

Abstract
Luminal B breast cancers represent a fraction of oestrogen receptor (ER)-positive tumours associated with poor recurrence-free and disease-specific survival in all adjuvant systemic treatment categories including hormone therapy alone. Identification of specific signalling pathways driving luminal B biology is paramount to improve treatment. We have studied 100 luminal breast tumours by combined analysis of genome copy number aberrations and gene expression. We show that amplification of the ZNF703 gene, located in chromosomal region 8p12, preferentially occurs in luminal B tumours. We explored the functional role of ZNF703 in luminal B tumours by overexpressing ZNF703 in the MCF7 luminal cell line. Using mass spectrometry, we identified ZNF703 as a co-factor of a nuclear complex comprising DCAF7, PHB2 and NCOR2. ZNF703 expression results in the activation of stem cell-related gene expression leading to an increase in cancer stem cells. Moreover, we show that ZNF703 is implicated in the regulation of ER and E2F1 transcription factor. These findings point out the prominent role of ZNF703 in transcription modulation, stem cell regulation and luminal B oncogenesis.
Keywords: 8p11 amplification, breast cancer, cancer stem cell, luminal B, ZNF703
→See accompanying Closeup by Vessela Kristensen DOI 10.1002/emmm201100128
INTRODUCTION
Despite improvements in the treatment of breast cancer, resistance to therapy is a major clinical problem. The molecular taxonomy of breast cancer based on transcriptional profiling has led to a better classification of tumours and pointed out new potential therapeutic targets. Breast cancers are classified into five molecular subtypes: luminal A, luminal B, basal, ERBB2-like and normal-like (Sorlie et al, 2001, 2003). Molecular subtypes are associated with various clinical outcomes, and treatment strategies are tailored to corresponding groups of patients (e.g. anti-ERBB2 therapies for patients with ERBB2-like tumours or hormone therapy for patients with luminal tumours). Although both luminal A and B tumours express hormone receptors (oestrogen and progesterone), the risk of relapse in women treated by hormone therapy is greater in women with a luminal B tumour than in women with a luminal A tumour. Moreover, luminal B tumours are associated with poor disease-specific survival when treated with chemotherapy (Cheang et al, 2009). Therefore, the elucidation of the molecular mechanisms that govern luminal B tumour biology should improve both our understanding of tumourigenesis and treatment of patients.
Little is known about specific signalling pathways deregulated in luminal B tumours. This subtype is characterized by the expression of hormone receptors combined with high proliferative index. The specific gene expression signature associated with luminal B tumours is enriched in genes that drive the proliferation of cancer cells, such as CCNE1, MKI67 or MYBL2 (Sorlie et al, 2001). Moreover, the functional loss of the retinoblastoma tumour suppressor gene (RB1) seems to be associated with the luminal B phenotype (Herschkowitz et al, 2008).
To identify master genes and molecular mechanisms implicated in luminal B biology, we used a strategy based on an integrated profiling study that combined high-resolution analysis of genome copy number aberrations (CNA) by array-comparative genomic hybridization (aCGH) and gene expression in luminal A and luminal B primary tumours. According to the number and features of CNAs, several types of genomic profiles have been identified in breast cancers, including simplex, complex and amplifier (also called firestorm). We and others have found that luminal B tumours are frequently associated with the ‘amplifier’ type, which shows several focal amplifications (Chin et al, 2006; Fridlyand et al, 2006; Geyer et al, 2009; Hicks et al, 2006). We show here that five genomic regions are frequently amplified in luminal B tumours compared to luminal A tumours. The amplification of chromosome region 8p12 is highly specific and among the most frequent alterations of the luminal B subtype. We identified ZNF703 as the most significant candidate oncogene of this amplification. The function of ZNF703 is not known. In this study, we identified and functionally validated ZNF703 protein interactors and gene expression programme activation. Our results suggest that ZNF703 plays a role in the regulation of the luminal B cancer stem cell (CSC) population via transcriptional control of key cellular processes.
RESULTS
8p12 amplification is a recurrent copy number abnormality in luminal B tumours
We had previously studied (Finetti et al, 2008) a series of 266 breast tumours by gene expression analysis and determined their molecular subtypes by SSP analysis (Hu et al, 2006). To identify molecular mechanisms and signalling pathways specifically deregulated in luminal B tumours, we compared the genomic profiles of 41 luminal B and 59 luminal A cases from this panel (Supplementary Table 1) by using high density aCGH. Because luminal B tumours have been associated with the amplifier genomic profile, we focused on recurrent amplifications. As shown in Fig 1A, five genomic regions of recurrent high-level amplification (8p12, 8q22, 11q13, 17q24, 20q13) were associated with luminal B tumours (Fisher's exact test, p < 0.01). The different genomic segments amplified and recurrently associated with luminal B tumours are reported in Supplementary Table 2. In contrast, with the same statistical cut-off we did not identify any genomic region specifically amplified in luminal A tumours; this was not surprising since luminal A tumours have been associated with the simplex phenotype. Several studies have already reported amplification of these genomic regions in luminal tumours (irrespective of A and B) as compared to basal tumours (Adelaide et al, 2007; Letessier et al, 2006).
Figure 1. ZNF703 is a target gene of the 8p12 amplification in luminal B breast tumours.
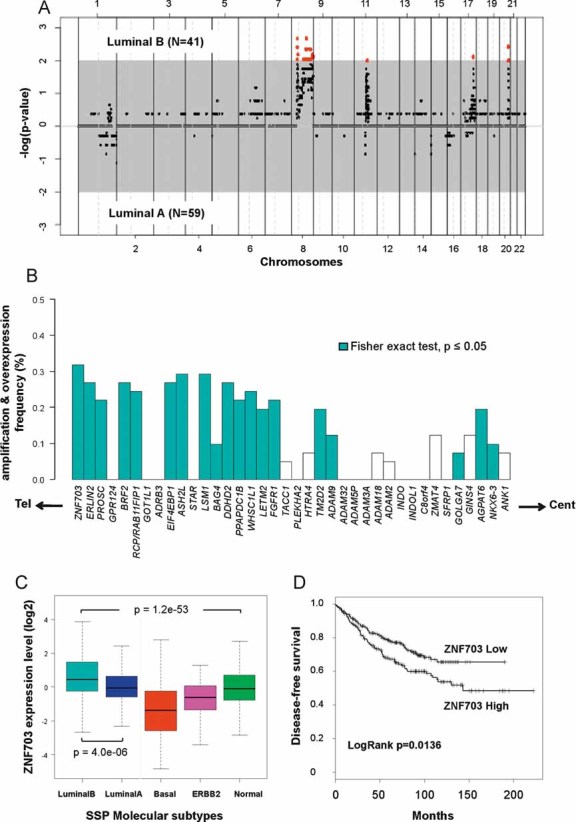
- Supervised analyses of aCGH data comparing amplifications in 41 luminal B tumours and 59 luminal A tumours. Genomic segments are ordered on the X-axis from chromosome 1 to 22 and on the Y-axis according to their association with luminal A or B molecular subtype (−log(p-value)). The grey zone contains genomic segments that are not significantly associated with any of the two molecular subtypes (p > 0.01). Red dots represent genomic segments significantly amplified in luminal B breast tumours (p < 0.01). These segments are contained in 8p12, 8q22, 11q13, 17q24, and 20q13 chromosomal regions. We did not identify genomic segments significantly amplified in luminal A tumours.
- Combined genomic and transcriptional analysis for each gene from the 8p12 amplicon identified in luminal B breast tumours. Genes are ordered from telomere (left) to centromere (right) and the frequency of luminal B tumours presenting an increase in gene copy number and mRNA level is represented. Blue bars label genes that present a statistically significant association between gene amplification and overexpression (Fisher's exact test, p-value ≤ 0.05).
- Box-plot representing ZNF703 expression distribution according to molecular subtypes of 1172 breast tumours. ZNF703 expression varies across the five molecular subtypes (ANOVA, p = 1.2 × 10−53) and is higher in luminal B tumours compared to luminal A tumours (t-test, p = 4 × 10−6).
- Kaplan–Meier survival curves according to ZNF703 gene expression status. ZNF703 gene expression is associated with DFS (p = 0.0136).
Luminal B tumours exhibit ZNF703 gene amplification and overexpression
We had previously characterized the amplified 8p12 region. To identify potential targeted genes that may specify luminal B biology, we focused on this 8p12 amplification. In the past, several genes, including RCP/RAB11FIP1, PPAPDC1B or FGFR1, have been proposed as candidate oncogenes targeted by the 8p12 amplification (Bernard-Pierrot et al, 2008; Turner et al, 2010; Zhang et al, 2009).
We found a strong association between 8p12 amplification and luminal B tumour subtype, with 32% of luminal B tumours displaying amplification (Fig 1A, Supplementary Table 3). We searched to identify the 8p11-12 genes targeted by amplification by comparing for each gene the association between copy number and messenger RNA (mRNA) expression level for the 38 genes comprised in the amplicon defined by the aCGH analysis. These genes are localized in a 4.6 Mb region delimited by ZNF703 towards the telomere and ANK1 towards the centromere. We found a hot spot of amplification frequency for the genomic segment that contains only the ZNF703 gene (Fisher's exact test, p < 0.01) (Supplementary Table 2). Nineteen genes showed amplification correlated with upregulation (Fisher's exact test, p < 0.01; Fig 1B). Among them, ZNF703 was the most frequently amplified and overexpressed gene. In 11 breast cancer cell lines, we did a similar integrated analysis of genomic amplification (using this time quantitative PCR) and gene overexpression by both DNA microarrays and reverse-transcribed (RT)-PCR for each of the 38 genes. ZNF703 expression levels measured by DNA microarrays and by quantitative RT-PCR were correlated (Pearson test, r = 0.96), and correlated with an increase in protein expression level (Supplementary Fig 1). The integrated analysis on cell lines confirmed the results obtained on luminal B primary tumours, with ZNF703 gene presenting the most significant correlation between DNA copy number and mRNA expression (Pearson test, r = 0.9) (Supplementary Fig 2). Amplification and overexpression of ZNF703 were especially high in MDA-MB-134 and HCC1500 cell lines, ZNF703 was overexpressed up to 15-fold in these cell lines as compared to cell lines derived from normal breast. Probably because of a constitutively high level of proliferation due to cell culture conditions, the distinction between luminal A and B is not possible in cell lines (Charafe-Jauffret et al, 2006; Neve et al, 2006). It was therefore not possible to show whether these two cell lines may have originated from a luminal B tumour.
