

Abstract
Oxidative stress in the central nervous system is strongly associated with neuronal cell death in the pathogenesis of several neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis. In order to overcome the oxidative damage, there are some protective signaling pathways related to transcriptional upregulation of antioxidant enzymes, such as heme oxygenase-1 (HO-1) and superoxide dismutase (SOD)-1/-2. Their expression is regulated by several transcription factors and/or cofactors like nuclear factor-erythroid 2 (NF-E2) related factor 2 (Nrf2) and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α). These antioxidant enzymes are associated with, and in some cases, prevent neuronal death in animal models of neurodegenerative diseases. They are activated by endogenous mediators and phytochemicals, and also by several gases such as carbon monoxide (CO), hydrogen sulphide (H2S), and hydrogen (H2). These might thereby protect the brain from severe oxidative damage and resultant neurodegenerative diseases. In this paper, we discuss how the expression levels of these antioxidant enzymes are regulated. We also introduce recent advances in the therapeutic uses of medical gases against neurodegenerative diseases.
1. Introduction
The brain consumes 20 to 50% of total body oxygen (O2) consumption, although it only accounts for 2% of the body weight, meaning that brain function is largely dependent on constitutive supply of O2 [1]. Compared to the normal physiological condition, in which 2 to 5% of total oxygen consumed by cells is converted into reactive oxygen species (ROS) as a byproduct of mitochondrial respiration, excessive and unregulated production of ROS can occur in pathological conditions [2, 3]. Therefore, scavenging and regulating the amount of ROS in the brain is important to maintain normal brain activity.
Although aberrant production of ROS in the central nervous system (CNS) is critically linked to several neurodegenerative diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and amyotrophic lateral sclerosis (ALS), a set of antioxidant defense system can save the brain from severe injuries [4–8]. Oxidative stress activates a stress response, and adaptation against ROS-derived cellular injury maintains the redox balance and protects cells from lethal damage [9]. This adaptive response often requires upregulation of endogenous antioxidant enzymes, and their expression levels can be regulated by several transcription factors. To date, the importance of transcriptional regulation of antioxidant enzymes is recognized as a route to the discovery of neuroprotective strategies. In this paper, we highlight two major transcriptional regulation factors, nuclear factor-erythroid 2 (NF-E2) related factor 2 (Nrf2), and peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α). Also, we focus on the role of heme oxygenase-1 (HO-1) and superoxide dismutase (SOD) in neurodegenerative diseases, because these are the key components of antioxidant mechanism Figure 1. Finally, we would like to introduce recent research on several gases such as CO, H2, and H2S (now called medical gases), suggesting a new therapeutic approach against oxidative damage and resultant neurodegenerative diseases, most notably PD.
Figure 1.
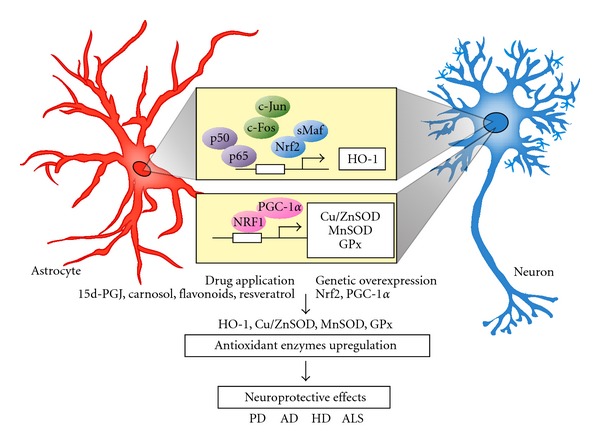
The transcriptional upregulation of antioxidant enzymes in neurodegenerative diseases. Both neurons and astrocytes can increase several antioxidant enzymes including heme oxygenase-1 (HO-1), copper and zinc-containing SOD (Cu/ZnSOD), manganese-containing SOD (MnSOD), and glutathione peroxidase (GPx). By drug application or genetic overexpression of transcription factor, the transcriptional responses via NF-κB (p50/p65), AP-1 (c-Jun/c-Fos), Nrf2/sMaf, and NRF1/PGC-1α in response to oxidative stress and related neurodegenerative disease are activated.
