was unknown, its conformation was predicted by ITASSER (1), resulting in five structures whose surface accessibilities were predicted by the GetArea program.
Fig. 6.

Structures for PARP-1 Domain C, including shortened PARP = PDB 2JVN and the full native PARP is predicted model 5 from ITASSER. Lys residues (dark gray) with high accessibility are labeled. Boxed sections show key changes in structure recognized based on NN reactivity.
Upon reaction of PARP-1 Domain C with NN, several residues are modified even at low molar ratios (Fig. 4B). The NN-modified protein was enzymatically digested using trypsin, GluC, or ArgC, and the resulting peptides were analyzed by the LCMS/MS strategy described above and with MS/MS examples shown in Fig. 3. Percent reactivities and surface accessibilities for all known and predicted structures were calculated and summarized in Table II.
Table II. Comparison of Surface Accessibility to Percent Reactivity for PARP-1 Domain C reacted with NN in a 1:5 molar ratio protein/NN. Surface accessibility for shortened structure was calculated using GetArea for pdb 2JVN and for the full native structure based on the top five structures predicted by ITASSER (IT1-IT5). Percent reactivity was calculated using equation 1. The bold box highlights the best model structure correlating with percent reactivity. “NA” indicates a peptide that was not found in either its modified or unmodified form. Standard deviations ranged from 10–20% for all percent reactivities.
| Residue | Surface accessibility % |
% Reactivity |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Shortened | IT1 | IT2 | IT3 | IT4 | IT5 | Tryptic | GluC | ArgC | |
| GLY 1 | – | 76 | 90 | 100 | 72 | 79 | 95 | 100 | 100 |
| LYS 4 | 100 | 87 | 78 | 91 | 76 | 88 | 93 | 85 | 95 |
| LYS 7 | 81 | 70 | 79 | 71 | 75 | 91 | 42 | 38 | 5 |
| LYS 10 | 85 | 76 | 63 | 64 | 71 | 77 | 31 | 0 | 10 |
| LYS 20 | 21 | 16 | 20 | 29 | 30 | 10 | 19 | 0 | 9 |
| LYS 24 | 65 | 56 | 48 | 37 | 70 | 40 | 19 | 0 | 0 |
| LYS 25 | 65 | 55 | 62 | 57 | 58 | 56 | 0 | 0 | 0 |
| LYS 33 | 49 | 35 | 33 | 34 | 34 | 56 | 0 | 0 | 0 |
| LYS 40 | 93 | 76 | 87 | 85 | 77 | 82 | 17 | 1 | 8 |
| LYS 76 | 95 | 62 | 45 | 45 | 48 | 62 | 0 | NA | 7 |
| LYS 91 | 54 | 67 | 55 | 63 | 67 | 45 | 0 | 0 | 0 |
| LYS 95 | 74 | 50 | 36 | 42 | 41 | 44 | 0 | 0 | 0 |
| LYS 102 | 65 | 69 | 66 | 63 | 52 | 37 | 88 | 0 | 0 |
| LYS 108 | 70 | 67 | 92 | 84 | 74 | 71 | 51 | 65 | 35 |
| LYS 117 | 25 | 37 | 42 | 42 | 37 | 13 | 42 | 0 | 0 |
| LYS 118 | 88 | 46 | 50 | 46 | 55 | 62 | 43 | 100 | 98 |
| LYS 120 | 25 | 41 | 53 | 50 | 52 | 26 | 20 | 0 | 0 |
| LYS 122 | 43 | 84 | 83 | 74 | 76 | 53 | 0 | 0 | 0 |
| LYS 123 | 100 | 84 | 83 | 84 | 80 | 54 | 36 | 0 | 0 |
DISCUSSION
This ETD-selectively cleavable surface accessibility reagent allows facile tracking of lysine-reactive sites. Enzymatic digestion of the surface modified proteins results in an array of peptides, some of which contain the hydrazone moiety. As illustrated for the applications involving wheat eIF4E and PARP-C, the hydrazine bond cleaves selectively and with high efficiency upon ETD to yield a unique and characteristic loss of 93 Da from the charge-reduced peptides. CID and/or ETD allows sequencing of the peptides and the locations of the modification sites of the peptides to be pinpointed.