We next examined ZNF703 expression measured by DNA microarrays according to the distribution of molecular subtypes in 1172 breast cancers from 11 different published datasets (Supplementary Tables 4 and 5). ZNF703 overexpression was associated with luminal B tumours when compared with luminal A tumours (t-test, p = 4 × 10−6) or with other subtypes (ANOVA, p = 1.2 × 10−53) (Fig 1C).
To determine whether ZNF703 overexpression had clinical impact, we investigated the association with clinical and pathologic features in 561 luminal tumours (A and B). ZNF703 overexpression was not correlated with any histoclinical factor (Supplementary Table 6). However, a high level of ZNF703 mRNA was associated with a decrease in disease-free survival (DFS) with a 5-years DFS of 68% versus 78% for a low level of ZNF703 (p = 0.0136) (Fig 1D).
Taken together, our results identified ZNF703 as a plausible oncogene candidate, prompting us to study its functional contribution to luminal B oncogenesis.
Oestrogen receptor regulates ZNF703 transcription
The oestrogen receptor (ER) is the master transcriptional regulator of luminal tumour phenotype (Hurtado et al, 2008). Interestingly, several studies have described ZNF703 as a target gene of ER transcriptional activity and have identified oestrogen response elements (ERE) in its promoter region (Lin et al, 2004, 2007). To firmly establish that ZNF703 expression is under the control of ER transcriptional activity, we cultured MDA-MB-134 and HCC1500 breast cancer cell lines in medium depleted or supplemented in 17β-estradiol (E2). In these cells, ZNF703 transcription and protein production were induced by the addition of E2 in a time-dependent manner (Supplementary Fig 3). Thus, ZNF703 is a potential oncogene under the control of ER activity in luminal B tumours.
Identification and characterization of ZNF703 in the nucleus
To identify signalling pathways regulated by ZNF703, we overexpressed ZNF703 in MCF7, a luminal breast cancer cell line that does not present 8p12 amplification, utilizing a GFP-ZNF703 fusion protein expression vector. As a control, we utilized a pEGFP-C1 vector that produces only a GFP protein (Fig 2A). Cellular fractionation showed that GFP-ZNF703 localized both to the nucleus and the cytoplasm (Supplementary Fig 4). Utilizing GFP expression, we monitored ZNF703 localization in the MCF7 GFP-ZNF703 cell line. GFP-ZNF703 was mainly localized in the nuclear matrix in subcellular dot-like structures (Fig 2A). To avoid potential artefactual subcellular location of ZNF703 due to the fusion protein product with GFP, we confirmed that endogenous ZNF703 is present in the same structures (Supplementary Fig 5A). The size and the morphology of these nuclear structures are reminiscent of promyelocytic leukaemia (PML), oncogenic domains (PODs) and RNA splicing bodies (SBs), which contain different sets of proteins (Dyck et al, 1994; Spector et al, 1991). To examine the potential overlap of ZNF703 localization with these nuclear structures, MCF7 GFP-ZNF703 cells were subjected to immunofluorescence staining with an antibody against PML and to DNase 1 or RNase A treatment. The staining pattern of PML did not overlap with that of ZNF703 (Supplementary Fig 5B). RNase A treatment did not affect ZNF703 nuclear structures (Fig 2B) suggesting that ZNF703 bodies are distinct from both PODs and SBs. In contrast, DNase 1 treatment completely disorganized ZNF703 nuclear localization (Fig 2B). Although ZNF703 protein has only one zinc finger motif (Nakamura et al, 2004), proscribing a direct DNA-binding, our results suggest that ZNF703 is part of nuclear structures that directly interact with DNA.
Figure 2. Identification and characterization of ZNF703 nuclear domain.
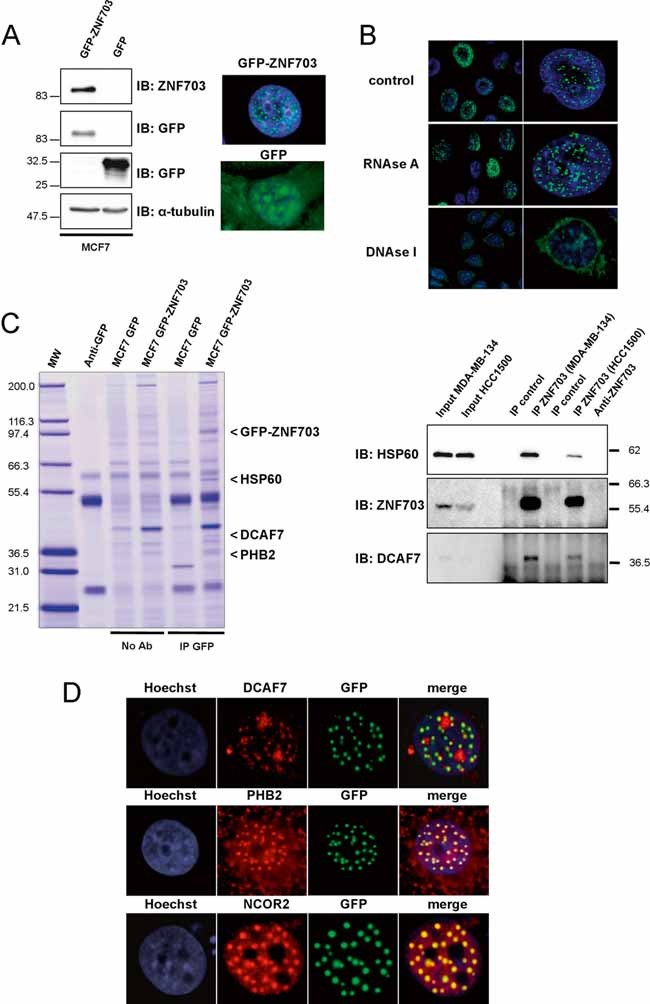
- Immunoblotting and immunofluorescence analyses of MCF7 GFP-ZNF703 and MCF7 GFP breast cancer cell lines. Immunoblotting with anti-ZNF703 and anti-GFP antibodies identifies GFP-ZNF703 fusion protein with a molecular weight of 83 kDa. α-tubulin protein expression is shown as a control. Immunofluorescence reveals nuclear dot-like structures in MCF7 GFP-ZNF703 cells, whereas GFP is detected in all subcellular structures in MCF7 GFP cells.
- GFP-ZNF703 interacts with DNA in the nucleus. MCF7 GFP-ZNF703 cells non-treated (control) or treated with RNAse A or DNAse I were processed for immunofluorescence with anti-GFP antibody. The GFP-ZNF703 signal in the nucleus is perturbed only when cells are treated with DNAse.
- MCF7 cells expressing GFP and GFP-ZNF703 proteins were subjected to immunoprecipitation with anti-GFP antibody (IP GFP) or beads only (No Ab), separated by SDS-PAGE, and stained with colloidal Coomassie blue. Three co-precipitated proteins were identified by mass spectrometry: HSP60 (accession number, NP 955472); DCAF7 (NP 005819) and PHB2 (NP 001138303), respectively. The lane on the left contains molecular weight standards (MW). The interaction between ZNF703 and HSP60 or DCAF7 was confirmed by ZNF703-immunoprecipitation, followed by Western blot analysis using anti-HSP60 or anti-DCAF7 antibody. Input represents whole-cell extract from HCC1500 and MDA-MB-134.
- MCF7 cells overexpressing GFP-ZNF703 were fixed and immunostained with anti-DCAF7, anti-PHB2 or anti-SMRT/NCOR2 antibody. GFP-ZNF703 (green) co-localizes in the nucleus with DCAF7, PHB2 and SMRT/NCOR2 (red). Nuclei are counterstained with Hoechst (blue).
ZNF703 is part of a nuclear repressor complex
We next sought to identify proteins that associate with ZNF703 in the nucleus. SDS-polyacrylamide gel electrophoresis (SDS-PAGE) analysis revealed three potential ZNF703-interacting proteins (Fig 2C). Their identity was determined by mass spectrometry (MS). All peptides identified by MS analysis corresponded to three different proteins: DCAF7, HSP60 and PHB2 (Prohibitin 2) (Supplementary Table 7). Co-immunoprecipitation experiments confirmed the interaction of ZNF703 with HSP60 and DCAF7 in HCC1500 and MDA-MB-134 (endogenous conditions) and in MCF7 GFP-ZNF703 (Fig 2C and Supplementary Fig 6). Moreover, DCAF7 and PHB2 co-localized with ZNF703 in the dot-like nuclear structures (Fig 2D). Despite the interaction between ZNF703 and HSP60, these two proteins did not co-localize in the nuclear matrix. HSP60 was only present in the cytoplasm and may interact with the cytoplasmic form of ZNF703 (Supplementary Fig 5C) and the ZNF703-HSP60 protein complex may have a distinct function from that of the ZNF703-DCAF7-PHB2 complex. PHB2 (also called REA for repressor of ER activity) is a nuclear co-repressor of ER activity in mammary gland development (Mussi et al, 2006; Umanskaya et al, 2007). Prohibitin mediates transcriptional repression through recruitment of additional nuclear co-repressors (NCOR1/NCOR2) and HDACs (Kurtev et al, 2004; Wang et al, 2002). We stained MCF7 GFP-ZNF703 cells with anti-NCOR2 antibody. The staining pattern of NCOR2 overlapped completely with the ZNF703 nuclear structures detected by GFP fluorescence (Fig 2D). Interestingly, when we questioned the multi-experiment matrix (MEM) web interface (http://biit.cs.ut.ee/mem/) for ZNF703 gene expression similarities across 432 published gene expression datasets analysed with Affymetrix GeneChip® from cell lines and primary tumours studies, we found that NCOR2 was the most co-expressed gene with ZNF703 (Supplementary Table 8).
Altogether, our results suggest that nuclear ZNF703 is a co-factor of a nuclear co-repressor complex and may contribute to transcriptional regulation.