2. Nrf2: a Master of Redox Homeostasis
Nuclear factor-erythroid 2 (NF-E2) related factor 2 (Nrf2) is an important transcription factor and is recognized as a major contributor to the upregulation of multiple antioxidant defense system in response to oxidative stress. Nrf2 belongs to the cap'n'collar (CNC) family of basic region-leucine zipper (bZip)-type transcription factors [10]. NF-E2 is a heterodimeric protein which contains a large p45 and small p18 subunit [11]. Cloning of its cDNA revealed that p45 contains a cap'n'collar-(CNC-) type bZip domain [12]. The p45 subunit utilizes its CNC-bZip domain to form a heterodimer with p18; the latter has been identified as MafK, one of the small musculoaponeurotic fibrosarcoma oncogene (Maf) transcription factors [12, 13]. The heterodimer binds to an NF-E2 motif; the small Maf protein p18 confers DNA-binding activity to p45, while p45 activates transcription via its transactivation domain [13, 14]. Nrf2 binds to the antioxidant-responsive element (ARE) or the electrophile-responsive element (EpRE) [15, 16]. ARE has been detected in the promoter or upstream promoter regions of the genes encoding phase II antioxidant enzymes including glutathione S-transferase subunits (GST-Ya, GST-P, GST-M1/M3, etc.), glutamate-cysteine ligase catalytic (GCLC) and glutamate-cysteine ligase modifier (GCLM) subunits, the thioredoxin (TRX) and peroxiredoxin (PRX) families, and NAD(P)H:quinone oxidoreductase (NQO-1) [17–21]. Heme oxygenase-1 (HO-1) is also one of the Nrf2-ARE pathway-derived upregulated factors [22, 23], and its transcriptional upregulation is also mediated by some other transcription factors such as AP-1, CREB, and NF-κB [24]. In the CNS, genetic or pharmacological activation of Nrf2-ARE pathway can confer resistance against neurodegenerative disease insults such as AD, PD, HD, and ALS. A lentiviral vector encoding human Nrf2 reduced spatial learning deficits in aged APP/PS1 mice, a mouse model of AD [25]. Compared to neurons, astrocytes have a much greater ability to increase Nrf2-ARE pathway-derived gene expressions, as shown by a study using cortical neuronal cultures obtained from ARE-human placental alkaline phosphatase (hPAP) reporter mice [26], grown under condition where the mixed cortical cultures consist of 30% astrocytes and 70% neurons. Genetic-overexpression of Nrf2 in astrocytes using the glial fibrillary acidic protein (GFAP) promoter (GFAP-Nrf2) has shown protective effects against several animal models of neurodegeneration, for example, motor neuron degeneration produced by expressing mutant human superoxide dismutase 1 in ALS model mice [27], and dopaminergic neuronal loss by a neurotoxin (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, MPTP) in PD model mice [28]. Transplantation of astrocytes infected with adenovirus-Nrf2 protects striatal medium-spiny neuron degeneration by a mitochondrial complex II inhibitor (3-nitropropionic acid or malonate) in HD model mice [29].
HO-1, one of the antioxidant enzymes upregulated by Nrf2, is thought to be highly associated with AD pathology. In AD brain, HO-1 is expressed both in neurons and in astrocytes; 86% of GFAP-positive astrocytes in AD hippocampus exhibit HO-1 immunoreactivity, whereas those in age-matched normal tissues are in the range of only 6-7% [30]. HO-1 overexpression in astrocyte by transient transfection of HMOX1 cDNA significantly decreased intracellular cholesterol concentrations and increased the level of at least four oxysterol species compared to untreated control cultures [30]. In mild cognitive impairment or early AD, enhanced HO-1 expression stimulated astrocyte cholesterol biosynthesis, oxysterol formation, and cholesterol efflux (to sites of neuronal repair and for egress across the blood-brain barrier). Glial cholesterol efflux exceeds biosynthesis, and total cholesterol levels in affected brains are normal or diminished. Regulation of sterol homeostasis is important in AD pathology because a massive increase in the free cholesterol pool saturates the sterol efflux mechanism, which results in an increase in brain cholesterol levels and exacerbates amyloid deposition and neurodegeneration in advanced AD. Upregulation of HO-1 has another therapeutic potential for clearance of tau protein by the ubiquitin-proteasome system (UPS) [30]. Proteasome activity is reduced in AD brain and amyloid beta (Aβ) inhibits the UPS in cultured cells [31, 32]. This influence on UPS, for which heme-derived iron and CO are responsible, promotes intracellular degradation of α-synuclein as observed in HMOX1-transfected M17 cells [33]. Therefore, HO-1 is highly associated with the therapeutic approach not only by its antioxidant function but also by its influence on proteasomal degradation of tau and α-synuclein.
