

Abstract
We examined the efficacy of unrelated cord blood (CB) transplantation in children with thalassemia (n = 35) and sickle cell disease (n = 16), using data reported to 3 registries. Donor-recipient pairs were matched at HLA-A and -B (antigen level) and DRB1 (allele level) in 7 or HLA mismatched at 1 (n = 18), 2 (n = 25), or 3 loci (n = 1). Transplant conditioning was myeloablative (n = 39) or reduced intensity (n = 12). Neutrophil recovery with donor chimerism was documented in 24 patients; 11 patients developed grade II-IV acute graft-versus-host disease (aGVHD) and 10 patients, chronic GVHD (cGVHD). Overall survival (OS) and disease-free survival (DFS) were 62% and 21% for thalassemia and 94% and 50% for sickle cell disease (SCD), respectively. In multivariate analysis, engraftment rate (hazard ratio [HR] 2.2, P =.05) and DFS (HR 0.4, P =.01) were higher with cell dose >5 × 107/kg. The 2-year probability of DFS was 45% in patients who received grafts with cell dose >5 × 107/kg and 13% with lower cell dose. Primary graft failure was the predominant cause of treatment failure occurring in 20 patients with thalassemia and 7 patients with SCD. Primary graft failure was fatal in 5 patients with thalassemia. These results suggest that only CB units containing an expected infused cell dose >5 × 107/kg should be considered for transplantation for hemoglobinopathy.
Keywords: Thalassemia, Sickle cell disease, Cord blood transplantation, Graft failure
INTRODUCTION
Transplantation of hematopoietic stem cells (HSC) from a HLA matched sibling including umbilical cord blood (CB) has been used successfully for thalassemia and sickle cell disease (SCD) [1,2]. Disease-free survival (DFS) rates approach 80% to 90% with transplantation of bone marrow (BM) for sickle cell disease [3,4] and 80% to 95% for beta-thalassemia [5,6]. Despite the availability of over 14 million potential unrelated adult donors registered worldwide, only approximately 17% of African Americans and 20% of Asians who may benefit from unrelated donor transplantation are able to identify a HLA matched unrelated adult donor [7]. As advances in supportive care after transplantation in recent years have led to improvements in survival for patients with hemoglobinopathy [8,9], alternative donor sources such as HLA mismatched family members or unrelated CB units are being explored for patients with hemoglobinopathy. Recent reports suggest DFS rates of approximately 70% for patients with thalassemia who received transplants of allele-level HLA matched unrelated donor BM [10], and similar results are also reported after haploidentical donor transplants [11].
Unrelated cord blood transplantation (CBT) has been explored as an alternative option for these patients without a suitably HLA matched unrelated adult donor. Tolerability of HLA mismatch with relatively low rates of acute and chronic graft-versus-host disease (aGVHD, cGVHD), make CBT a potentially attractive option for those with a hemoglobinopathy. However, CBT may be limited by less than adequate cell dose and higher rates of primary graft failure [12-14]. In a recent report from Taiwan, 27 of 32 patients with thalassemia were disease free at 2 years after CBT with myeloablatve conditioning regimens [15]. In contrast, 4 of 7 patients with SCD after CBT had primary graft failure; importantly, 3 of these 4 patients received reduced-intensity conditioning (RIC) regimens [16]. In the absence of conclusive recommendations on CBT for thalassemia and SCD, we examined outcomes after CBT using data reported to Eurocord, the National Cord Blood Program (NCBP), New York Blood Center, and the Center for International Blood and Marrow Transplant Registry (CIBMTR) to determine the state of the field at the present time.
PATIENTS, MATERIALS, AND METHODS
Patients
Patients with hemoglobinopathy who received transplants of a single unmanipulated CB unit and who were without a history of prior HSC transplantation were eligible; 35 patients had thalassemia, and 16 patients had SCD. Patients reported to 2 or more registries (n = 6) were identified, and each patient included in the current analysis represents a “unique case.” CBT were performed at 32 transplant centers between 1996 and 2009. The study was approved by the institutional review board, Medical College of Wisconsin and the Eurocord-Netcord scientific committee. Myeloablative conditioning regimen was defined as oral (p.o.) busulphan ≥8 mg/kg or intravenous (i.v.) busulphan ≥6.4 mg/kg, or melphalan ≥150 mg/m2, or total-body irradiation (TBI) (≥6 Gy) were used.
Endpoints
Neutrophil recovery was defined as achieving absolute neutrophil count (ANC) ≥0.5 × 109/L for 3 consecutive days with sustained donor engraftment assessed by chimerism assay (≥95% donor). The timing and method of assay were at the discretion of the transplant center. Primary graft failure was defined as having never achieved ANC ≥0.5 × 109/L or ANC ≥0.5 × 109/L without donor engraftment (autologous recovery). Secondary graft failure was defined as having achieved ANC ≥0.5 × 109/L with subsequent decline to <0.5 × 109/L or loss of donor engraftment. Platelet recovery was defined as achieving platelets ≥20 × 109/L unsupported by platelet transfusions for 7 days. The diagnosis and grading of aGVHD and cGVHD was assigned by the transplant center using standard criteria [17,18]. DFS was defined as being alive with donor chimerism. Graft failure, second transplant, and death from any cause were considered events.
Statistical Analysis
Median value and ranges are reported for continuous variables and percentages for categoric variables (Table 1). The probabilities of overall survival (OS) and DFS were calculated using the Kaplan-Meier estimator and the log-rank test for univariate comparisons [19]. The probabilities of neutrophil engraftment, grade acute 2-4 and cGVHD were calculated with the cumulative incidence estimator. Multivariate analyses were performed using Cox proportional hazards regression model for DFS and OS, and Fine and Gray’s proportional hazards regression model [20,21].