ZNF703 expression regulates cell proliferation
To evaluate the role of ZNF703 overexpression as a potential oncogene, we measure the kinetic of cell proliferation in MCF7 GFP compared to MCF7 GFP-ZNF703 as assessed by an MTT assay. This test was done in the presence or absence of E2. As expected, MCF7 GFP cell proliferation was highly stimulated by the presence of E2. ZNF703 overexpression induced a significant cell proliferation independently of E2 stimulation (Fig 3A). Using a ZNF703-siRNA, we inhibited ZNF703 expression in the MDA-MB-134 cell line, which presents an amplification and overexpression of ZNF703 (Supplementary Fig 7) and measured the kinetic of cell proliferation. Depletion of ZNF703 expression in MDA-MB-134 cells reduced cell proliferation (Fig 3B). These observations indicate that ZNF703 is implicated in the regulation of cell proliferation.
Figure 3. ZNF703 modulation regulates cancer cell proliferation.
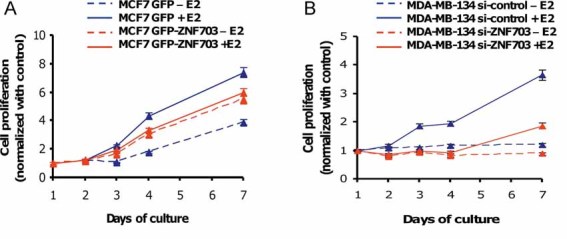
- MCF7 GFP and MCF7 GFP-ZNF703 cells were cultured in the presence or absence of 17-β estradiol (E2) and cell number was measured during 7 days. GFP-ZNF703 overexpression increases cell proliferation independently of E2 stimulation compared to MCF7 GFP cells cultured in the absence of E2, whereas E2 treatment boosts cell proliferation in MCF7 GFP only.
- To confirm the role of ZNF703 in the proliferation process, we inhibited ZNF703 expression via a ZNF703-siRNA in the MDA-MB-134 cell line, which presents amplification and overexpression of the ZNF703 gene. An empty siRNA was used as control (si-control). We started to measure cell number 30 h after siRNA lipofection and over 7 days of culture. MDA-MB-134 cells proliferate only in the presence of E2 and ZNF703 knock-down reduces MDA-MB-134 proliferation.
ZNF703 overexpression increases cancer stem cell population
To further characterize the effect of ZNF703 overexpression, we analysed the modification of gene expression programmes in MCF7 GFP-ZNF703 cells compared to MCF7 GFP cells using DNA microarrays. A total of 189 genes were differentially expressed between the two types of cells, with 160 genes induced and 29 genes downregulated by ZNF703 overexpression (Fig 4A and Supplementary Table 9). Among the 160 genes induced by ZNF703, several genes were related to WNT (such as LEF1, TCF12, WNT4) or NOTCH (such as ASCL1, HEY2, TLE4) signalling pathways. Activation of these pathways is known to regulate the self-renewal programme in breast CSC (Harrison et al, 2010; Korkaya and Wicha, 2009; Ponti et al, 2005).
Figure 4. ZNF703 overexpression regulates CSC biology in MCF7 cell line.
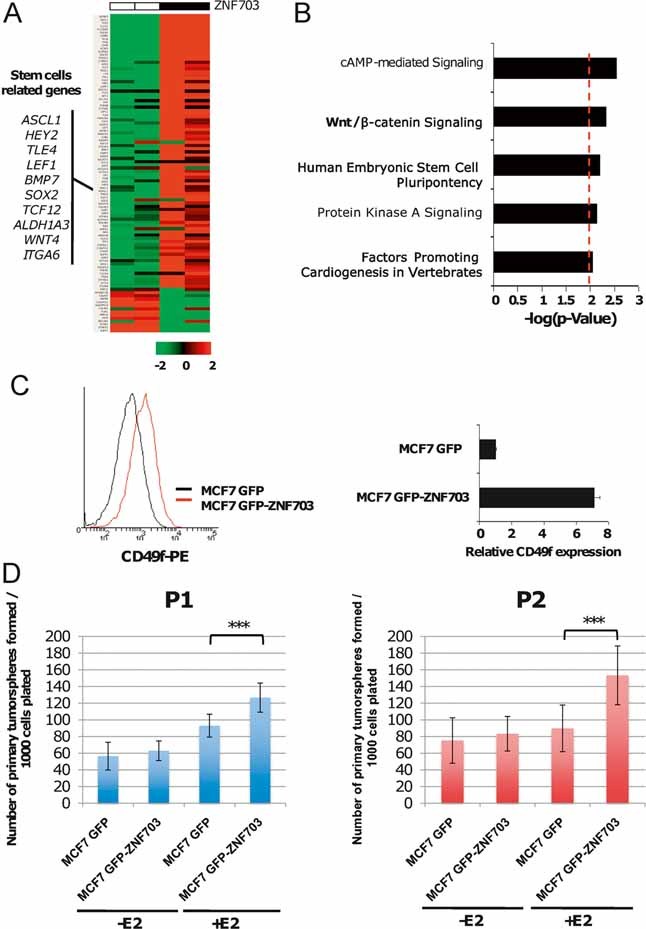
- Gene expression supervised analysis identified ZNF703 co-regulated genes. Gene expression profiles of two independent MCF7 GFP and two MCF7 GFP-ZNF703 stably expressing clones were compared. The 100 top genes regulated by ZNF703 overexpression are represented on hierarchical clustering. This gene list is highly enriched in stem cell-related genes.
- To determine whether genes induced by ZNF703 overexpression were related to stem cell biology, we utilized IPA using Ingenuity® software. Bar plot represents enrichment for each of the network components identified, where the strength of the association is represented by the −log10 (p-value). IPA identified five significant networks associated with ZNF703 overexpression, including three directly related to stem cell biology and development process (in bold).
- Using FACS analysis, we measured CD49f expression in MCF7 GFP compared to MCF7 GFP-ZNF703 cells. We found seven times more CD49f-positive cells in MCF7 GFP-ZNF703 cells compared to MCF7 GFP cells.
- MCF7 GFP-ZNF703 and GFP cells were cultivated in suspension and tumoursphere formation was evaluated in the presence or absence of 17-β estradiol (E2). ZNF703 overexpression increases primary (t-test, p = 2.8 × 10−6) [left part] and secondary tumourosphere (t-test, p = 1.4 × 10−6) [right part] formation under E2 treatment.
To confirm that genes induced by ZNF703 overexpression were related to stem cell biology, we utilized an Ingenuity pathway analysis (IPA) using Ingenuity® software (Fig 4B). IPA identified five significant networks associated with ZNF703 overexpression, including three directly related to stem cell biology and development process. Moreover, to better characterize the association between genes induced by ZNF703 and stem cell-related genes, we measured the enrichment of 26 published stem cell-related gene sets in genes differentially expressed in MCF7 GFP-ZNF703. Ten out of twenty-six gene sets were enriched in the ZNF703-related genes (Supplementary Table 10).
These results suggest that ZNF703 overexpression may be implicated in the regulation of breast CSCs. To test this hypothesis, we first measured the proportion of CD49f-positive cells in MCF7 GFP and MCF7 GFP-ZNF703 cell lines. The CD49f-positive population has been described to contain the CSC population in the MCF7 cell line (Cariati et al, 2008). Overexpression of ZNF703 induces an expansion of the CD49f-positive population with seven times more CD49f-positive cells in the MCF7 GFP-ZNF703 cell line than in the MCF7 GFP cell line (Fig 4C). Second, we used an in vitro tumoursphere assay, a powerful surrogate method to evaluate cancer stem/progenitor cells (Ponti et al, 2005). We cultivated MCF7 GFP and MCF7 GFP-ZNF703 cells in adherent conditions in the presence or absence of E2, then in suspension culture at clonogenic density. When stimulated by E2, MCF7 GFP-ZNF703 cells showed an increase in primary tumoursphere formation. Similar results were observed for secondary tumoursphere formation (Fig 4D). These results suggest that ZNF703 overexpression increases the CSC population and may act as a regulator of the self-renewal programme in ER-positive cells.
ZNF703 overexpression regulates ER and E2F1 transcriptional activity
The above observations suggested that ZNF703 could regulate breast CSC self-renewal activity by interacting with the PHB2/NCOR2 nuclear co-repressor complex. Nuclear co-repressors modulate the activity of master transcriptional regulators, such as ER or E2F1, and thus regulate key cell processes (Battaglia et al, 2010; Perissi et al, 2010). Both ER and E2F1 have been implicated in the control of differentiation and self-renewal programmes in stem cells (Dontu et al, 2004; Galderisi et al, 2006; Orford and Scadden, 2008). Therefore, we speculated that ZNF703 overexpression may regulate breast CSC biology through the regulation of ER and E2F1 transcriptional activities. To test this hypothesis, we first studied ER protein expression by immunostaining in MCF7 GFP-ZNF703 cells. We observed a decrease of ER expression in cells with ZNF703 dot-like structures: 80% of cells with ZNF703 dot-like structures displayed low ER expression, whereas 60% of cells with diffuse cytoplasmic ZNF703 expression displayed high ER expression (Fig 5A). Second, we measured ER transcriptional activity using an ER reporter system that consisted in a vector (p17M-ERE-Luc) coding for the luciferase protein under the control of 17 ERE. Once again, we observed a decrease of ER transcriptional activity in MCF7 GFP-ZNF703 compared to MCF7 GFP cells, independently of E2 induction (Fig 5B). Third, we found that MCF7 GFP-ZNF703 cells presented a decrease in expression for the two main ER-associated proteins, FOXA1 and GATA3 (Fig 5C), and we confirmed a lower ER expression comparatively to MCF7 GFP cells. These results indicate that ZNF703 modulates ER transcriptional activity and may control breast CSC differentiation process through this regulatory mechanism.
Figure 5. ZNF703 overexpression regulates ER and E2F1 transcriptional activity.