3. The Role of PGC-1α in Neurodegenerative Diseases
Since its discovery over a decade ago, peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) has been implicated in energy homeostasis, adaptive thermogenesis, β-oxidation of fatty acids, and glucose metabolism [34]. The activity of PGC-1α depends on its ability to form heteromeric complexes with a variety of transcription factors including nuclear respiratory factor 1 and 2 (NRF-1 and NRF-2), and the nuclear hormone receptors [35]. In particular, NRF-1 and NRF-2 are transcriptional regulators that act on nuclear genes encoding for constituent subunits of the oxidative phosphorylation system including cytochrome c, complex I-V, and mitochondrial transcription factor A (Tfam) [36]. Tfam, a transcription factor that acts on the promoters within the D-loop region of mitochondrial DNA and regulates the replication and transcription of the mitochondrial genome, contains consensus-binding sites for both NRF-1 and NRF-2 [37].
Mice lacking PGC-1α show a profound spongiform pattern of lesions in the striatum together with hyperactivity, which are the features of human HD [38]. In response to hydrogen peroxide (H2O2), there is over a 6-fold increase in PGC-1α expression in mouse embryonic fibroblasts, as well as an increase of the transcription of mRNA encoding ROS defense enzymes such as copper/zinc superoxide dismutase (SOD1), manganese SOD (SOD2), catalase, and glutathione peroxidase (GPx) in association with the transcription factor, cAMP-responsive element binding protein (CREB). PGC-1α expression is reduced by overexpression of mutant Huntingtin through its interference with formation of the CREB/TAF4 complex [39]. The HD striatal cell line, STHdhQ111 also shows reduced expression of PGC1-α target genes encoding mitochondrial cytochrome c and cytochrome oxidase IV. On the other hand, lentiviral overexpression of mitogen- and stress-activated protein kinase-1 increased PGC-1α and protected against striatal lentiviral expression of polyglutamine expansion in huntingtin protein (Exp-Htt) [40]. Moreover, in a postmortem brain tissue study of HD patients, expression levels of 24 out of 26 PGC-1α target genes were reduced, which implies that targeting PGC-1α would be beneficial as a therapeutic approach for HD. PGC-1α might also be beneficial for other neurodegenerative diseases such as PD, as reported in MPTP-induced PD model animals. PGC-1α-deficient mice were more sensitive to neurotoxic insult by MPTP [38], whereas overexpression of PGC-1α protected neurons against MPTP neurotoxicity [41].
PGC-1α activity is regulated by posttranslational modification including direct phosphorylation or deacetylation, which increases PGC-1α activity or expression [42–46]. One such protein is NAD-dependent deacetylase Sir2. Its mammalian or human homologue, SIRT1, has been focused on as a prospective candidate for neuroprotective strategies against AD, PD, HD, and ALS [47–51]. One of the important roles of SIRT1 lies in its deacetylase activity, and its deacetylase substrates such as PGC-1α and forkhead box O3A (Foxo3a) are involved in antioxidant responses and gene transcription [52]. Most especially, overexpression of SIRT1 deacetylase and suppression of GCN5 acetylase increase the transcriptional activity of PGC-1α and prevent mitochondrial loss in neurons induced by expanded Huntingtin protein [53]. A recent study by Martin et al. has revealed that mitogen- and stress-activated kinase (MSK-1), a nuclear protein kinase involved in chromatin remodeling through histone H3 phosphorylation, is linked to the nucleosomal response at the PGC-1α promoter, and transcription via CREB phosphorylation [40].