Table 1.
Patient, Disease, and Transplant Characteristics
| Thalassemia | Sickle Cell Disease | |
|---|---|---|
| Number evaluation | 35 | 16 |
| Sex male, % | 58% | 75% |
| Interval from diagnosis to transplant, months | 29 (3-169) | 76 (7-191) |
| Conditioning regimen | ||
| Myeloablative regimens, n | 30 | 9 |
| Busulfan + cyclophosphamide (Cy) + ATG | 27 | 6 |
| Busulfan + fludarabine + Cy | 2 | — |
| Busulfan + fludarabine | — | — |
| Busulfan + Cy +Thiotepa + ATG | 1 | — |
| Busulfan + fludarabine + alemtuzumab | — | 1 |
| Busulfan + melphalan | — | 1 |
| Busulfan + melphalan + ATG | — | 1 |
| Reduced-intensity regimens, n | 5 | 7 |
| Busulfan + fludarabine + TLI + ATG/alemtuzumab | 1 | 2 |
| Melphalan + fludarabine + ATG/alemtuzumab | 1 | 2 |
| Cy + fludarabine ± TLI + ATG/alemtuzumab | 2 | 3 |
| Fludarabine ± TB 200 Gy | 1 | — |
| Use of ATG/Campath before day 0, % | 86% | 94% |
| GVHD prophylaxis | ||
| Cyclosporine based,n, % | 27 (80%) | 9 (54%) |
| Tacrolimus based, n, % | 8 (20%) | 7 (46%) |
| Donor-recipient HLA match, n | ||
| 6/6 matched | 5 | 2 |
| 5/6 matched | 14 | 4 |
| 4/6 matched | 15 | 10 |
| 3/6 matched | 1 | — |
| Total nucleated cell dose, at collection/kg (107) | 6 (2-32) | 6 (2-12) |
| Total nucleated cell dose, infused/kg (107) | 5 (2.4-23) | 4.9 (1.1-9) |
| Median follow-up of surviving patients | 21 (3-138) | 37 (3-80) |
GVHD indicates graft-versus-host disease; ATG, antithymocyte globulin; YLI, total lymphoid irradiation.
The variables evaluated included recipient age, donor sex, disease duration before UCBT, ABO compatibility, HLA matching high-resolution DNA typing, number of total nucleated cells (TNCs) at the time of freezing and infusion, conditioning regimen, and year of UCBT. Each potential risk factor was tested independently. All factors that reached a P value = .10 in the univariate analysis were included in the multivariate model. All models were built using a forward stepwise method. Only factors that reached P value ≤.05 were retained in the final model. Patients were censored at death if they underwent a second transplant or for surviving patients, at last follow-up. P values are 2 sided. Statistical analyses were performed with SPSS (Inc., Chicago, IL) and Splus (MathSoft, Inc, Seattle, WA).
RESULTS
Patient, Disease, and Transplant Characteristics
Patient, disease, and transplant characteristics are shown in Table 1. The indications for transplantation were a history of stroke (n = 12) and acute chest syndrome or vaso-occlusive crisis (n = 4) in patients with SCD. As transplantations occurred worldwide and consistent with published reports, the Pesaro risk score was not assigned pretransplant for all patients with thalassemia; only 15 of 35 patients had sufficient data for assignment of the Pesaro classification: class 1 (n = 9), class 2 (n = 2), and class 3 (n = 4). Patients with thalassemia were younger (median, 4 years) compared to those with SCD (median, 6 years). Only 7 donor-recipient pairs were matched at HLA-A and -B (antigen level) and DRB1 (allele level) with the remaining mismatched at 1 (n = 18), 2 (n = 25), or 3 HLA loci (n = 1). Myeloablative conditioning regimens were used in 30 of 35 patients with thalassemia and 9 of 16 with SCD. Busulfan and cyclophosphamide with or without antityhmocyte globulin (ATG) was the most common myeloablative-conditioning regimen. Twelve patients received RIC and all except 1 received in vivo T cell depletion (ATG or alemtuzumab). All patients received calcinuerin inhibitor containing GVHD prophylaxis, and cyclosporine was the most common agent (65%). The median follow-up of surviving patients with thalassemia and SCD was 21 and 37 months, respectively.
Engraftment and Graft Failure
Twenty-four of 51 patients (n = 15 thalassemia; n = 9 SCD) achieved neutrophil recovery with complete donor chimerism. The median time to neutrophil recovery was 22 days (range: 10-62 days). Two patients had mixed chimerism prior to day 100 but subsequently achieved complete donor chimerism. None of the patients experienced secondary graft failure. In multivariate analysis, engraftment (neutrophil recovery with full donor chimerism) was higher in recipients who received CB units with infused total nucleated cell dose of >5 × 107 nucleated cells/kg (hazard ratio [HR] 2.2, 95% confidence interval [CI], 0.96-3.6, P = .05). The day 60 cumulative incidence of engraftment was 63% ± 9% with CB units containing >5 × 107 nucleated cells/kg and 32% ± 8% with lower cell dose CB units.
The median time to platelet recovery was 40 days (range: 15-127 days). As with neutrophil recovery, platelet recovery was higher with transplantation of CB units containing total nucleated cells >5 × 107/kg (HR 3.4, 95% CI, 1.2-6.2, P = .02). The day 180 cumulative incidence of platelet recovery was 68% ± 9% with CB units containing >5 × 107 nucleated cells/kg compared to 25% ± 9% with CB units containing a lower dose.