eIF4E is a crucial protein for the initiation of protein synthesis (49). In comparing the experimental percent reactivities to the predicted surface accessibilities of the dimer, mutant, and ITASSER-predicted monomer of eIF4E (Table I), a few key variations are noted. In both the dimer and mutant forms, K18 and K122 are much less accessible whereas in the predicted monomer form, these residues have surface accessibilities of 96 and 92% respectively, making them two of the most accessible sites. This notable difference mirrors the percent reactivities obtained from the NN protein modification results, confirming that both of these sites are two of the most reactive in wheat eIF4E. K69 is 100% accessible in the putative dimer but is completely unreactive with NN based on analysis of the various protein digests, indicating it resides in the interior of the monomer. K172 for the 7-Me-GDP mutant is expected to be located on the protein surface as a highly accessible residue, yet the experimental percent reactivity value is zero. The site of this amino acid coincides with the binding location of the ligand 7-methyl-GDP in the mutant, thus suggesting that the bound ligand induces a significant structural change that does not occur in the native monomer. In light of these differences in predicted accessibilities and experimentally determined reactivities, the NN reactivity results support the ITASSER predicted structure for the native monomer.
PARP-1 is a multi-modular protein consisting of six domains (domains A-F) that is involved in several key biological processes including DNA repair and cell death (50, 51). Domain C has been shown to be vital to these functions and without it, PARP-1 ABDEF exhibits no activity by itself. Neither does domain C bind to DNA, confirming that it is the interactions of the entire multi-domain protein that stimulates biological activity. Because the tertiary structure of the full sequence monomeric version of domain C was unknown, its conformation was predicted by ITASSER, resulting in five structures whose surface accessibilities were predicted by the GetArea program. The surface accessibilities for the shortened domain C structures are shown in Table II. The percent reactivities obtained for the NN-modified peptides do not uniformly parallel the surface accessibilities predicted from the NMR structure, as reflected by the differences in reactivities of a few key residues, including the highly reactive N-terminal G1 and the nonreactive K123 which was predicted to be 100% accessible based on the NMR structure. Of the structures predicted by ITASSER, the fifth model (IT5) showed the best correlation with the experimental surface accessibility results. This structure showed a highly reactive N-terminal portion of the protein with G1, K4, K7, and K10 all having surface accessibility values above 75% whereas the K123 showed somewhat lower accessibility at 54%. Upon examination of the structure of IT5, the low reactivity of K123 may be rationalized by steric blocking by the chain of amino acids on the adjacent C terminus (Fig. 6). The percent reactivities and predicted surface accessibilities of the other regions of the protein show good agreement with K40, K76, K108, and K118 all predicted to be more than 60% accessible, in line with the values from the NN modification reactions. The only residue that does not correlate between the percent reactivity and surface accessibility is K7. It was predicted to be one of the more accessible residues but in all cases exhibited low reactivity in comparison to G1 and K4. We hypothesize that this deviation is because of structural effects caused upon modification. Since G1 and K4 are so highly reactive, we suspect that upon their modification by NN, the K7 site is blocked, therefore hindering its reaction with the NN reagent. The modeled structure has not been verified by NMR or crystallographic measurements, thus reinforcing the need for alternative experimental methodologies for probing protein structures.
As reported herein, this selective ETD/CID method has proven to be efficient and robust for several proteins, ultimately providing maps of surface accessibility that are consistent with the predicted structures of the proteins. A decision-tree could be used to automate this method in the future, in which the selective cleavage triggers a subsequent CID scan for only the modified peptides.
Acknowledgments
We thank Laura Mayberry for the preparation of wheat eIF4E.
Footnotes
* This work was supported by grants from the NSF (CHE-1012622) and the Welch Foundation (F1155 to J.S.B. and F1511 to H.-W.L.), and the Texas Institute for Drug and Diagnostic Development (TI-3D) of the University of Texas at Austin (to H.-W.L.).
 This article contains supplemental Figs. S1 to S5 and Tables S1 to S4.
This article contains supplemental Figs. S1 to S5 and Tables S1 to S4.
1 The abbreviations used are:
- NMR
- nuclear magnetic resonance
- CID
- collision induced dissociation
- LC-MS/MS
- liquid chromatography-tandem MS
- NHS
- N-hydroxysuccinimide
- eIF4E
- eukaryotic translation initiation factor-4E
- PARP-1
- Poly(ADP-ribose) polymerase-1.