Among the genes regulated by NRF-1 and/or PGC-1α, SOD1 and 2 are dominant and the first lines of defense against ROS, especially the superoxide anion radical (O2 •−), are catalyzed to molecular oxygen and hydrogen peroxide [54]. In humans, three different forms of SOD are reported: SOD1, SOD2 in mitochondria, and extracellular Cu/ZnSOD (SOD3) [55–57]. SOD1 and SOD2 are abundant in the CNS, whereas SOD3 is less abundant than SOD1 and SOD2 [58]. The expression levels of SOD1 and SOD2 are associated with human amyloid precursor protein (hAPP)-/Aβ-induced impairments in aged mouse brain. SOD1 overexpression protects against the in vitro neurotoxicity induced by Aβ [59]. In vivo, coexpression of hSOD1 with an APP transgene protects against the lethal effects of APP [60]. SOD2 is enriched around amyloid plaques [61, 62] and brain microvessels [63] in hAPP transgenic mice but decreased in AD brains overall [64]. Esposito et al. has shown that partial reduction in the main mitochondrial superoxide scavenger SOD2 using SOD2+/− mice accelerates the onset of hAPP/Aβ-dependent behavioral abnormalities and worsens a range of AD-related molecular and pathological alterations [65]. On the other hand, overexpression of SOD2 reduces hippocampal superoxide and prevents memory deficits in the Tg2576 mouse model of AD that overexpresses the hAPP carrying the Swedish mutation (K670N:M671L) [66].
4. Reducing ROS by Medical Gases
The generation of ROS and related oxidative damage are believed to be involved in the pathogenesis of neurodegenerative diseases. The main ROS involved in the pathogenesis of neurodegeneration are O2 •−, H2O2, and the highly reactive hydroxyl radical (HO•). Recently, there have been increasing reports showing that medical gases, such as carbon monoxide (CO), nitric oxide (NO), and hydrogen sulfide (H2S) as well as molecular hydrogen (H2), might overcome the harmful damage produced by oxidative stress [67, 68]. These gases directly eliminate ROS, or induce resistant proteins and antioxidant enzymes to antagonize oxidative stress Table 1.
Table 1.
Reducing ROS by medical gases, hydrogen (H2), carbon monoxide (CO), and hydrogen sulphide (H2S). These gases can increase endogenous antioxidant enzymes and stress response protein such as HO-1, SOD, and Hsp72. Hydrogen and hydrogen sulphide can directly react with ROS and show radical scavenging effects.
| Medical gases | Direct reaction to ROS | Reference | Endogenous cytoprotective protein induction | Reference |
|---|---|---|---|---|
| Hydrogen (H2) |
Radical scavenging HO• + H2→H2O + H• |
[67] | Increase superoxide dismutase (SOD) | [81] |
| Increase bilirubin ≒ induction of HO-1 |
[81] | |||
|
| ||||
| Activation of p38 MAPK signaling p38→HSF1→Hsp72 |
[59] | |||
| Carbon monoxide (CO) | Activation of Nrf2 pathway Nrf-2→HO-1 |
[61] | ||
| Induce superoxide dismutase (SOD2) and HO-1 | [62] | |||
|
| ||||
| Hydrogen sulphide (H2S) | Radical scavenging | [64] | Upregulation of cytoprotective genes including HO-1 | [65] |
| 2O2 •− + H2S→HS–SH + O2 + 2OH− | [63] | [66] | ||
4.1. Carbon Monoxide
CO is a diatomic molecule and is soluble in aqueous media and organic solvents [69]. Not only exogenous environmental exposure but also endogenous production during heme metabolism are major sources of CO from primitive prokaryocytes to human [69, 70]. Endogenous production of CO is highly associated with HO-1 activity which induces enzymatic degradation of heme. HO breaks the alpha-methylene carbon bond of the porphyrin ring using NADPH and molecular O2 in a reaction that releases equimolar amount of biliverdin, iron, and CO [71, 72].
Recent studies have revealed that CO serves as an intrinsic signaling molecule and shows anti-inflammatory and antiapoptotic effects. These effects of CO are mediated by p38 mitogen-activated protein kinase (MAPK) signaling, which is activated in response to physical and chemical stress inducers including oxidative stress, UV light, ischemia, and proinflammatory cytokines [73]. Activation of p38 also mediates the induction of heat shock protein (Hsp72) via its transcriptional factor, heat shock factor-1, leading to the cytoprotective effects [74].