The characteristics and outcome of the 24 patients who engrafted are shown in Table 2A. Nineteen patients received myeloablative conditioning regimens and 5, RIC regimens. The characteristics of the 27 patients with primary graft failure and their outcome are shown in Table 2B. The cumulative incidence of graft failure was 52% at day 60 and at 1 year. Twenty patients received myeloablative conditioning regimens and 7, RIC regimens. Of these patients with graft failure, 18 had autologous reconstitution, 3 patients received their autologous back-up for prolonged neutropenia and absence of chimerism, 4 patients died without neutrophil recovery, and 2 patients underwent a second allogeneic transplant. Both recipients of second allogeneic HSC transplant are alive with complete donor chimerism with the donor of the second transplant.
Table 2.
A. Characteristics of Patients Who Engrafted with Donor Chimerism
| Patient | Age, Years | Diagnosis | Median Disease Duration, Months | Conditioning Regimen | GVHD Prophylaxis | Donor-Recipient HLA Match | TNC Infused, (107/kg) | Follow-Up, Months | Status | Cause of Death |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 8 | Sickle cell disease | 20 | Cy+Bu+ATG | FK506 | 5/6 matched | — | 23.93 | alive | |
| 2 | 9 | Sickle cell disease | 106 | Cy+Bu+ATG | CsA+Pred | 5/6 matched | 6.9 | 63.50 | alive | |
| 3 | 6 | Sickle cell disease | 76 | Bu+Fluda+Alemtuzumab* | FK506+MMF | 4/6 matched | 3.5 | 37.22 | alive | |
| 4 | 8 | Sickle cell disease | 97 | Fluda+Melph+Alemtuzumab* | CsA+MTX+Pred | 4/6 matched | — | 11.64 | alive | |
| 5 | 5 | Sickle cell disease | Cy+Bu+Fluda | CsA+Pred | 5/6 matched | 11 | 2.41 | dead | GVHD | |
| 6 | 3 | Sickle cell disease | 39 | Cy+Bu+ATG | FK506+Pred | 4/6 matched | 9.3 | 80.50 | alive | |
| 7 | 3 | Sickle cell disease | 37 | Cy+Bu+ATG | CsA+Pred | 6/6 matched | 5.1 | 20.53 | alive | |
| 8 | 12 | Sickle cell disease | 151 | Cy+Bu+ATG | Other | 4/6 matched | 1.7 | 79.44 | alive | |
| 9 | 4 | Sickle cell disease | 54 | Cy+Fluda+Alemtuzumab* | FK506+MMF | 4/6 matched | 9.1 | 29.62 | alive | |
| 10 | 4 | Thalassemia | 26 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 7.1 | 10.74 | dead | Unknown |
| 11 | 1 | Thalassemia | 15 | Cy+Bu+Fluda | CsA+MMF | 5/6 matched | 15.2 | 11.57 | alive | |
| 12 | 0.3 | Thalassemia | 4 | Cy+Fluda* | CsA+Pred | 6/6 matched | 3.9 | 3.01 | dead | Veno-occlusive disease |
| 13 | 15 | Thalassemia | — | Cy+Bu+ATG | CsA | 4/6 matched | 3.3 | 5.88 | dead | MOF |
| 14 | 2 | Thalassemia | 20 | Cy+Bu+ATG | CsA+Pred | 5/6 matched | 4.9 | 44.36 | alive | |
| 15 | 8 | Thalassemia | 101 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 4.3 | 14.21 | dead | Infection |
| 16 | 1 | Thalassemia | — | Fluda+Melph+ATG* | Other | 6/6 matched | 7.6 | 7.40 | alive | |
| 17 | 5 | Thalassemia | 3 | Cy+Bu+ATG | FK506 | 5/6 matched | 8.7 | 4.43 | alive | |
| 18 | 4 | Thalassemia | 64 | Cy+Bu+ATG | Other | 5/6 matched | 7.4 | 5.88 | dead | Hemorrhage |
| 19 | 2 | Thalassemia | 26 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 1.4 | 5.88 | dead | Interstitial pneumonitis |
| 20 | 3 | Thalassemia | 43 | Cy+Bu+ATG | FK506+MMF | 6/6 matched | 11 | 5.72 | alive | |
| 21 | 0.2 | Thalassemia | 7 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 1.9 | 91.07 | alive | |
| 22 | 1 | Thalassemia | — | Cy+Bu+ATG | CsA+MTX | 5/6 matched | 9.6 | 1.49 | alive | |
| 23 | 5 | Thalassemia | 29 | Cy+Bu+ATG | CsA+MMF+ Pred | 5/6 matched | 7.1 | 5.02 | dead | Infection |
| 24 | 2 | Thalassemia | 19 | Cy+Bu+ATG | FK506 | 5/6 matched | 5.1 | 37.26 | alive | |
|
| ||||||||||
| B. Characteristics of Patients with Primary Graft Failure