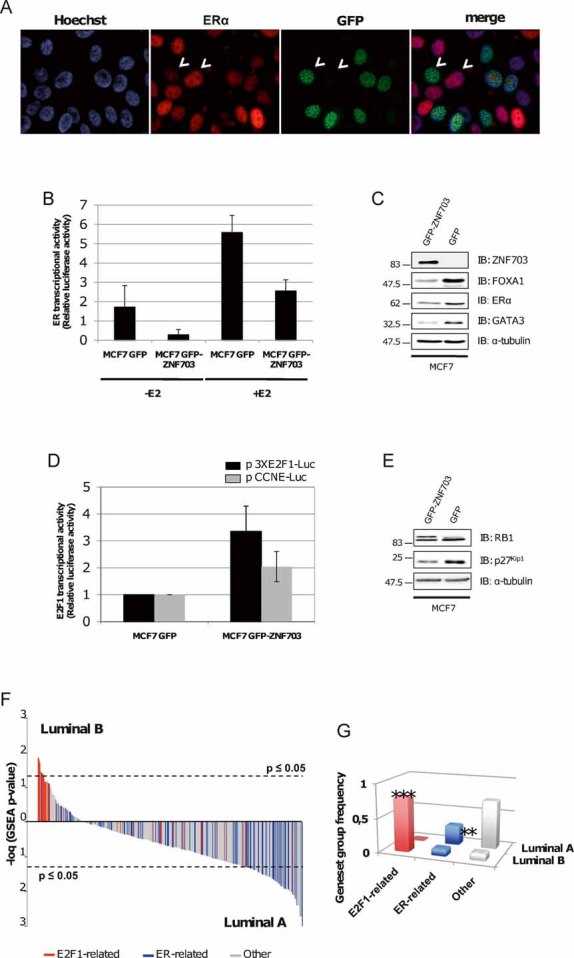
- MCF7 GFP-ZNF703 cells were fixed and immunostained with anti-ER. ZNF703 (green) and ER (red) do not co-localize and are expressed in different cells (white arrowheads). Nuclei were counterstained with Hoechst (blue).
- Oestrogen receptor-dependent transcriptional activity was evaluated using a luciferase reporter system (p17M-ERE-Luc) in the presence and absence of 17-β estradiol (E2) in MCF7 GFP and GFP-ZNF703 breast cancer cell lines. ER-dependent transcriptional activity decreases in MCF7 GFP-ZNF703 cells as compared to GFP expressing cells, independently of E2 induction.
- Analysis of ER, FOXA1, and GATA3 protein expression by immunoblotting in MCF7 GFP-ZNF703 and MCF7 GFP breast cancer cell lines. MCF7 GFP-ZNF703 cells show a decrease in ER-associated proteins.
- E2F1-dependent transcriptional activity was evaluated using two different luciferase reporter systems. Both vectors code for the luciferase protein under the control of either three E2F1 responsive elements (pGL2-3XE2F1-Luc) or CCNE1 promoter (pGL3-CCNE1-Luc), which is a direct E2F1 target gene. E2F1-dependent transcriptional activity decreases with ZNF703 overexpression.
- Analysis of RB1 and P27Kip1 protein expression by immunoblotting in MCF7 GFP-ZNF703 and MCF7 GFP cells. ZNF703 overexpression induces a hyperphosphorylation of RB1 protein and a decrease in P27Kip1 protein expression. α-Tubulin protein expression was used as a control.
- To test E2F1 or ER transcriptional activity in luminal B primary tumours compared to luminal A, we performed a GSEA to screen the activated transcription factors from the Broad Institute (MSigDB C3: motif gene sets TFT). We represented the different gene sets that share a transcription factor binding site defined in the TRANSFAC database enriched in luminal B tumours, compared to luminal A tumours. Each gene sets is plotted according to its association to either luminal A or B tumours (−log(diff GSEA p-value)); E2F1-related gene sets are in red, ER-related gene sets are in blue and other gene sets are in grey (F). A total of 117 gene sets were differentially regulated between luminal B and luminal A primary tumours (FDR q-value < 0.25; Nominal p-value < 0.05). E2F1-related gene sets are significantly enriched in luminal B tumours, whereas luminal A tumours, gene expression programme is enriched in ER-related gene sets (G).
We next examined the effect of ZNF703 overexpression on the other classical nuclear target of co-repressors, E2F1. We used two different E2F1 reporter systems (p3XE2F1-Luc and pCCNE-Luc). The vectors code, respectively, for the luciferase protein under the control of three E2F1 responsive elements or under the control of Cyclin E (CCNE1) promoter, a direct E2F1 target. We found similar results for both reporter assays with an increase of E2F1 transcriptional activity induced by ZNF703 overexpression (Fig 5D). We further tested the functional activation of E2F1 by western blot analysis of RB1 and P27Kip1 protein status. Both proteins are implicated in E2F1 regulation. RB1 binds E2F1 and represses cell cycle progression; phosphorylations inactivate RB1 that releases E2F1 leading to proteolysis of P27Kip1 and to transcription of several target genes implicated in cell cycle progression from G1 to S (Orford and Scadden, 2008). We found an increase of RB1 phosphorylation and a decrease of P27Kip1 protein expression in MCF7 GFP-ZNF703 cells, attesting of E2F1 activation induced by ZNF703 overexpression (Fig 5E). It is interesting to note that the PHB1/NCOR1 complex is known to repress E2F1 transcriptional activity (Wang et al, 2002), whereas the ZNF703/PHB2/NCOR2 complex seems to positively regulate E2F1 activity.
Altogether, these results suggest that ZNF703 may be implicated in the regulation of ER and E2F1 transcriptional regulators and thus may modulate gene expression programmes implicated in breast CSC biology through these two major pathways.
Luminal B tumours present an activation of E2F1 transcriptional programme and a decrease in the activity of ER-related transcription factors
We sought to extend these results to clinical samples of luminal B primary tumours. We used the GSEA algorithm to screen the activated transcription factors from the Broad Institute (MSigDB C3: motif gene sets TFT). We evaluated the different gene sets that share a transcription factor binding site defined in the TRANSFAC database enriched in luminal B tumours compared to luminal A tumours. A total of 117 gene sets were differentially regulated between luminal B and luminal A tumours (FDR q-value < 0.25; Nominal p-value < 0.05) (Fig 5F, Supplementary Table 11). Among these gene sets, 10 were associated with luminal B tumours and 107 with luminal A tumours. Eight out of the ten gene sets (80%) associated with luminal B tumours were related to genes containing E2F1 cis-regulatory motifs. Among the 107 gene sets associated with luminal A tumours, 38 (35.5%) were related to genes containing EREs or to ER-associated transcription factors whereas none of these gene sets was related to E2F1 transcription factor (Fig 5G). These results are consistent with our results suggesting that ZNF703 is a specific luminal B oncogene regulating E2F1 and ER transcriptional activity. It seems that in luminal B cancer cells the balance between E2F1 and ER activation is tilted towards an E2F1 transcriptional programme, in which ZNF703 may act as a key modulator.
ZNF703 expression regulates cell cycle progression
Taken together, our observations suggest a strong implication of ZNF703 in the transcriptional control of cell cycle progression. The E2F1/RB1/CCNE1 protein network controls cell cycle entry and cell cycle progression from G1 to S. This cell cycle stage has been proposed to control stem cell fate by allowing an equilibrium between self-renewal and committed cell fate decision (Orford and Scadden, 2008). In self-renewing cells, CCNE is constitutively active throughout the cell cycle, which allows the transition of stem cells from M phase directly to late G1. The resulting absence of the Cyclin D-dependent early G1 phase shortens the G1 phase and the entire cell cycle. Thus, ZNF703 overexpression may modify the balance between self-renewal and differentiation by shortening the G1 phase.
To evaluate the role of ZNF703 overexpression in the control of cell cycle progression, we analysed the cell cycle status of synchronized MCF7 GFP and MCF7 GFP-ZNF703 cells in the presence or absence of E2. This analysis was done every 2 h over 24 h. Results are summarized in Supplementary Fig 8. MCF7 GFP cells needed E2 stimulation to enter the cell cycle, whereas ZNF703 overexpression was sufficient to induce cell cycle entry independently of E2 (Fig 6A). Interestingly, in the presence of E2, the G0/G1 phase was shorter (4 h) in MC7 GFP-ZNF703 cells compared to cells cultured in the absence of E2 (6 h). These results suggest that ZNF703 may regulate CSC biology by activating the E2F1/RB/CCNE1 network and inducing a short G1 phase that may favour self-renewal over differentiation. Consistent with our results of tumoursphere assay, combined E2 stimulation and ZNF703 overexpression seem to increase both self-renewal and proliferation in MCF7 cells, whereas cell proliferation seems only to need ZNF703 overexpression.
Figure 6. Role of ZNF703 in the regulation of cell cycle progress.
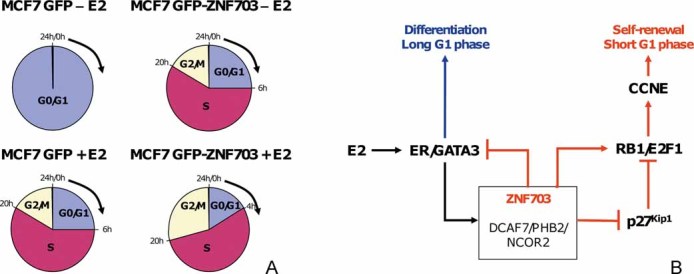
- To determine the role of ZNF703 in cell cycle regulation, we analysed the cell cycle status of synchronized cells from MCF7 GFP and MCF7 GFP-ZNF703 cell lines in the presence or absence of E2. This analysis was done every 2 h over 24 h after FBS stimulation. Results are summarized in Supplementary Fig 8. Pie charts represent cell cycle progress of synchronized cells during 24 h after FBS stimulation. MCF7 GFP cells need E2 stimulation to enter the cell cycle, whereas ZNF703 overexpression is sufficient to induce cell cycle entry independently of E2 stimulation. Interestingly, in the presence of E2, G0/G1 phase is significantly shorter (4 h) in MC7 GFP-ZNF703 cells compared to cells cultured in the absence of E2 (6 h).
- Hypothetical representation of the putative ZNF703-associated network in luminal B tumours presenting an 8p12 amplification. ZNF703 is part of a nuclear complex comprising DCAF7, PHB2 and NCOR2. This complex may act as a nuclear co-repressor modulating CSC self-renewal and differentiation. ZNF703 expression is under the control of ER/GATA3, and potentially CCND1, and activates E2F1 transcriptional activity through RB1 and P27Kip1 inactivation. Interaction with DCAF7 should induce P27Kip1 degradation. E2F1 activation induces CCNE1 transcription that controls cell cycle entry in late G1 phase. When ZNF703 gene is amplified and overexpressed, it induces a shortened G1 phase supporting CSC self-renewal; CCNE1 is constitutively active and ER-dependent differentiation and proliferation is repressed (red lines).