Exogenous CO also activates Nrf2 pathway and decreased infarct size in an ischemia/reperfusion model [75]. Nrf2 activation can coordinately upregulate expression of several antioxidative enzymes recognized to play important roles in combating oxidative stress, including HO-1. Endogenous CO, which is produced by heme degradation, induces ROS-dependent signal transduction in the mitochondrial SOD2 and in HO-1 itself [76].
4.2. Hydrogen Sulfide
H2S is a flammable, water-soluble gas with a smell of rotten eggs and is known as a toxic gas and as an environmental hazard. The production of H2S from L-cysteine is catalysed primarily by two enzymes, cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS). Although exposure to higher levels (~mM) of H2S is cytotoxic (free radical generation, glutathione depletion, intracellular iron increase, and mitochondrial cell death signal), lower concentration (~μM) of H2S shows cytoprotective (antinecrotic and antiapoptotic) effects.
Biochemical analysis has revealed that sulphide shows a direct antioxidant reaction with one- or two-electron molecules (one-electron molecules: •NO2, •OH, CO3 •−, two-electron molecules: peroxynitrite, hydrogen peroxide, hypochlorite, taurine, and chloramine) as well as other low-molecular-weight thiol molecules such as cysteine and glutathione. Although sulphide is not a preferential target for radicals or oxidants due to its low concentration in vivo, it can serve as a direct antioxidant [77, 78]. H2S can also induce upregulation of transcription for anti-inflammatory and cytoprotective genes including HO-1 [79, 80]. By upregulating HO-1 expression, H2S can trigger the production of CO, which shows anti-inflammatory and antiapoptotic effects.
4.3. Hydrogen
Since the first striking evidence indicating that molecular hydrogen acts as an antioxidant and inhalation of hydrogen-containing gas reduces ischemic injury in brain [81], there have been increasing numbers of reports which support the therapeutic properties of hydrogen against oxidative stress-related diseases and damages in brain [82, 83], liver [84], intestinal graft [85], myocardial injury [86, 87], and atherosclerosis [88]. Hydrogen can be taken up by inhalation of hydrogen-containing air (hydrogen gas) or drinking hydrogen-containing water (hydrogen water). One hour after the start of inhalation of hydrogen gas, hydrogen can be detected in blood, at levels of 10 μM in arterial blood [81]. The content of hydrogen can be measured even after intake of hydrogen water by a catheter, which yields 5 μM in arterial blood calculated after 3 min of hydrogen water incorporation [82]. Taking into account its continuous intake, it is easier and safer to drink hydrogen water than inhaling hydrogen gas.
We have previously showed that H2 in drinking water reduced the loss of dopaminergic neurons in MPTP-induced Parkinson's disease (PD) mice [89]. The therapeutic effects of H2 water were observed in another PD model, 6-OHDA-treated rats [90]. In these animal models, administration of neurotoxins decreased the number of dopaminergic neurons in the substantia nigra pars compacta (SNpc), as well as dopaminergic nerve terminal fibers in the striatum. However, taking H2 water significantly reduced the loss of both neuronal cell bodies and fibers compared with the controls drinking normal water. Mice chronically treated with MPTP using an osmotic pump also showed behavioral impairments observed by open-field test [91], and rats administered with 6-OHDA showed behavioral impairments assessed by the rotarod test. Hydrogen improved these behavioral impairments in both of these animal models of PD.
In the first report [81], H2 selectively reduced cytotoxic •OH radicals, whereas the production of other radicals such as superoxide, hydrogen peroxide, and nitric oxide was not altered. This selectivity was verified in a cell-free system, and in particular, the preference for scavenging •OH rather than superoxide was confirmed in PC12 cell culture system. According to Setsukinai et al. [92], both •OH and peroxynitrite (ONOO−) are much more reactive than other ROS. This would explain why H2 shows a selective reaction with only the strongest radicals, both in the cell-free system and in PC12 cells.