| ||||||||||
| 1 | 6 | Sickle cell disease | 77 | Cy+Bu+ATG | FK506 | 4/6 matched | 5.6 | 72.36 | alive | |
| 2 | 5 | Sickle cell disease | 59 | Bu+Fluda+TLI+ATG‡ | CsA+MMF | 6/6 matched | 5.8 | 29.52 | alive | |
| 3 | 9 | Sickle cell disease | 107 | Bu+Melph+ATG | CsA | 5/6 matched | 3 | 41.22 | alive | |
| 4 | 1 | Sickle cell disease | 7 | Cy+Fluda‡ | FK506+MMF | 4/6 matched | 4.7 | 22.12 | alive | |
| 5 | 5 | Sickle cell disease | 62 | Cy+Bu+ATG | FK506 | 4/6 matched | 4.2 | 3.57 | alive | |
| 6 | 8 | Sickle cell disease | 100 | Cy+Fluda+TLI+ATG‡ | CsA+Pred | 4/6 matched | 4 | 45.29 | alive | |
| 7 | 17 | Sickle cell disease | 191 | Fluda+Melph+ATG‡ | CsA+Pred | 4/6 matched | 1.4 | 56.23 | alive | |
| 8* | 0/5 | Thalassemia | 6 | Bu+Fluda | FK506+MMF | 5/6 matched | 9.52 | 75.57 | alive | |
| 9 | 15 | Thalassemia | 73 | Cy+Bu | Other | 3/6 matched | 3.7 | 24.33 | alive | |
| 10 | 10 | Thalassemia | 96 | Cy+Bu | CsA+MTX | 5/6 matched | 3.2 | 16.66 | alive | |
| 11 | 6 | Thalassemia | 69 | Cy+Bu+ATG | CsA+MMF+Pred | 5/6 matched | 3.3 | 7.07 | alive | |
| 12 | 2 | Thalassemia | 22 | Cy+Bu+ATG | CsA | 5/6 matched | 15 | 21.36 | alive | |
| 13† | 11 | Thalassemia | 124 | Cy+Bu+ATG | CsA+Pred | 5/6 matched | 2.6 | 45.75 | alive | |
| 14 | 6 | Thalassemia | 57 | Cy+Bu+ATG | Other | 6/6 matched | 5.8 | 17.95 | alive | |
| 15* | 1 | Thalassemia | 9 | Bu+Fluda+TLI+ATG§ | CsA+MMF | 4/6 matched | 5.3 | 43.54 | alive | |
| 16 | 4 | Thalassemia | 46 | Fluda+Melph+ATGh‡ | FK506 | 6/6 matched | 3.7 | 35.37 | alive | |
| 17 | 7 | Thalassemia | 33 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 4.5 | 0.07 | dead | ARDS |
| 18 | 6 | Thalassemia | 28 | Cy+Bu+ATG | CsA+MMF | 5/6 matched | 9.2 | 0.46 | dead | ARDS |
| 19 | 1 | Thalassemia | 5 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 23.3 | 0.5 | dead | ARDS |
| 20 | 2 | Thalassemia | 23 | Cy+Bu+ATG | CsA+MMF | 4/6 matched | 6.5 | 12.1 | alive | |
| 21 | 2 | Thalassemia | 30 | Cy+Bu+ATG | Other | 4/6 matched | 4.9 | 6.41 | alive | |
| 22 | 14 | Thalassemia | 169 | Fludarabine‡ | CsA+MMF | 4/6 matched | 3.7 | 2.51 | dead | Infection |
| 23 | 8 | Thalassemia | 97 | Cy+Bu+ATG | CsA+Pred | 4/6 matched | 4.8 | 27.93 | alive | |
| 24† | 2 | Thalassemia | 25 | Cy+Bu+ATG | CsA+Pred | 5/6 matched | 4.6 | 137.88 | alive | |
| 25 | 9 | Thalassemia | 98 | Cy+Bu+ATG | Other | 4/6 matched | 2.4 | 0.79 | dead | Infection |
| 26 | 6 | Thalassemia | 74 | Cy+Bu+ATG | CsA | 4/6 matched | 3.7 | 25.62 | alive | |
| 27† | 1 | Thalassemia | 4 | Cy+Bu+Thio+ATG | CsA | 4/6 matched | 12 | 13.65 | alive | |
ATG indicates serum antilymphocyt; Bu, busulfan; CsA, cyclosporine A; Cy cyclophosfamide; Fluda, fludarabine; Melph, melphalan; MMF, mycophenolate mofetil; MTX, methotrexate; MOF, multiorgan failure; Pred, prednisone.
Reduced-intensity conditioning.
aGVHD indicates acute graft-versus-host disease; ARDS, acute respiratory distress syndrome; ATG, serum antilymphocyt; Bu, busulfan; CsA, cyclosporine A; Cy, cyclophosfamide; Fluda, fludarabine; Melph, melphalan; MMF, mycophenolate mofetil; MTX, methotrexate; Thio, Thiotepa; Pred, prednisone; TNC, total nuclease cell; TLI, total lymphoid irradiation.
Second hematopoietic stem cell transplantation (HSCT).
Autologous backup.
Reduced-intensity conditioning (RIC).
Acute GVHD and cGVHD
Eleven patients developed grade 2-4 aGVHD (grade 2 in 8; grade 3 in 2; grade 4 in 1). The cumulative incidence of grade 2-4 aGVHD at day 100 was 23% ± 2%. Ten patients developed cGVHD. Chronic GVHD was extensive in 2 patients and limited in the remaining 8 patients. The cumulative incidence of cGVHD at 2 years was 16% ± 4%.