DISCUSSION
We describe here the identification of a potential breast cancer oncogene, ZNF703, implicated in the definition of the luminal B subtype. Recent molecular portraits of breast cancer have consistently revealed biologically distinct subtypes associated with different prognosis (Sorlie et al, 2001). Luminal B tumours are associated with poor breast cancer recurrence-free and disease-specific survival in all adjuvant systemic treatment categories including hormone therapy alone (Cheang et al, 2009). The identification of key mechanisms that regulate luminal B biology will thus lead to a better diagnostic and treatment of luminal tumours.
We have shown that luminal B tumours display specific amplifications, including amplification of the 8p12 region. Interestingly, breast cancer cell lines with 8p12 amplification show resistance to 4-hydroxytamoxifen (Turner et al, 2010). We (Gelsi-Boyer et al, 2005) and others (Kwek et al, 2009; Ray et al, 2004) have previously dissected the 8p12 amplification and identified up to four subamplicons, A1–A4. We have shown here that ZNF703, which is included in A1, is significantly amplified in luminal B tumours. Moreover, ZNF703 mRNA expression is associated with luminal B tumours and defines patients with poor clinical outcome. A different study has been performed on a large series of breast tumours and has similarly identified ZNF703 as the most prominent gene in the 8p12 amplication occurring frequently in luminal B samples (Holland et al, 2011).
ZNF703 satisfies the main criterion for being a driver of the A1 amplicon, i.e. it is overexpressed when amplified. We found it was the most regularly amplified and overexpressed 8p11-12 gene in both our cell lines and tumour panels. This was also observed by previous studies (Letessier et al, 2006; Ray et al, 2004). Other potential oncogenes of 8p11-12 have been identified, including FGFR1 (Turner et al, 2010), TC-1/C8orf4 (Yang et al, 2007; Zhang et al, 2009), PPAPDC1P (Bernard-Pierrot et al, 2008), RCP/RAB11FIP1 (Zhang et al, 2009) and WHS1L1 (Yang et al, 2010). Various assays, mostly proliferation assays, have shown that these genes may function as oncogenes. They could act in collaboration with ZNF703.
ZNF703 has four important features that may explain its involvement in mammary oncogenesis. First, we observed an induction of proliferation when we overexpressed ZNF703 gene in MCF7 cells independently of E2 stimulation, whereas non-modified MCF7 cells need E2 stimulation to boost their proliferation. Reciprocally, when we used siRNA to deplete ZNF703 in MDA-MB-134, and tested the capacity of the ZNF703-depleted cells to proliferate, we observed a reduction of the capacity to proliferate of ZNF703-depleted cells. Second, ZNF703 transcription is induced by oestrogen stimulation. In agreement, ZNF703 has been identified as an ER-regulated gene (Lin et al, 2004, 2007). In contrast, the other 8p12 potential oncogenes have not been shown to be regulated by ER. This could explain the association of A1 (ZNF703) amplification with the luminal B subtype. The 8p11-12 region is often co-amplified with the 11q13 region (Kwek et al, 2009; Ollendorff et al, 1992; Szepetowski et al, 1992), which comprises the Cyclin D-encoding locus CCND1. CCND1 is also under the control of ER (Cicatiello et al, 2004). ZNF703 and CCND1 may thus function in the same pathway and potentiate each other. Indeed, CCND1 overexpression increases ZNF703 expression (Kwek et al, 2009). Third, the ZNF703 protein may be a cofactor in a nuclear co-repressor complex composed of DCAF7, PHB2 and NCOR2. Co-repressors control the action of many transcription factors including ER (Battaglia et al, 2010; Perissi et al, 2010). Interestingly, ZNF703 overexpression represses the expression of luminal markers and ER-associated proteins such as FOXA1 and GATA3. ZNF703 would uncouple ER-related differentiation and proliferation programmes. Moreover, ZNF703-overexpressing cells display RB1 phosphorylation, P27Kip1 downregulation, E2F1 activation and CCNE1 activation. ZNF703 interaction with DCAF7 may explain its role on E2F1 signalling. DCAF7 is a CUL4/DDB1 interacting protein (Jin et al, 2006) that activates SKP2 to induce P27Kip1 proteolysis (Bondar et al, 2006). Interestingly, the DCAF7 gene is located in chromosomal region 17q23, which we found often gained or amplified in luminal B tumours (Supplementary Table 2). GSEA showed that the E2F1 activation/ER repression balance is prominent in luminal B tumours. Fourth, ZNF703 overexpression increases both the CD49f-positive cell population and the formation of primary and secondary tumourspheres suggesting a role in the control of CSC self-renewal. Moreover, the gene expression programme activated by ZNF703 overexpression is enriched in genes related to stem cell biology. It has been proposed that luminal tumours might arise through transformation of an ER-positive luminal progenitor (Dontu et al, 2004). The oncogenic capacity of ZNF703 could thus be exerted on ER-positive mammary progenitors and contribute to re-activation of a self-renewal programme.
All these specific features may explain both the association of ZNF703 amplification with the luminal B subtype and its frequent association with CCND1. We propose that ZNF703 and Cyclin D1 are two ER-regulated components of a pathway leading to activation of CCNE and cell cycle progression from G1 to S past the R restriction point (Fig 6B). This latter has been defined as an important cell cycle stage controlling stem cell fate allowing equilibrium between self-renewal and committed cell-fate decision (Orford and Scadden, 2008). In self-renewing cells, CCNE is constitutively active throughout the cell cycle, which allows the transition of stem cells from M phase directly to late G1. The resulting absence of the Cyclin D-dependent early G1 phase shortens the G1 phase and the entire cell cycle. Thus, tempering with the R point would modify the balance between self-renewal and differentiation (Orford and Scadden, 2008). In luminal tumours, the ‘tempering pathway’ would include ZNF703 and CCND1. It could be set into action and kept overfunctioning by the amplification of 8p12 ZNF703 or 11q13 CCND1, or by 8p12/11q13 co-amplification. Alterations in other components of the pathway, such as loss of function of RB1 (Herschkowitz et al, 2008) or overexpression of CCNE1 (Keyomarsi et al, 2002) can also activate this pathway but can occur in any subtype since these proteins are not regulated by ER. We therefore propose that ZNF703 amplification represses luminal differentiation and increases self-renewal, thus sustaining a luminal B phenotype. This does not prevent other genes of the amplified regions to exert an effect. The number of 8p11-12 subamplicons and validated candidates (e.g. DDHD2, ERLIN2, FGFR1, PPAPDC1P, RCP/RAB11FIP1, TC-1/C8orf4 and WHS1L1) indicates that the 8p11-12 region contains several closely-linked oncogenic targets that function in cooperation after amplification. This cooperative effect could be to sustain both self-renewal and proliferation of the luminal progenitor. Thus, our results explain all the three enigmatic features of the 8p11-12 amplification that are the association with luminal B tumours, the plethora of candidate oncogenes and the frequent co-amplification with CCND1 at 11q13.
From a more general point of view, our study describes another type of cancer gene. If amplified genes may be considered as potential oncogenes, not all of these oncogenes should be viewed as solely proliferation boosters but some may be considered as transcriptional re-programmers controlling self-renewal capacity. Association of proliferation genes and self-renewal regulators constitute an ideal combination to initiate and sustain tumour progression.
MATERIALS AND METHODS
Ethics statement
Each patient gave informed consent and the study was approved by our institutional review board.
Cell culture
A total of eleven breast cell lines (BCL) derived from normal or malignant epithelium were analysed: 184B5, CAMA-1, HCC1500, HME-1, MCF7, MCF10A, MDA-MB-134, T47D, ZR-75–1 obtained from ATCC (http://www.atcc.org/), SUM52 obtained from Dr S. Ethier (http://www.asterand.com/asterand/BIOREPOSITORY/hbreastcancercelllines.aspx), and S68 obtained from V. Catros (Université de Rennes, Rennes, France). BCLs were grown using the recommended culture conditions. For experiments with oestrogen induction, cells were washed with PBS and precultured in phenol red-free RPMI 1640 medium supplemented with 10% charcoal-filtered FCS (Hyclone, Thermo Fisher Scientific) for 24 h. For rescue experiments, MDA-MB-134 and HCC1500 cells were treated with 17β-estradiol (1 or 10 nM) (E2, Sigma–Aldrich, E8875).
Tumour samples
Tumour tissues were collected from 266 patients with invasive adenocarcinoma who underwent initial surgery at the Institut Paoli-Calmettes (Marseille, France) between 1987 and 2007, and from 22 samples from the Jules Bordet Institute previously described (Loi et al, 2007). Samples were macrodissected and frozen in liquid nitrogen within 30 min of removal.
DNA and RNA extraction
DNA and RNA were extracted from frozen tumour samples by using guanidium isothiocynanate and cesium chloride gradient as previously described (Bertucci et al, 2006). RNA and DNA integrity was controlled on Agilent Bioanalyzer (Agilent Technologies) and agarose gel electrophoresis, respectively. DNA and RNA form BCLs were isolated utilizing RNeasy Mini Kit (QIAGEN) and used for QPCR and RTQPCR in a LightCycler FastStart DNA Masterplus SYBR Green I (Roche, Mannheim, Germany) (supplementary method).
Gene expression analysis
Expression profiles were analysed on a set of 266 tumours, 4 normal breast samples and 31 cell lines using Affymetrix U133 Plus 2.0 human oligonucleotide microarrays as previously described (Charafe-Jauffret et al, 2006; Irizarry et al, 2003). Scanning was done with Affymetrix GeneArray scanner. Data were analysed by ‘Robust Multichip Average’ (RMA) with the non-parametric quantile algorithm as normalization parameter in R/Bioconductor and associated packages (Benito et al, 2004). The five molecular subtypes related to the intrinsic breast cancer tumour classification were determined for the 266 samples using the single sample predictor (SSP) classifier based on list of 306 genes and 315 samples (Adler et al, 2009) associated to ‘distance weighted discrimination’ (DWD) as data set adjustment (Subramanian et al, 2005). Co-expression search for ZNF703 (probeset 222760_at) was done using MEM web tool (Chen and Sharp, 2004) as described in supplementary methods. Pathway-based analysis was done using IPA (Ingenuity® Systems, www.ingenuity.com). Gene expression data are deposited in Gene expression Omnibus (GEO) public database (www.ncbi.nlm.nih.gov/geo) and sample accession number are reported in supplementary Table 12.