REFERENCES
- 1. Roy A., Kucukural A., Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mendoza V. L., Vachet R. W. (2009) Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom. Rev. 28, 785–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jørgensen T. D., Gårdsvoll H., Ploug M., Roepstorff P. (2005) Intramolecular migration of amide hydrogens in protonated peptides upon collisional activation. J. Am. Chem. Soc. 127, 2785–2793 [DOI] [PubMed] [Google Scholar]
- 4. Kaltashov I. A., Bobst C. E., Abzalimov R. R. (2009) H/D Exchange and Mass Spectrometry in the Studies of Protein Conformation and Dynamics: Is There a Need for a Top-Down Approach? Anal. Chem. 81, 7892–7899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rand K. D., Adams C. M., Zubarev R. A., Jørgensen T. J. D. (2008) Electron Capture Dissociation Proceeds with a Low Degree of Intramolecular Migration of Peptide Amide Hydrogens. J. Am. Chem. Soc. 130, 1341–1349 [DOI] [PubMed] [Google Scholar]
- 6. Rand K. D., Zehl M., Jensen O. N., Jørgensen T. J. D. (2009) Protein Hydrogen Exchange Measured at Single-Residue Resolution by Electron Transfer Dissociation Mass Spectrometry. Anal. Chem. 81, 5577–5584 [DOI] [PubMed] [Google Scholar]
- 7. Petrotchenko E. V., Borchers C. H. (2010) Crosslinking combined with mass spectrometry for structural proteomics. Mass Spectrom. Rev. 29, 862–876 [DOI] [PubMed] [Google Scholar]
- 8. Suckau D., Mak M., Przybylski M. (1992) Protein surface topology-probing by selective chemical modification and mass spectrometric peptide mapping. Proc. Natl. Acad. Sci. U.S.A. 89, 5630–5634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Glocker M. O., Borchers C., Fiedler W., Suckau D., Przybylski M. (1994) Molecular characterization of surface topology in protein tertiary structures by amino-acylation and mass spectrometric peptide mapping. Bioconjug. Chem. 5, 583–590 [DOI] [PubMed] [Google Scholar]
- 10. Zappacosta F., Ingallinella P., Scaloni A., Pessi A., Bianchi E., Sollazzo M., Tramontano A., Marino G., Pucci P. (1997) Surface topology of Minibody by selective chemical modifications and mass spectrometry. Protein Sci. 6, 1901–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Izumi S., Kaneko H., Yamazaki T., Hirata T., Kominami S. (2003) Membrane Topology of Guinea Pig Cytochrome P450 17α Revealed by a Combination of Chemical Modifications and Mass Spectrometry. Biochemistry 42, 14663–14669 [DOI] [PubMed] [Google Scholar]
- 12. Turner B. T., Jr., Sabo T. M., Wilding D., Maurer M. C. (2004) Mapping of Factor XIII Solvent Accessibility as a Function of Activation State Using Chemical Modification Methods. Biochemistry 43, 9755–9765 [DOI] [PubMed] [Google Scholar]
- 13. Scholten A., Visser N. F., van den Heuvel R. H., Heck A. J. (2006) Analysis of Protein-Protein Interaction Surfaces Using a Combination of Efficient Lysine Acetylation and nanoLC-MALDI-MS/MS Applied to the E9:Im9 Bacteriotoxin-Immunity Protein Complex. J. Am. Soc. Mass Spectrom. 17, 983–994 [DOI] [PubMed] [Google Scholar]
- 14. Liu X., Reilly J. P. (2009) Correlating the Chemical Modification of Escherichia coli Ribosomal Proteins with Crystal Structure Data. J. Proteome Res. 8, 4466–4478 [DOI] [PubMed] [Google Scholar]
- 15. Running W. E., Reilly J. P. (2010) Variation of the chemical reactivity of Thermus thermophilus HB8 ribosomal proteins as a function of pH. Proteomics 10, 3669–3687 [DOI] [PubMed] [Google Scholar]
- 16. Lehman J. A., Hoelz D. J., Turchi J. J. (2008) DNA-Dependent Conformational Changes in the Ku Heterodimer. Biochemistry 47, 4359–4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shell S. M., Hess S., Kvaratskhelia M., Zou Y. (2005) Mass Spectrometric Identification of Lysines Involved in the Interaction of Human Replication Protein A with Single-Stranded DNA. Biochemistry 44, 971–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sharp J. S., Nelson S., Brown D., Tomer K. B. (2006) Structural characterization of the E2 glycoprotein from Sindbis by lysine biotinylation and LC-MS/MS. Virology 348, 216–223 [DOI] [PubMed] [Google Scholar]