Especially, •OH overproduction in oxidative and neurotoxic reaction by MPTP leads to lipid peroxidation in nigral dopaminergic neurons prior to cell death, observed by 4-hydroxynonenal (4-HNE) immunostaining, the markers of membrane lipid peroxidation. Immunoreactivity of 4-HNE in MPTP-treated mice is increased by three times compared to that in saline-treated mice [89], which is similar to the previous report of 4-HNE protein levels in substantia nigra observed at the same time after MPTP administration using HPLC [93]. H2 water significantly reduces the formation of 4-HNE in dopaminergic neurons in substantia nigra to the level of control [89]. MPTP-induced loss of dopaminergic neuron is associated with the accumulation of 8-oxo-7, 8-dihydroguanine (8-oxoG) in the dopaminergic neurons. H2 water significantly decreased this accumulation in the striatum [89]. 8-oxoG is the major oxidized form of guanine in DNA, RNA and nucleotide pool by •OH and is accumulated in both mitochondrial and nuclear DNA; their nomenclature are mt8oxo-G and nu8-oxoG, respectively. Mt8oxoG accumulates in the striatum which is rich in mitochondria in nerve terminals of dopaminergic neurons projecting from the substantia nigra. Although nu8oxoG was not detected in the nigral cell nucleus, H2 water might prevent the mt8oxoG-induced cellular apoptotic signals, not just reduce •OH in the dopaminergic nerve terminal. On the other hand, the increase in O2 •−, which was detected by the O2 •− indicator, dihydroethidium (DHE), was not significantly decreased by H2 water [89]. Although H2 prevented superoxide formation in brain slices in hypoxia/reperfusion injury [94], H2 water might show a preferential reduction of •OH during the protection of dopaminergic neurons.
Initial evidence suggests that H2 protects cells and tissues against strong oxidative stress by scavenging •OH [81]. Also, H2 was effective when it was inhaled during reperfusion; when H2 was inhaled just during the initial ischemia (not in the reperfusion stage), infarct volume was not significantly decreased. It was shown that hydrogen in the brain decreased immediately after stopping inhalation and completely disappeared within 10 min [89], indicating that the effect of hydrogen can be observed only during the period when the oxidative insults occur. According to a previous report [82], H2 could be detected in the blood 3 minutes after administration of H2 water into the stomach. However, unpublished data showed that the half-life of H2 in the muscle in rats was approximately 20 minutes after the administration of H2 gas. Taking these reports into consideration, H2 in the brain and other tissues does not stay long enough to exert its ability as an antioxidant to ROS directly. Therefore, it is unlikely that direct reaction of H2 itself with ROS plays a major role in the neuroprotection, especially with H2 in drinking water, even though H2 itself has the ability to reduce •OH preferentially. In accordance with this hypothesis, previous reports from Nakao et al. has demonstrated that drinking hydrogen water increased the amount of antioxidant enzyme, superoxide dismutase (SOD) [95], an endogenous defensive system against ROS-induced cellular damage. It was also reported that H2-water increases total bilirubin for four to eight weeks compared to baseline. Bilirubin is produced by the catalytic reaction of HO-1, and degradation of heme generates bilirubin as well as CO and free iron. Therefore, taking these observations into consideration, there seem to be other mechanisms for protective effect of H2 in drinking water, different from that exerted by H2 inhalation. It is possible that drinking H2 water has not only the ability to reduce cytotoxic radicals, but also brings into play novel mechanisms which are related to antioxidative defense system.
5. Conclusion
Recent advance in understanding of the regulation of antioxidant enzyme expression by transcriptional factors has given us the possibility that we can overcome several diseases mediated or induced by oxidative stress. As discussed above, transcriptional upregulation of several antioxidant enzymes like HO-1 and SOD might be beneficial for several neurodegenerative diseases such as AD, PD, HD, and ALS. Several phytochemicals (resveratrol, curcumin, flavonoids, carnosol, etc.) and endogenous mediators (15-deoxy-Δ12,14-PGJ2) can upregulate the antioxidants via transcription factors such as Nrf2, NF-κB, and AP-1 [24]. Surprisingly, HO-1 and SOD are increased by CO, H2 and H2S, although we cannot say whether these gases accelerate the stress response signaling or some transcriptional regulation system mediated by Nrf2 and PGC-1α. Together with the fact that H2, and H2S themselves have the ability to react with ROS directly, we strongly suggest that these gases can buffer the ROS and in addition might prevent and/or protect the neurons from oxidative stress damages in neurodegenerative diseases.