DFS and OS
Only 16 of 51 patients are alive and disease free (SCD, n = 8; thalassemia, n = 8). DFS was lower in patients with thalassemia (21% ± 7% compared to 50% ± 9% for SCD, P = .05) (Figure 1). In multivariate analysis, DFS was higher with CB units containing total nucleated cell dose >5 × 107/kg (HR 0.4, 95% CI, 0.2-0.8, P =.01). The effect of TNC on DFS is independent of disease. The 2-year probability of DFS was 45% ± 9% in recipients of CB units containing a cell dose >5 × 107/kg compared to 13% ± 7% for lower cell dose. Although only 16 patients are alive and cured of their disease, 38 of 51 patients are alive largely because of autologous reconstitution after transplantation. All SCD patients with autologous recovery are alive, whereas there were 5 deaths in the thalassemia group. The probabilities of OS for patients with thalassemia and SCD are 62% ± 9% and 94% ± 6%, respectively (Figure 2).
Figure 1.
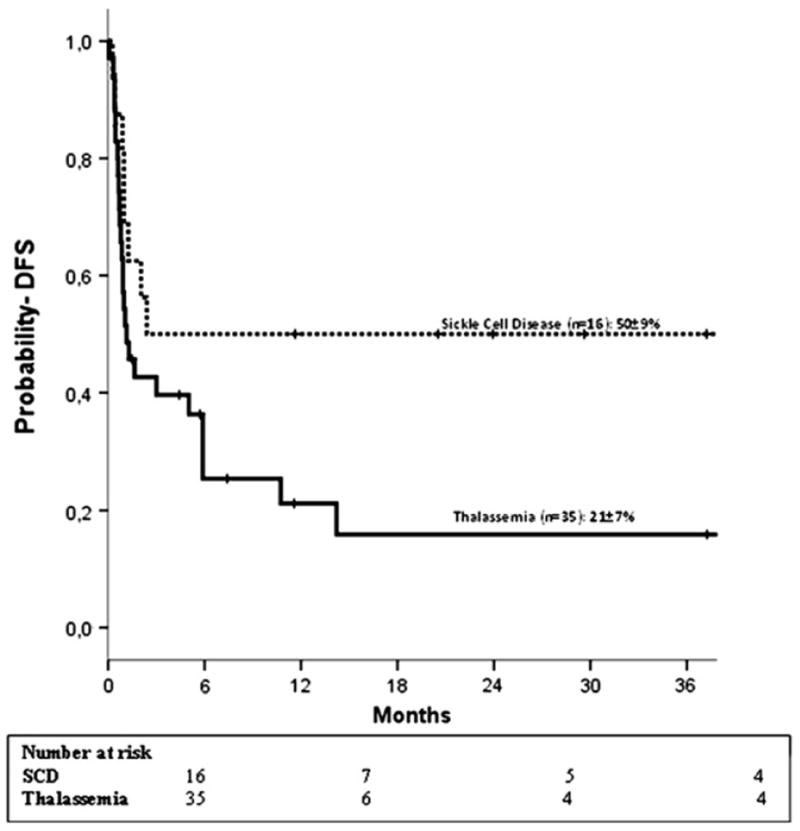
Estimated 2-year DFS according to diagnosis (••• sickle cell disease; — thalassemia).
Figure 2.
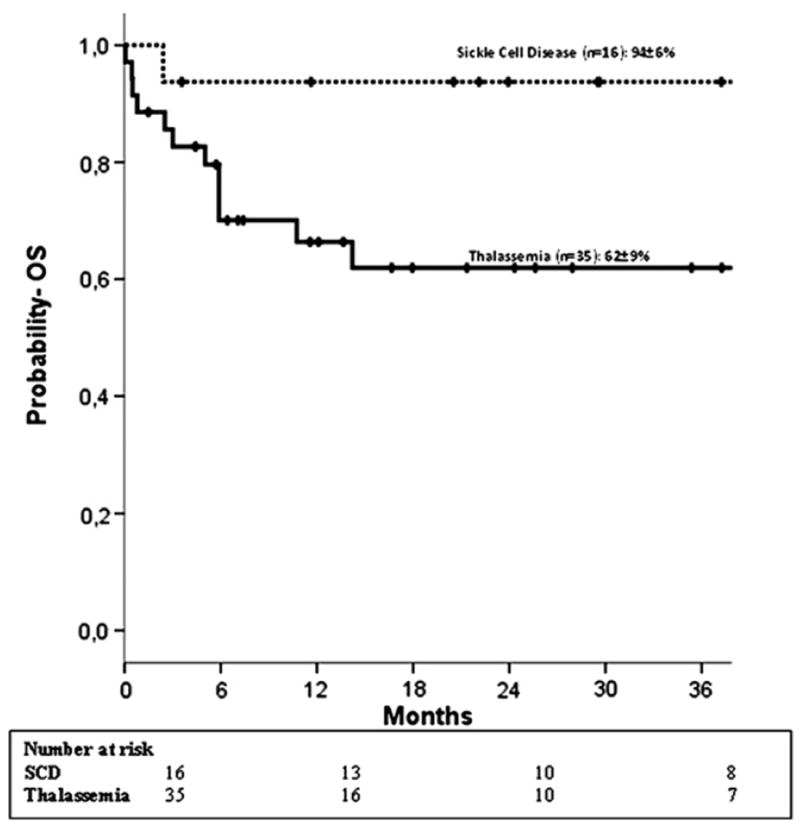
Estimated 2-year OS according to diagnosis (••• sickle cell disease; — thalassemia.