- 19. Blodgett D. M., De Zutter J. K., Levine K. B., Karim P., Carruthers A. (2007) Structural basis of GLUT1 inhibition by cytoplasmic ATP. J. Gen. Physiol. 130, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Azim-Zadeh O., Hillebrecht A., Linne U., Marahiel M. A., Klebe G., Lingelbach K., Nyalwidhe J. (2007) Use of Biotin Derivatives to Probe Conformational Changes in Proteins. J. Biol. Chem. 282, 21609–21617 [DOI] [PubMed] [Google Scholar]
- 21. Vasilescu J., Guo X., Kast J. (2004) Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 4, 3845–3854 [DOI] [PubMed] [Google Scholar]
- 22. Kosower E. M., Kosower N. S. (1995) Bromobimane probes for thiols. Methods Enzymol. 251, 133–148 [DOI] [PubMed] [Google Scholar]
- 23. Alley S. C., Ishmael F. T., Jones A. D., Benkovic S. J. (2000) Mapping Protein-Protein Interactions in the Bacteriophage T4 DNA Polymerase Holoenzyme Using a Novel Trifunctional Photo-cross-linking and Affinity Reagent. J. Am. Chem. Soc. 122, 6126–6127 [Google Scholar]
- 24. Sinz A., Kalkhof S., Ihling C. (2005) Mapping Protein Interfaces by a Trifunctional Cross-Linker Combined with MALDI-TOF and ESI-FTICR Mass Spectrometry. J. Am. Soc. Mass Spectrom. 16, 1921–1931 [DOI] [PubMed] [Google Scholar]
- 25. Hurst G. B., Lankford T. K., Kennel S. J. (2004) Mass spectrometric detection of affinity purified crosslinked peptides. J. Am. Soc. Mass Spectrom. 15, 832–839 [DOI] [PubMed] [Google Scholar]
- 26. Fujii N., Jacobsen R. B., Wood N. L., Schoeniger J. S., Guy R. K. (2004) A novel protein crosslinking reagent for the determination of moderate resolution protein structures by mass spectrometry (MS3-D). Bioorg. Med. Chem. Lett. 14, 427–429 [DOI] [PubMed] [Google Scholar]
- 27. Gardner M. W., Vasicek L. A., Shabbir S., Anslyn E. V., Brodbelt J. S. (2008) Chromogenic Cross-Linker for the Characterization of Protein Structure by Infrared Multiphoton Dissociation Mass Spectrometry. Anal. Chem. 80, 4807–4819 [DOI] [PubMed] [Google Scholar]
- 28. Pearson K. M., Pannell L. K., Fales H. M. (2002) Intramolecular cross-linking experiments on cytochrome c and ribonuclease A using an isotope multiplet method. Rapid Commun. Mass Spectrom. 16, 149–159 [DOI] [PubMed] [Google Scholar]
- 29. Müller D. R., Schindler P., Towbin H., Wirth U., Voshol H., Hoving S., Steinmetz M. O. (2001) Isotope-Tagged Cross-Linking Reagents. A New Tool in Mass Spectrometric Protein Interaction Analysis. Anal. Chem. 73, 1927–1934 [DOI] [PubMed] [Google Scholar]
- 30. Collins C. J., Schilling B., Young M., Dollinger G., Guy R. K. (2003) Isotopically labeled crosslinking reagents: resolution of mass degeneracy in the identification of crosslinked peptides. Bioorg. Med. Chem. Lett. 13, 4023–4026 [DOI] [PubMed] [Google Scholar]
- 31. Huang B. X., Kim H. Y., Dass C. (2004) Probing three-dimensional structure of bovine serum albumin by chemical cross-linking and mass spectrometry. J. Am. Soc. Mass Spectrom. 15, 1237–1247 [DOI] [PubMed] [Google Scholar]
- 32. Taverner T., Hall N. E., O'Hair R. A., Simpson R. J. (2002) Characterization of an Antagonist Interleukin-6 Dimer by Stable Isotope Labeling, Cross-linking, and Mass Spectrometry. J. Biol. Chem. 277, 46487–46492 [DOI] [PubMed] [Google Scholar]
- 33. Yang L., Tang X., Weisbrod C. R., Munske G. R., Eng J. K., von Haller P. D., Kaiser N. K., Bruce J. E. (2010) A Photocleavable and Mass Spectrometry Identifiable Cross-Linker for Protein Interaction Studies. Anal. Chem. 82, 3556–3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Davidson W. S., Hilliard G. M. (2003) The spatial organization of apolipoprotein A-I on the edge of discoidal high density lipoprotein particles: A mass spectrometry study. J. Biol. Chem. 278, 27199–27207 [DOI] [PubMed] [Google Scholar]