Acknowledgments
The authors thank Professor D. A. Brown (University College London, UK) for valuable suggestion and reading the paper. This work was supported by Grants-in-Aid for Scientific Research of Japan Society for Promotion of Science.
References
- 1.Papa S. Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochimica et Biophysica Acta. 1996;1276(2):87–105. doi: 10.1016/0005-2728(96)00077-1. [DOI] [PubMed] [Google Scholar]
- 2.Boveris A, Oshino N, Chance B. The cellular production of hydrogen peroxide. Biochemical Journal. 1972;128(3):617–630. doi: 10.1042/bj1280617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stowe DF, Camara AKS. Mitochondrial reactive oxygen species production in excitable cells: modulators of mitochondrial and cell function. Antioxidants and Redox Signaling. 2009;11(6):1373–1414. doi: 10.1089/ars.2008.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy PH. Mitochondrial medicine for aging and neurodegenerative diseases. NeuroMolecular Medicine. 2008;10(4):291–315. doi: 10.1007/s12017-008-8044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Medical Hypotheses. 2004;63(1):8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF. Mitochondria take center stage in aging and neurodegeneration. Annals of Neurology. 2005;58(4):495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 7.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annual Review of Genetics. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145(4):1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- 9.Miura Y, Endo T. Survival responses to oxidative stress and aging. Geriatrics and Gerontology International. 2010;10(supplement 1):S1–S9. doi: 10.1111/j.1447-0594.2010.00597.x. [DOI] [PubMed] [Google Scholar]
- 10.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ney PA, Andrews NC, Jane SM, et al. Purification of the human NF-E2 complex: cDNA cloning of the hematopoietic cell-specific subunit and evidence for an associated partner. Molecular and Cellular Biology. 1993;13(9):5604–5612. doi: 10.1128/mcb.13.9.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrews NC, Erdjument-Bromage H, Davidson MB, Tempst P, Orkin SH. Erythroid transcription factor NF-E2 is a haematopoietic-specific basic-leucine zipper protein. Nature. 1993;362(6422):722–728. doi: 10.1038/362722a0. [DOI] [PubMed] [Google Scholar]
- 13.Igarashi K, Kataoka K, Itoh K, Hayashi N, Nishizawa M, Yamamoto M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature. 1994;367(6463):568–572. doi: 10.1038/367568a0. [DOI] [PubMed] [Google Scholar]
- 14.Igarashi K, Itoh K, Motohashi H, et al. Activity and expression of murine small Maf family protein MafK. The Journal of Biological Chemistry. 1995;270(13):7615–7624. doi: 10.1074/jbc.270.13.7615. [DOI] [PubMed] [Google Scholar]
- 15.Rushmore TH, Pickett CB. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. The Journal of Biological Chemistry. 1990;265(24):14648–14653. [PubMed] [Google Scholar]
- 16.Friling RS, Bensimon A, Tichauer Y, Daniel V. Xenobiotic-inducible expression of murine glutathione S-transferase Ya subunit gene is controlled by an electrophile-responsive element. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(16):6258–6262. doi: 10.1073/pnas.87.16.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Advances in Enzyme Regulation. 2006;46(1):113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 18.Johnson JA, Johnson DA, Kraft AD, et al. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Annals of the New York Academy of Sciences. 2008;1147:61–69. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Innamorato NG, Rojo AI, García-Yagüe ÁJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. Journal of Immunology. 2008;181(1):680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 20.Hintze KJ, Keck AS, Finley JW, Jeffery EH. Induction of hepatic thioredoxin reductase activity by sulforaphane, both in Hepa1c1c7 cells and in male Fisher 344 rats. Journal of Nutritional Biochemistry. 2003;14(3):173–179. doi: 10.1016/s0955-2863(02)00282-6. [DOI] [PubMed] [Google Scholar]
- 21.Ishii T, Itoh K, Sato H, Bannai S. Oxidative stress-inducible proteins in macrophages. Free Radical Research. 1999;31(4):351–355. doi: 10.1080/10715769900300921. [DOI] [PubMed] [Google Scholar]
- 22.Favreau LV, Pickett CB. The rat quinone reductase antioxidant response element. Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines. The Journal of Biological Chemistry. 1995;270(41):24468–24474. doi: 10.1074/jbc.270.41.24468. [DOI] [PubMed] [Google Scholar]