Thirteen patients are dead. Twelve of these patients were transplanted for thalassemia; 7 of these patients had engrafted and died of transplant-related complications. The remaining 5 patients had primary graft failure. Deaths in patients with graft failure may be attributed to patient selection rather than the graft type, a hypothesis that is difficult to test when using data collected by registries. One patient with SCD is dead as a result of severe aGVHD. There were 7 early deaths (≤100 days after transplantation) because of infection (n = 2), adult respiratory distress syndrome (n = 3), veno-occlusive disease (n = 1), and GVHD (n = 1). Causes of death beyond 100 days were because of opportunistic infection (n = 2), hemorrhage (n = 1), interstitial pneumonitis (n = 1), and multiorgan failure (n = 1). Cause of death was not reported for 1 patient.
DISCUSSION
Allogeneic HSC transplantation offers the only chance of cure for patients with thalassemia major and SCD. However, during the last 3 decades, the medical management of thalassemia or SCD, based primarily on hypertransfusion programs and appropriate chelation treatment or on the use of hydroxyurea, has improved survival of those patients [9,22]. Conventional therapy requires strict compliance of the patients, and therefore the real benefit of an allogeneic transplantation should take these developments into consideration, before indication of such a procedure. In fact, transplantation of BM from a HLA matched sibling remains the standard of care when such a donor is available, with cure rates of 85% for both diseases [4,5,23]. OS after related HLA-matched sibling donor CB transplants are similar to OS after BM transplants in children with malignant and nonmalignant diseases [24], and the same results were observed when analyzing only patients with thalassemia and SCD transplanted with CB or BM from HLA identical sibling donors [25]. Yet another recent approach in the setting of HLA matched sibling transplantation, in order to decrease mortality related to the transplantation procedure, is the use of CD34-selected peripheral blood and a RIC [26]. Of 10 patients treated, 9 patients achieved engraftment with mixed chimerism. Longer follow-up is required to determine whether “mixed chimerism” is stable and capable of curing these patients and eliminating the end-organ effects of the underlying disease. Interestingly, mixed chimerism was not associated with an increased risk of graft failure in a series of 27children with beta-thalassemia who received transplants of CB from a related donor [27].
However, as few patients have a HLA matched sibling donor, most rely on alternative donor transplantation for cure. Transplantation of BM from a HLA matched unrelated adult donor coupled with myeloablative transplant conditioning for thalassemia major approach 80% of survival rate [10,28]. In a recent report, using maternal donors, 14 of 22 patients were reported alive and disease-free with a median follow-up of 40 months [11].
Another alternative donor possibility is the use of cord blood cells; however, there are very few reports on the outcomes after this procedure in patients with hemoglobinopathies. To our knowledge, the current analysis is the largest to date on the impact of UCBT in the treatment of thalassemia and SCD. Engraftment, DFS, and OS for thalassemia are inferior to that reported after unrelated adult donor BM transplantation, and primary graft failure was the most common cause of treatment failure. Graft failure accounted for 25% of deaths in patients with thalassemia. Graft failure was also the most common cause of treatment failure for SCD. However, all patients with SCD who experienced graft failure had autologous reconstitution and are alive. Our findings are consistent with that of others [3,29]. Death from treatment-related complications was common in patients with thalassemia; infection and pulmonary complications were the most common causes of treatment-related mortality. The current analysis is limited in that we could not assign the Pesaro risk score for most patients with thalassemia. It is possible that the 7 patients with thalassemia major who engrafted and died of a treatment-related complication were “high risk.” However, the most common cause of treatment failure, although not necessarily of mortality, was graft failure, which cannot be predicted with the Pesaro risk score. Therefore, the lack of assignment of this risk score does not underscore the validity of our observation for patients with thalassemia after CBT.
The observed high rate of graft failure after unrelated CB transplantation is related to the number of nucleated cells infused. As observed for other nonmalignant diseases, the cell dose of the UCB unit was a major factor associated with engraftment and DFS as well as HLA [30,31]. In the current analysis, engraftment and DFS were higher after transplantation of UCB units containing >5 × 107 nucleated cells/kg at the time of infusion, similar to that observed in patients with Fanconi anemia [32].
We know that in the setting of HLA matched sibling and unrelated adult donor BM transplants, increasing pretransplant immune suppression to a myeloablative conditioning regimen is associated with a lower graft failure rate of 5% [4,33]. For UCB, only 1 group has reported 80% DFS at 2 years specifically in recipients of busulfan, cyclophosphamide, and ATG limited to patients with thalassemia [15]. Although most patients in the current analysis received a myeloablative transplant conditioning regimen with ATG or alemtuzumab, DFS was not as good as previously reported by Jaing and colleagues [15]. However, the heterogeneity of transplant conditioning regimens in the current analysis precludes definitive recommendations other than increasing pretransplant immune suppression to facilitate engraftment. New conditioning regimens more likely to effect sustained engraftment are needed for this patient population.