Additional expression data set
To determine ZNF703 gene expression in luminal breast cancer, we collected gene expression data from several public databases. Eleven public data sets were included, collected from eleven independent studies (Supplementary Table 4) representing a total of 1172 different samples. For each sample, molecular subtypes were defined as described above (Supplementary Table 5). To compare ZNF703 gene expression across data sets and to exclude bias from population heterogeneity, expression levels were standardized within data sets using the luminal A population as referential. Student's t-test and one-way ANOVA test were used to evaluate ZNF703 expression level difference between luminal B and luminal A population and across the five molecular subtypes, respectively.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) {http://www.broadinstitute.org/gsea/} (Subramanian et al, 2005) was used to identify a priori defined sets of genes that were differentially expressed between luminal A and luminal B samples. GSEA was applied on the IPC series with 138 luminal BCs (i.e. the 49 luminal B and 89 luminal A). Prior the GSEA, data were filtered to remove low and poorly measured expression probe sets as defined by an expression value inferior to 6.64 log2 units across the 138 BCs. GSEA was done using signal-to-noise metric for ranking genes, weighted enrichment statistic to computed enrichment score (ES) of each gene sets tested and 1000 phenotype permutations to evaluate significance. As gene sets database, we chose the transcription factor targets list (C3, TFT) from the Molecular Signatures database (http://www.broadinstitute.org/gsea/msigdb). The C3 TFT list contains gene sets that share a transcription factor-binding site defined in the TRANSFAC database {http://www.gene-regulation.com}. Gene sets were defined as significant at the 5% level with a false discovery rate (FDR) under 25%. Relation between each transcription factor and E2F1 or ESR1/ERα was established by mining PubMed abstracts using Chilibot (Chen and Sharp, 2004).
Array-comparative genomic hybridization
Genome profiling was established on a subset of 100 luminal breast tumours (59 luminal A and 41 luminal B) for which RNA and DNA samples were both available (Supplementary Table 1). Genomic imbalances of 100 breast tumours were analysed by aCGH using 244K CGH Microarrays (Hu-244A, Agilent Technologies) following a described protocol (Etienne et al, 2007). The final data set contained 225 388 unique probes covering 22 509 genes and intergenic part following the hg17 human genome mapping. aCGH data processing is described in Supplementary method section.
Plasmids constructs and clone selection
Human ZNF703 cDNA full length (IMAGE: 5527569) was obtained by ImaGenes (Berlin, Germany). cDNA sequence was PCR-amplified using attB1-TCACCATGAGCGATTCGCCCGCTGG-3′ and attB2-CCTGGTATCCCAGCGCCGAGG-5′ oligonucleotides. The resulting PCR fragment was inserted into the vector pDONR™/Zeo using BP-reaction gateway technology (Invitrogen). pDONR™/Zeo-ZNF703 was then subcloned in pEGFP-C1-DEST using LR-reaction (Invitrogen). MCF7 cells were transfected with pEGFP-ZNF703 or pEGFP by electroporation as described by the manufacturer (Amaxa, Lonza Cologne, Germany). Individual clones stably expressing GFP-ZNF703 fusion protein or GFP were obtained by constant geneticin selection at 0.5 mg/ml, the resulting clones were confirmed by Western blotting.
The paper explained
PROBLEM
Despite improvements in the treatment of breast cancer, resistance to conventional therapy is a major clinical problem. The heterogeneity of breast cancer renders prognosis and treatment difficult. Heterogeneity may result from various cell-of-origins and genetic alterations. The latter are numerous in breast cancer but few have been characterized and even less can be used as markers or therapeutic targets. Although both luminal A and B tumours express hormone receptors (oestrogen and progesterone), the risk of relapse in women treated by hormone therapy is greater in women with a luminal B tumour than in women with a luminal A tumour. Therefore, the elucidation of the molecular mechanisms that govern luminal B tumour biology should improve both our understanding of tumourigenesis and treatment of patients.
RESULTS
We have studied 100 luminal breast tumours by combined analysis of genome CNAs and gene expression. We show that amplification of the ZNF703 gene, located in chromosomal region 8p12, preferentially occurs in luminal B tumours.
We identified ZNF703 as a nuclear protein whose expression is stimulated by oestrogen and which represses ER-associated proteins. ZNF703 is part of a protein complex including DCAF7 and PHB2 and its expression both stimulates cell proliferation and increases the stem cell population. Moreover, ZNF703 expression in the MCF7 cell line is associated with a shift from an ER-associated gene expression programme to an E2F1-associated programme. This transcriptional shift may lead to the activation of stem cell-related gene expression, which induces an increase in CSCs.
IMPACT
We identified a new type of oncogene. ZNF703 should be not be viewed solely as a proliferation booster but may be considered as a transcriptional re-programmer controlling self-renewal capacity. Defining the role of ZNF703 in the regulation of luminal B oncogenesis may help to understand luminal B tumour biology and hormone-resistance: the association of ZNF703 amplification with the luminal B type may serve as a marker for this subtype of poor prognosis; the cross-talk between ER and ZNF703 explains features of luminal B tumours; the role of ZNF703 on self-renewal may be used as a handle to characterize the luminal B cell-of-origin; finally, the ZNF703-associated protein complex may be used as a therapeutic target to treat luminal B tumours with 8p12 amplification.
siRNA lipofection
Three stealth siRNA targeted to the ZNF703 mRNA (Invitrogen) were designed along with the corresponding stealth siRNA control. MDA-MB-134 cells were lipofected with these four oligonucleotides utilizing lipofectamine RNAiMAX (Invitogen). Twenty-five thousand cells were plated in triplicate in 24 well plates with 10 pmol of siRNA in 600 µl of Opti-MEM I Medium. Cell proliferation was evaluated in triplicate at 30-h after lipofection.
Cell proliferation assay
For MTS assays, cells were plated in adherent conditions in 96-well plates at 5000 cells per well in the presence or absence of E2. The cell growth kinetic was estimated at different time points by addition of 20 µl MTS solution (5 mg/ml in PBS) in each well. Cells were then incubated for 1 h at 37°C, followed by addition of 50 µl DMSO to each well. Absorbance was measured at 560 nm in a fluorescence plate reader (Spectrafluor; Tecan). For MDA-MB-134 cells were first lipofected with siRNA and plated for MTS assays after 30 h.
Nuclease treatments
MCF7 GFP-ZNF703 culture cells were grown on labtek chamber, washed briefly in PBS and fixed in cold methanol/acetone for 7 min at −20°C. Fixed cells were then treated, at room temperature, with RNAse A (80 µg/ml) or DNAse I (100 µg/ml) for 30 and 15 min, respectively. Cells were then washed with PBS and immunostained with an anti-GFP antibody for 1 h (Roche 11814460001, 1:500). The nuclei were counterstained with DAPI/antifade (Invitrogen) and coverslipped. Sections were examined with a fluorescent microscope (Leica).
Co-immunoprecipitation
Cells were lysed in extraction buffer (1% v/v Triton X-100, 50 mM Hepes, pH 7, 1 mM EDTA, 1 mM EGTA, 150 mM NaCl, 100 mM sodium fluoride, 1 mM Na3VO4 and one tablet of Complete™ inhibitor mix (Roche Applied Science) per 25 ml of buffer and loaded onto SDS-polyacrylamide gels. For co-immunoprecipitation experiments 12 mg of protein lysates were produced, with 2 mg for SDS-PAGE analysis and 10 mg for mass spectroscopy analysis. First, lysates were precleared 1 h with 70 µl of sepharose beads at 4°C. After centrifugation, 5 µg of antibody were added to the precleared lysate. Immunoprecipitation was performed overnight at 4°C under constant rotation. 70 µl of protein G-sepharose or A-sepharose beads were added and incubated 1 h at 4°C. Beads were collected by centrifugation (1500 rpm, 5 min) and washed once in lysis buffer and three times in HNTG buffer (Hepes 20 mM, NaCl 150 mM, Glycerol 10%, Triton X-100 0.1%). Beads were then resuspended in 35 µl of Sample Buffer 2X (Tris pH 6, 8; 120 mM, Glycerol 20%, SDS 4%, β-Mercapto-ethanol), boiled at 99°C. After centrifugation, immunoprecipitated complexes were resolved by SDS-PAGE.
Mass spectrometry
Immunocomplexes were separated on NuPage 4–12% Bis–tris acrylamide gels in Mops buffer according to the manufacturer's instructions (Invitrogen, Groningen, Netherlands). SDS-PAGE gels were stained with colloidal Coomassie blue (Imperial protein stain, Pierce) and proteins of interest were isolated as previously described (Goncalves et al, 2008). Extracted peptides were subjected to mass spectrometric analysis for identification. Proteins were identified by nanoLC-MS/MS using a nano-LC Ultimate system (Dionex, LC Packing) on line with an ion trap analyser (HCT, Ultraflex, Bruker Daltonics Inc. Bremen, Germany). MS/MS Raw data were processed using DataAnalysis 3.1 and Biotools 3.1 softwares (Bruker Daltonics Inc.). Protein searches were done using a local Mascot search engine (Version 2.2.03) (Matrix Science Ltd., London, UK) against SwissProt database (release 57.12, release date December 17, 2009; 513877 sequences; Homo sapiens: 20401 sequences) using the following parameters: human for taxonomy, 0.5 Da mass tolerance for parent and fragments search, one missed cleavage side for trypsin was allowed. Fixed carbamidomethylated cystein and potential methionine oxidation were selected as modifications. Protein identification was based on at least three unique peptides for each protein. Mascot Ions score was set to >30 and each score was further validated against ions score corresponding to sequence identity. Proteins identified were validated by immunoblotting as described in supplementary methods.