- 35. Bennett K. L., Kussmann M., Björk P., Godzwon M., Mikkelsen M., Sørensen P., Roepstorff P. (2000) Chemical cross-linking with thiol-cleavable reagents combined with differential mass spectrometric peptide mapping-A novel approach to assess intermolecular protein contacts. Protein Sci. 9, 1503–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Back J. W., Hartog A. F., Dekker H. L., Muijsers A. O., de Koning L. J., de Jong L. (2001) A new crosslinker for mass spectrometric analysis of the quaternary structure of protein complexes. J. Am. Soc. Mass Spectrom. 12, 222–227 [DOI] [PubMed] [Google Scholar]
- 37. Tang X., Munske G. R., Siems W. F., Bruce J. E. (2005) Mass spectrometry identifiable cross-linking strategy for studying protein-protein interactions. Anal. Chem. 77, 311–318 [DOI] [PubMed] [Google Scholar]
- 38. Gardner M. W., Brodbelt J. S. (2010) Preferential Cleavage of N-N Hydrazone Bonds for Sequencing Bis-arylhydrazone Conjugated Peptides by Electron Transfer Dissociation. Anal. Chem. 82, 5751–5759 [DOI] [PubMed] [Google Scholar]
- 39. Petrotchenko E. V. S., Sherpa J. J., Borchers C. H. (2011) An isotopically coded CID-cleavable biotinylated cross-linker for structural proteomics. Mol. Cell Proteomics 10, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kao A., Chiu C. I., Vellucci D., Yang Y., Patel V. R., Guan S., Randall A., Baldi P., Rychnovsky S. D., Huang L. (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics 10, 1–17, 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reid G. E., Roberts K. D., Simpson R. J., O'Hair R. A. J. (2005) Selective Identification and Quantitative Analysis of Methionine Containing Peptides by Charge Derivatization and Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 16, 1131–1150 [DOI] [PubMed] [Google Scholar]
- 42. Roberts K. D., Reid G. E. (2007) Leaving group effects on the selectivity of the gas-phase fragmentation reactions of side chain fixed-charge-containing peptide ions. J. Mass Spectrom. 42, 187–198 [DOI] [PubMed] [Google Scholar]
- 43. Zhou X., Lu Y., Wang W., Borhan B., Reid G. E. (2010) ‘Fixed Charge’ Chemical Derivatization and Data Dependant (sic) Multistage Tandem Mass Spectrometry for Mapping Protein Surface Residue Accessibility. J. Am. Soc. Mass Spectrom. 21, 1339–1351 [DOI] [PubMed] [Google Scholar]
- 44. Madsen J. A., Kaoud T. S., Dalby K. N., Brodbelt J. S. (2011) 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics 11, 1329–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fraczkiewicz R., Braun W. (1998) Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Computational Chem. 19, 319–333 [Google Scholar]
- 46. Zhu H., Fraczkiewicz R., Braun W. (1999) Solvent accessible surface areas, atomic solvation energies, and their gradients for macromolucules http://curie.utmb.edu/area_man.html
- 47. Janecki D. J., Beardsley R. L., Reilly J. P. (2005) Probing protein tertiary structure with amidination. Anal. Chem. 77, 7274–7281 [DOI] [PubMed] [Google Scholar]
- 48. Novak P., Kruppa G. H., Young M. M., Schoeniger J. (2004) A top-down method for the determination of residue-specific solvent accessibility in proteins. J. Mass Spectrom. 39, 322–328 [DOI] [PubMed] [Google Scholar]
- 49. Monzingo A. F., Dhaliwal S., Dutt-Chaudhuri A., Lyon A., Sadow J. H., Hoffman D. W., Robertus J. D., Browning K. S. (2007) The structure of eukaryotic translation initiation factor-4E from wheat reveals a novel disulfide bond. Plant Physiol. 143, 1504–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Satoh M. S., Lindahl T. (1992) Role of poly(ADP-ribose) formation in DNA repair. Nature 356, 356–358 [DOI] [PubMed] [Google Scholar]
- 51. Yu S. W., Wang H., Poitras M. F., Coombs C., Bowers W. J., Federoff H. J., Poirier G. G., Dawson T. M., Dawson V. L. (2002) Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297, 259–263 [DOI] [PubMed] [Google Scholar]
- 52. Tao Z., Gao P., Hoffman D. W., Liu H. w. (2008). Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry 47, 5804–5813 [DOI] [PubMed] [Google Scholar]