Indeed, graft failure after umbilical CBT is multi-factorial including known factors such as disease, HLA-disparity, cell dose, conditioning regimen, as well as several unknown and unmeasured factors that may include matching at HLA-C locus and presence of HLA antibodies [34]. Recently, the Japanese group [35] reported on the impact of anti-HLA antibodies on the engraftment rate. In this series among 386 UCBT, 89 tested positive for anti-HLA antibodies, and 20 cases had specificity against the cord blood HLA. Patients with antibodies against the cord blood graft had significant higher graft failure rate. We do not have donor-recipient samples, which prevent us from examining for the impact of HLA antibodies on graft failure in our population. Additionally, all patients in the current analysis were transfused prior to transplantation, and we cannot exclude the possibility of alloimmunization and its effect on engraftment. Taken together, the transplantation strategies using unrelated CB as the stem cell source is suboptimal for patients with hemoglobinopathy. Graft failure remains a major limitation to success, and the continuing use of CB for this disease must be discouraged outside of well-designed novel clinical trials. One potential strategy may be the use of 2 CB units in order to achieve the desired cell dose as has been done in patients with malignancy [36]. However, early reports suggest that the double CB transplant approach may be associated with a higher rate of aGVHD, which may add to the burden of morbidity and mortality for such a patient population [37]. Indeed, no data have yet been reported on double CB transplant for hemoglobinopathies. An alternative is HSC transplantation [11]. Results are encouraging, and a larger series is needed to confirm the observed success after this strategy. Other strategies such as preimplantation genetics and embryo selection [38] or gene therapy to correct the beta-globin synthesis [39] for those with access to this technology are currently under investigation and may optimize outcomes.
Acknowledgments
This work was funded in part by the National Cancer Institute, National Heart Lung and Blood Institute and the National Institute of Allergy and Infectious Diseases (U24-CA76518).
Footnotes
Financial disclosure: The authors have no conflict of interest to disclose.
A.R., M.E., E.G., and V.R. designed the study, prepared and analyzed data, and wrote the article. A.S. gathered cases from New York Blood Center and verified data, M.S.C., M.B., J.K., J.W., A.F., M.A., L.L.N., M.W., and J.W. provided cases for the study. D.P., K.B., and W.C. prepared and verified data. All authors edited and approved the manuscript.
References
- 1.Pinto FO, Roberts I. Cord blood stem cell transplantation for haemoglobinopathies. Br J Haematol. 2008;141:309–324. doi: 10.1111/j.1365-2141.2008.07016.x. [DOI] [PubMed] [Google Scholar]
- 2.Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008;22:53–63. doi: 10.1016/j.blre.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Walters MC, Storb R, Patience M, et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood. 2000;95:1918–1924. [PubMed] [Google Scholar]
- 4.Bernaudin F, Socie G, Kuentz M, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007;110:2749–2756. doi: 10.1182/blood-2007-03-079665. [DOI] [PubMed] [Google Scholar]
- 5.Lucarelli G, Clift RA, Galimberti M, et al. Marrow transplantation for patients with thalassemia: results in class 3 patients. Blood. 1996;87:2082–2088. [PubMed] [Google Scholar]
- 6.Lawson SE, Roberts IA, Amrolia P, Dokal I, Szydlo R, Darbyshire PJ. Bone marrow transplantation for beta-thalassaemia major: the UK experience in two paediatric centres. Br J Haematol. 2003;120:289–295. doi: 10.1046/j.1365-2141.2003.04065.x. [DOI] [PubMed] [Google Scholar]
- 7.Eapen M, Rocha V, Sanz G, et al. Effect of graft source on unrelated donor haemopoietic stem-cell transplantation in adults with acute leukaemia: a retrospective analysis. Lancet Oncol. 2010;11:653–660. doi: 10.1016/S1470-2045(10)70127-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Telfer P, Coen P, Chakravorty S, et al. Clinical outcomes in children with sickle cell disease living in England: a neonatal cohort in East London. Haematologica. 2007;92:905–912. doi: 10.3324/haematol.10937. [DOI] [PubMed] [Google Scholar]
- 9.Telfer P, Coen PG, Christou S, et al. Survival of medically treated thalassemia patients in Cyprus. Trends and risk factors over the period 1980-2004. Haematologica. 2006;91:1187–1192. [PubMed] [Google Scholar]
- 10.La Nasa G, Caocci G, Argiolu F, et al. Unrelated donor stem cell transplantation in adult patients with thalassemia. Bone Marrow Transplant. 2005;36:971–975. doi: 10.1038/sj.bmt.1705173. [DOI] [PubMed] [Google Scholar]
- 11.Sodani P, Isgro A, Gaziev J, et al. Purified T-depleted, CD34+ peripheral blood and bone marrow cell transplantation from haploidentical mother to child with thalassemia. Blood. 2010;115:1296–1302. doi: 10.1182/blood-2009-05-218982. [DOI] [PubMed] [Google Scholar]
- 12.Gluckman E, Rocha V, Arcese W, et al. Factors associated with outcomes of unrelated cord blood transplant: guidelines for donor choice. Exp Hematol. 2004;32:397–407. doi: 10.1016/j.exphem.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Rocha V, Locatelli F. Searching for alternative hematopoietic stem cell donors for pediatric patients. Bone Marrow Transplant. 2008;41:207–214. doi: 10.1038/sj.bmt.1705963. [DOI] [PubMed] [Google Scholar]
- 14.Niehues T, Rocha V, Filipovich AH, et al. Factors affecting lymphocyte subset reconstitution after either related or unrelated cord blood transplantation in children—a Eurocord analysis. Br J Haematol. 2001;114:42–48. doi: 10.1046/j.1365-2141.2001.02900.x. [DOI] [PubMed] [Google Scholar]