Immunofluorescence
MCF7 GFP and GFP-ZNF703 cells were grown on labtek chambers. After 48 h, cells were fixed 20 min in 3.7% paraformaldehyde, PBS; permeabilized 5 min with PBS, 0.1% Triton X-100; blocked with protein block serum-free (Dako, X0909); and incubated 1 h with primary antibodies (anti-PHB2; Abcam ab71970, 1:100), anti-SMRT (BD Biosciences 611386, 1:100), anti-HSP60 (Abcam ab46798, 1:50), anti-PML (Santa-Cruz Sc-966, 1:20), anti-GFP (Roche 11814460001, 1:250), anti-ERα (Neomarkers Fremont CA, RB-9016-P1, 1:100) and anti-DCAF7 (Sigma–Aldrich, HPA022962, 1:50), and with corresponding secondary antibody Alexa Fluor 594 (Invitrogen) conjugated for 20 min incubation. Images of the nuclei were counterstained with DAPI/antifade (Invitrogen) and coverslipped. Sections were examined with a fluorescent microscope (Leica). For ER-α and ZNF703 expression scoring, around 100 cells were scored for three independent chambers.
Flow cytometry analysis
Staining for CD49f was done using a PE-labelled anti-CD49f antibody (diluted 1:100; BD Biosciences). Fresh cells were stained with 1 µg/ml PI (Sigma–Aldrich) for 5 min for viability. Analysis of CD49f expression was done on FACS LSRII (BD biosciences).
Luciferase reporter assays
We utilized three different luciferase reporter assays for the detection of oestrogen transcriptional activity (p17M-ERE-Luc, a gift from Dr. D. Metzger, IGBMC, Strasbourg, France) and for detection of E2F1 transcriptional activity (pGL2-3XE2F1-Luc and pGL3-CCNE-Luc, a gift from Dr. C. Sardet, IGM, Montpellier, France). MCF7 GFP and GFP-ZNF703 stable cell lines were electroporated (Amaxa) with 250 ng TK-R-Luc (pGL3; Promega, France) as an internal control and 1 µg of one luciferase reporter vector according to the manufacturer's protocol. For ER-related transcriptional activity, cells were seeded overnight in 60-mm dishes containing phenol red-free RPMI 1640 (Invitrogen) supplemented with 10% charcoal dextran-treated fetal bovine serum (Hyclone). 16 h later, 10 nM of 17-β estradiol (E2) were added to the medium and luciferase activity was determined after 24 h. For luciferase activity detection, we prepared transfected cells lysate and utilized the Dual-luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer's instructions. We normalized each luciferase activity value by protein concentration determined by bradford assay and TK-renilla luciferase activity value. Each experiment was done in duplicate.
Tumoursphere assay
MCF7 GFP-ZNF707 and MCF7 GFP cell lines were cultured in adherent conditions in the presence or absence of E2 for 3 days then plated as single cells in 1% agarose coated plates at a density of 1000 cells/ml and grown for 5 days. The number of primary spheres was counted under microscope. Subsequent cultures after dissociation of primary spheres were plated in 1% coated plates at a density of 1000 cells/ml. As for primary spheres, the number of secondary spheres was determined under microscope. Experiments were done in triplicate. Tumoursphere cultures were grown in a serum-free mammary epithelium basal medium as previously described (Ginestier et al, 2009).
Cell cycle progression analysis
Cell cycle synchronization was achieved by a non-pharmacological method using cell-to-cell contact inhibition in conjunction with serum depletion to induce cell quiescence. In brief, individual cells were spread out in 24-well plates and were grown to 80% confluence. Then FBS-rich medium (10%) was removed and cells were incubated in high density conditions with charcoal FBS-poor medium (0.5%) for 72 h at 37°C in a 5% CO2 atmosphere. Cells were subsequently released from the G1/S arrest by addition of serum-rich medium in the presence or absence of E2. Subsequent synchronization was analysed by flow cytometry. Briefly, supernatant and adherent cells were harvested, washed and suspended in 0.5 ml medium containing propidium iodide (40 µg/ml) and RNase A (40 µg/ml). Analysis of the cell cycle was done on the FACScan (BD biosciences) using CellQuest analysis software. Cell cycle progression was evaluated every 2 h over 24 h after release. It is important to note that a large proportion of cells arrested in G1/S phase will not enter into cell cycle due to senescence induced by the starvation.
Statistical analyses
Correlations between sample groups and histoclinical factors were calculated with the Fisher's exact test for qualitative variables with discrete categories, and the Mann-Whitney test for continuous variables. DFS from 510 luminal breast tumours collected from the 11 public datasets was calculated from the date of diagnosis until date of first relapse whatever its location (local, regional or distant) or date of death (when the relapse data was not available). Univariate and multivariate analyses were done using Cox regression analysis. The p-values were based on the Wald test, and patients with one or more missing data were excluded. All statistical tests were two-sided at the 5% level of significance. Statistical analysis was done using the survival package (Version 2.30), in the R software (Version 2.9.1).
Acknowledgments
We thank C. Chabannon for biobanking, and F. Birg, P. Viens and L. Xerri for help and discussions. We thank D. Metzger (IGBMC, Strasbourg, France) for the gift of the ER reporter as well as C. Sardet (IGMM, Montpellier, France) for the gift of the E2F reporter. This work has been supported by Inserm, Institut Paoli-Calmettes, and grants from Ligue Nationale Contre le Cancer (Label DB), Institut National du Cancer (Recherche translationnelle 2009, DB), and Marie Curie Actions (IRG 2005, NN). FS, IB and ER have been supported by the Association pour la Recherche contre le Cancer (2009), the Algerian Government (2005–2009), and the Institut National du Cancer (2008), respectively.
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that there is no conflict of interest.
Author contributions
FS, NN, IB, ER, AL, JA, EB, SA, SE, JW performed the experiments. AF, PF, ML, JPB, CP, FB analysed the data, ECJ, JJ, CS contributed materials and analyses, DB, MC, CG designed the study and CG wrote the manuscript.
For more information
Broad Institute: Gene set enrichment matrix
BIIT research group, University of Tartu, Estonia: Multi Experiment Matrix
Cancer stem cell group, Centre de Recherche en Cancérologie de Marseille, Inserm-U891, France
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Adelaide J, Finetti P, Bekhouche I, Repellini L, Geneix J, Sircoulomb F, Charafe-Jauffret E, Cervera N, Desplans J, Parzy D, et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007;67:11565–11575. doi: 10.1158/0008-5472.CAN-07-2536. [DOI] [PubMed] [Google Scholar]
- Adler P, Kolde R, Kull M, Tkachenko A, Peterson H, Reimand J, Vilo J. Mining for coexpression across hundreds of datasets using novel rank aggregation and visualization methods. Genome Biol. 2009;10:R139. doi: 10.1186/gb-2009-10-12-r139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia S, Maguire O, Campbell MJ. Transcription factor co-repressors in cancer biology: roles and targeting. Int J Cancer. 2010;126:2511–2519. doi: 10.1002/ijc.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito M, Parker J, Du Q, Wu J, Xiang D, Perou CM, Marron JS. Adjustment of systematic microarray data biases. Bioinformatics. 2004;20:105–114. doi: 10.1093/bioinformatics/btg385. [DOI] [PubMed] [Google Scholar]
- Bernard-Pierrot I, Gruel N, Stransky N, Vincent-Salomon A, Reyal F, Raynal V, Vallot C, Pierron G, Radvanyi F, Delattre O. Characterization of the recurrent 8p11-12 amplicon identifies PPAPDC1B, a phosphatase protein, as a new therapeutic target in breast cancer. Cancer Res. 2008;68:7165–7175. doi: 10.1158/0008-5472.CAN-08-1360. [DOI] [PubMed] [Google Scholar]
- Bertucci F, Finetti P, Cervera N, Charafe-Jauffret E, Mamessier E, Adelaide J, Debono S, Houvenaeghel G, Maraninchi D, Viens P, et al. Gene expression profiling shows medullary breast cancer is a subgroup of basal breast cancers. Cancer Res. 2006;66:4636–4644. doi: 10.1158/0008-5472.CAN-06-0031. [DOI] [PubMed] [Google Scholar]
- Bondar T, Kalinina A, Khair L, Kopanja D, Nag A, Bagchi S, Raychaudhuri P. Cul4A and DDB1 associate with Skp2 to target p27Kip1 for proteolysis involving the COP9 signalosome. Mol Cell Biol. 2006;26:2531–2539. doi: 10.1128/MCB.26.7.2531-2539.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariati M, Naderi A, Brown JP, Smalley MJ, Pinder SE, Caldas C, Purushotham AD. Alpha-6 integrin is necessary for the tumourigenicity of a stem cell-like subpopulation within the MCF7 breast cancer cell line. Int J Cancer. 2008;122:298–304. doi: 10.1002/ijc.23103. [DOI] [PubMed] [Google Scholar]
- Charafe-Jauffret E, Ginestier C, Monville F, Finetti P, Adelaide J, Cervera N, Fekairi S, Xerri L, Jacquemier J, Birnbaum D, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene. 2006;25:2273–2284. doi: 10.1038/sj.onc.1209254. [DOI] [PubMed] [Google Scholar]
- Cheang MC, Chia SK, Voduc D, Gao D, Leung S, Snider J, Watson M, Davies S, Bernard PS, Parker JS, et al. Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J Natl Cancer Inst. 2009;101:736–750. doi: 10.1093/jnci/djp082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Sharp BM. Content-rich biological network constructed by mining PubMed abstracts. BMC Bioinformatics. 2004;5:147. doi: 10.1186/1471-2105-5-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, DeVries S, Fridlyand J, Spellman PT, Roydasgupta R, Kuo WL, Lapuk A, Neve RM, Qian Z, Ryder TC, et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell. 2006;10:529–541. doi: 10.1016/j.ccr.2006.10.009. [DOI] [PubMed] [Google Scholar]