- 15.Jaing TH, Chen SH, Tsai MH, Yang CP, Hung IJ, Tsay PK. Transplantation of unrelated donor umbilical cord blood for nonmalignant diseases: a single institution’s experience with 45 patients. Biol Blood Marrow Transplant. 2010;16:102–107. doi: 10.1016/j.bbmt.2009.09.009. [DOI] [PubMed] [Google Scholar]
- 16.Adamkiewicz TV, Szabolcs P, Haight A, et al. Unrelated cord blood transplantation in children with sickle cell disease: review of 4-center experience. Pediatr Transplant. 2007;11:641–644. doi: 10.1111/j.1399-3046.2007.00725.x. [DOI] [PubMed] [Google Scholar]
- 17.Martin PJ, Weisdorf D, Przepiorka D, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: VI. Design of Clinical Trials Working Group report. Biol Blood Marrow Transplant. 2006;12:491–505. doi: 10.1016/j.bbmt.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 18.Glucksberg H, Storb R, Fefer A, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation. 1974;18:295–304. doi: 10.1097/00007890-197410000-00001. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 20.Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94 [Google Scholar]
- 21.Cox DR. Regression models and life tables. J R Stat Soc. 1972;34:187. [Google Scholar]
- 22.Voskaridou E, Christoulas D, Bilalis A, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS) Blood. 115:2354–2363. doi: 10.1182/blood-2009-05-221333. [DOI] [PubMed] [Google Scholar]
- 23.Lucarelli G, Galimberti M, Polchi P, et al. Marrow transplantation in patients with thalassemia responsive to iron chelation therapy. N Engl J Med. 1993;329:840–844. doi: 10.1056/NEJM199309163291204. [DOI] [PubMed] [Google Scholar]
- 24.Rocha V, Wagner JE, Jr, Sobocinski KA, et al. Graft-versus-host disease in children who have received a cord-blood or bone marrow transplant from an HLA-identical sibling. Eurocord and International Bone Marrow Transplant Registry Working Committee on Alternative Donor and Stem Cell Sources. N Engl J Med. 2000;342:1846–1854. doi: 10.1056/NEJM200006223422501. [DOI] [PubMed] [Google Scholar]
- 25.Kabbara N. A multicentric comparative analysis of outcomes of HLA identical related cord blood and bone marrow transplantation in patients with beta-thalassemia or sickle cell disease. Bone Marrow Transplant. 2008;41:29–30. Abs184. [Google Scholar]
- 26.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009;361:2309–2317. doi: 10.1056/NEJMoa0904971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lisini D, Zecca M, Giorgiani G, et al. Donor/recipient mixed chimerism does not predict graft failure in children with beta-thalassemia given an allogeneic cord blood transplant from an HLA-identical sibling. Haematologica. 2008;93:1859–1867. doi: 10.3324/haematol.13248. [DOI] [PubMed] [Google Scholar]
- 28.La Nasa G, Giardini C, Argiolu F, et al. Unrelated donor bone marrow transplantation for thalassemia: the effect of extended haplotypes. Blood. 2002;99:4350–4356. doi: 10.1182/blood.v99.12.4350. [DOI] [PubMed] [Google Scholar]
- 29.Iannone R, Casella JF, Fuchs EJ, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant. 2003;9:519–528. doi: 10.1016/s1083-8791(03)00192-7. [DOI] [PubMed] [Google Scholar]
- 30.Rubinstein P, Carrier C, Scaradavou A, et al. Outcomes among 562 recipients of placental-blood transplants from unrelated donors. N Engl J Med. 1998;339:1565–1577. doi: 10.1056/NEJM199811263392201. [DOI] [PubMed] [Google Scholar]
- 31.Gluckman E, Rocha V, Boyer-Chammard A, et al. Outcome of cord-blood transplantation from related and unrelated donors. Eurocord Transplant Group and the European Blood and Marrow Transplantation Group. N Engl J Med. 1997;337:373–381. doi: 10.1056/NEJM199708073370602. [DOI] [PubMed] [Google Scholar]
- 32.Gluckman E, Rocha V, Ionescu I, et al. Results of unrelated cord blood transplant in Fanconi anemia patients: risk factor analysis for engraftment and survival. Biol Blood Marrow Transplant. 2007;13:1073–1082. doi: 10.1016/j.bbmt.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 33.Locatelli F, Rocha V, Reed W, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003;101:2137–2143. doi: 10.1182/blood-2002-07-2090. [DOI] [PubMed] [Google Scholar]
- 34.Spellman S, Bray R, Rosen-Bronson S, et al. The detection of donor-directed, HLA-specific alloantibodies in recipients of unrelated hematopoietic cell transplantation is predictive of graft failure. Blood. 2010;115:2704–2708. doi: 10.1182/blood-2009-09-244525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takanashi M, Atsuta Y, Fujiwara K, et al. The impact of anti-HLA antibodies on unrelated cord blood transplantations. Blood. 2010;116:2839–2846. doi: 10.1182/blood-2009-10-249219. [DOI] [PubMed] [Google Scholar]
- 36.Barker JN, Weisdorf DJ, DeFor TE, et al. Transplantation of 2 partially HLA-matched umbilical cord blood units to enhance engraftment in adults with hematologic malignancy. Blood. 2005;105:1343–1347. doi: 10.1182/blood-2004-07-2717. [DOI] [PubMed] [Google Scholar]
- 37.MacMillan ML, Weisdorf DJ, Brunstein CG, et al. Acute graft-versus-host disease after unrelated donor umbilical cord blood transplantation: analysis of risk factors. Blood. 2009;113:2410–2415. doi: 10.1182/blood-2008-07-163238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grewal SS, Kahn JP, MacMillan ML, Ramsay NK, Wagner JE. Successful hematopoietic stem cell transplantation for Fanconi anemia from an unaffected HLA-genotype-identical sibling selected using preimplantation genetic diagnosis. Blood. 2004;103:1147–1151. doi: 10.1182/blood-2003-02-0587. [DOI] [PubMed] [Google Scholar]
- 39.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature. 467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]