- Cicatiello L, Addeo R, Sasso A, Altucci L, Petrizzi VB, Borgo R, Cancemi M, Caporali S, Caristi S, Scafoglio C, et al. Estrogens and progesterone promote persistent CCND1 gene activation during G1 by inducing transcriptional derepression via c-Jun/c-Fos/estrogen receptor (progesterone receptor) complex assembly to a distal regulatory element and recruitment of cyclin D1 to its own gene promoter. Mol Cell Biol. 2004;24:7260–7274. doi: 10.1128/MCB.24.16.7260-7274.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dontu G, El-Ashry D, Wicha MS. Breast cancer, stem/progenitor cells and the estrogen receptor. Trends Endocrinol Metab. 2004;15:193–197. doi: 10.1016/j.tem.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Dyck JA, Maul GG, Miller WH, Jr, Chen JD, Kakizuka A, Evans RM. A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein. Cell. 1994;76:333–343. doi: 10.1016/0092-8674(94)90340-9. [DOI] [PubMed] [Google Scholar]
- Etienne A, Carbuccia N, Adelaide J, Bekhouche I, Remy V, Sohn C, Sainty D, Gastaut JA, Olschwang S, Birnbaum D, et al. Rearrangements involving 12q in myeloproliferative disorders: possible role of HMGA2 and SOCS2 genes. Cancer Genet Cytogenet. 2007;176:80–88. doi: 10.1016/j.cancergencyto.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Finetti P, Cervera N, Charafe-Jauffret E, Chabannon C, Charpin C, Chaffanet M, Jacquemier J, Viens P, Birnbaum D, Bertucci F. Sixteen-kinase gene expression identifies luminal breast cancers with poor prognosis. Cancer Res. 2008;68:767–776. doi: 10.1158/0008-5472.CAN-07-5516. [DOI] [PubMed] [Google Scholar]
- Fridlyand J, Snijders AM, Ylstra B, Li H, Olshen A, Segraves R, Dairkee S, Tokuyasu T, Ljung BM, Jain AN, et al. Breast tumor copy number aberration phenotypes and genomic instability. BMC Cancer. 2006;6:96. doi: 10.1186/1471-2407-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galderisi U, Cipollaro M, Giordano A. The retinoblastoma gene is involved in multiple aspects of stem cell biology. Oncogene. 2006;25:5250–5256. doi: 10.1038/sj.onc.1209736. [DOI] [PubMed] [Google Scholar]
- Gelsi-Boyer V, Orsetti B, Cervera N, Finetti P, Sircoulomb F, Rouge C, Lasorsa L, Letessier A, Ginestier C, Monville F, et al. Comprehensive profiling of 8p11-12 amplification in breast cancer. Mol Cancer Res. 2005;3:655–667. doi: 10.1158/1541-7786.MCR-05-0128. [DOI] [PubMed] [Google Scholar]
- Geyer FC, Marchio C, Reis-Filho JS. The role of molecular analysis in breast cancer. Pathology. 2009;41:77–88. doi: 10.1080/00313020802563536. [DOI] [PubMed] [Google Scholar]
- Ginestier C, Wicinski J, Cervera N, Monville F, Finetti P, Bertucci F, Wicha MS, Birnbaum D, Charafe-Jauffret E. Retinoid signaling regulates breast cancer stem cell differentiation. Cell Cycle. 2009;8:3297–3302. doi: 10.4161/cc.8.20.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves A, Charafe-Jauffret E, Bertucci F, Audebert S, Toiron Y, Esterni B, Monville F, Tarpin C, Jacquemier J, Houvenaeghel G, et al. Protein profiling of human breast tumor cells identifies novel biomarkers associated with molecular subtypes. Mol Cell Proteomics. 2008;7:1420–1433. doi: 10.1074/mcp.M700487-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–718. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, He X, Fan C, Perou CM. The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res. 2008;10:R75. doi: 10.1186/bcr2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks J, Krasnitz A, Lakshmi B, Navin NE, Riggs M, Leibu E, Esposito D, Alexander J, Troge J, Grubor V, et al. Novel patterns of genome rearrangement and their association with survival in breast cancer. Genome Res. 2006;16:1465–1479. doi: 10.1101/gr.5460106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, Burleigh A, Git A, Chin S-F, Hurtado A, Bruna A, Ali R, Greenwood W, Dunning MJ, Samarajiwa S, et al. ZNF703, a Luminal B breast cancer oncogene, is a transcriptional repressor and differentially regulates luminal and basal progenitors in human mammary epithelium. EMBO Mol Med. 2011 doi: 10.1002/emmm.201100122. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;7:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado A, Holmes KA, Geistlinger TR, Hutcheson IR, Nicholson RI, Brown M, Jiang J, Howat WJ, Ali S, Carroll JS. Regulation of ERBB2 by oestrogen receptor-PAX2 determines response to tamoxifen. Nature. 2008;456:663–666. doi: 10.1038/nature07483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Keyomarsi K, Tucker SL, Buchholz TA, Callister M, Ding Y, Hortobagyi GN, Bedrosian I, Knickerbocker C, Toyofuku W, Lowe M, et al. Cyclin E and survival in patients with breast cancer. N Engl J Med. 2002;347:1566–1575. doi: 10.1056/NEJMoa021153. [DOI] [PubMed] [Google Scholar]
- Korkaya H, Wicha MS. HER-2, notch, and breast cancer stem cells: targeting an axis of evil. Clin Cancer Res. 2009;15:1845–1847. doi: 10.1158/1078-0432.CCR-08-3087. [DOI] [PubMed] [Google Scholar]
- Kurtev V, Margueron R, Kroboth K, Ogris E, Cavailles V, Seiser C. Transcriptional regulation by the repressor of estrogen receptor activity via recruitment of histone deacetylases. J Biol Chem. 2004;279:24834–24843. doi: 10.1074/jbc.M312300200. [DOI] [PubMed] [Google Scholar]
- Kwek SS, Roy R, Zhou H, Climent J, Martinez-Climent JA, Fridlyand J, Albertson DG. Co-amplified genes at 8p12 and 11q13 in breast tumors cooperate with two major pathways in oncogenesis. Oncogene. 2009;28:1892–1903. doi: 10.1038/onc.2009.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Sircoulomb F, Ginestier C, Cervera N, Monville F, Gelsi-Boyer V, Esterni B, Geneix J, Finetti P, Zemmour C, et al. Frequency, prognostic impact, and subtype association of 8p12, 8q24, 11q13, 12p13, 17q12, and 20q13 amplifications in breast cancers. BMC Cancer. 2006;6:245. doi: 10.1186/1471-2407-6-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, et al. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome Biol. 2004;5:R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Vega VB, Thomsen JS, Zhang T, Kong SL, Xie M, Chiu KP, Lipovich L, Barnett DH, Stossi F, et al. Whole-genome cartography of estrogen receptor alpha binding sites. PLoS Genet. 2007;3:e87. doi: 10.1371/journal.pgen.0030087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007;25:1239–1246. doi: 10.1200/JCO.2006.07.1522. [DOI] [PubMed] [Google Scholar]
- Mussi P, Liao L, Park SE, Ciana P, Maggi A, Katzenellenbogen BS, Xu J, O'Malley BW. Haploinsufficiency of the corepressor of estrogen receptor activity (REA) enhances estrogen receptor function in the mammary gland. Proc Natl Acad Sci USA. 2006;103:16716–16721. doi: 10.1073/pnas.0607768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Runko AP, Sagerstrom CG. A novel subfamily of zinc finger genes involved in embryonic development. J Cell Biochem. 2004;93:887–895. doi: 10.1002/jcb.20255. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollendorff V, Szepetowski P, Mattei MG, Gaudray P, Birnbaum D. New gene in the homologous human 11q13-q14 and mouse 7F chromosomal regions. Mamm Genome. 1992;2:195–200. doi: 10.1007/BF00302877. [DOI] [PubMed] [Google Scholar]
- Orford KW, Scadden DT. Deconstructing stem cell self-renewal: genetic insights into cell-cycle regulation. Nat Rev Genet. 2008;9:115–128. doi: 10.1038/nrg2269. [DOI] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, Pilotti S, Pierotti MA, Daidone MG. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–5511. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- Ray ME, Yang ZQ, Albertson D, Kleer CG, Washburn JG, Macoska JA, Ethier SP. Genomic and expression analysis of the 8p11-12 amplicon in human breast cancer cell lines. Cancer Res. 2004;64:40–47. doi: 10.1158/0008-5472.can-03-1022. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de RM, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector DL, Fu XD, Maniatis T. Associations between distinct pre-mRNA splicing components and the cell nucleus. EMBO J. 1991;10:3467–3481. doi: 10.1002/j.1460-2075.1991.tb04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szepetowski P, Ollendorff V, Grosgeorge J, Courseaux A, Birnbaum D, Theillet C, Gaudray P. DNA amplification at 11q13.5-q14 in human breast cancer. Oncogene. 1992;7:2513–2517. [PubMed] [Google Scholar]
- Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A, et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umanskaya K, Radke S, Chander H, Monardo R, Xu X, Pan ZQ, O'Connell MJ, Germain D. Skp2B stimulates mammary gland development by inhibiting REA, the repressor of the estrogen receptor. Mol Cell Biol. 2007;27:7615–7622. doi: 10.1128/MCB.01239-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Fusaro G, Padmanabhan J, Chellappan SP. Prohibitin co-localizes with Rb in the nucleus and recruits N-CoR and HDAC1 for transcriptional repression. Oncogene. 2002;21:8388–8396. doi: 10.1038/sj.onc.1205944. [DOI] [PubMed] [Google Scholar]
- Yang ZQ, Moffa AB, Haddad R, Streicher KL, Ethier SP. Transforming properties of TC-1 in human breast cancer: interaction with FGFR2 and beta-catenin signaling pathways. Int J Cancer. 2007;121:1265–1273. doi: 10.1002/ijc.22831. [DOI] [PubMed] [Google Scholar]
- Yang ZQ, Liu G, Bollig-Fischer A, Giroux CN, Ethier SP. Transforming properties of 8p11-12 amplified genes in human breast cancer. Cancer Res. 2010;70:8487–8497. doi: 10.1158/0008-5472.CAN-10-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu X, Datta A, Govindarajan K, Tam WL, Han J, George J, Wong C, Ramnarayanan K, Phua TY, et al. RCP is a human breast cancer-promoting gene with Ras-activating function. J Clin Invest. 2009;119:2171–2183. doi: 10.1172/JCI37622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.