- 23.Prestera T, Talalay P, Alam J, Ahn YI, Lee PJ, Choi AM. Parallel induction of heme oxygenase-1 and chemoprotective phase 2 enzymes by electrophiles and antioxidants: regulation by upstream antioxidant-responsive elements (ARE) Molecular Medicine. 1995;1(7):827–837. [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrandiz ML, Devesa I. Inducers of heme oxygenase-1. Current Pharmaceutical Design. 2008;14(5):473–486. doi: 10.2174/138161208783597399. [DOI] [PubMed] [Google Scholar]
- 25.Kanninen K, Heikkinen R, Malm T, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(38):16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. Journal of Neurochemistry. 2002;81(6):1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- 27.Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. The Journal of Neuroscience. 2008;28(50):13574–13581. doi: 10.1523/JNEUROSCI.4099-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen PC, Vargas MR, Pani AK, et al. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: critical role for the astrocyte. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2933–2938. doi: 10.1073/pnas.0813361106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Calkins MJ, Jakel RJ, Johnson DA, Chan K, Yuen WK, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(1):244–249. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schipper HM, Song W, Zukor H, Hascalovici JR, Zeligman D. Heme oxygenase-1 and neurodegeneration: expanding frontiers of engagement. Journal of Neurochemistry. 2009;110(2):469–485. doi: 10.1111/j.1471-4159.2009.06160.x. [DOI] [PubMed] [Google Scholar]
- 31.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40(2):427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 32.Shastry BS. Neurodegenerative disorders of protein aggregation. Neurochemistry International. 2003;43(1):1–7. doi: 10.1016/s0197-0186(02)00196-1. [DOI] [PubMed] [Google Scholar]
- 33.Song W, Patel A, Qureshi HY, Han D, Schipper HM, Paudel HK. The Parkinson disease-associated A30P mutation stabilizes α-synuclein against proteasomal degradation triggered by heme oxygenase-1 over-expression in human neuroblastoma cells. Journal of Neurochemistry. 2009;110(2):719–733. doi: 10.1111/j.1471-4159.2009.06165.x. [DOI] [PubMed] [Google Scholar]
- 34.Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): transcriptional coactivator and metabolic regulator. Endocrine Reviews. 2003;24(1):78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 35.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 36.Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes and Development. 2004;18(4):357–368. doi: 10.1101/gad.1177604. [DOI] [PubMed] [Google Scholar]
- 37.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiological Reviews. 2008;88(2):611–638. doi: 10.1152/physrev.00025.2007. [DOI] [PubMed] [Google Scholar]
- 38.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 39.Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1α by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127(1):59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Martin E, Betuing S, Pagès C, et al. Mitogen- and stress-activated protein kinase 1-induced neuroprotection in Huntington’s disease: role on chromatin remodeling at the PGC-1-alpha promoter. Human Molecular Genetics. 2011;20(12):2422–2434. doi: 10.1093/hmg/ddr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mudo G, Makela J, Di Liberto V, et al. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cellular and Molecular Life Sciences. 2012;69(7, article 1153) doi: 10.1007/s00018-011-0850-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lagouge M, Argmann C, Gerhart-Hines Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α . Cell. 2006;127(6):1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 43.Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(25):15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α . Proceedings of the National Academy of Sciences of the United States of America. 2007;104(29):12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Outeiro TF, Marques O, Kazantsev A. Therapeutic role of sirtuins in neurodegenerative disease. Biochimica et Biophysica Acta. 2008;1782(6):363–369. doi: 10.1016/j.bbadis.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 46.Ho DJ, Calingasan NY, Wille E, Dumont M, Beal MF. Resveratrol protects against peripheral deficits in a mouse model of Huntington’s disease. Experimental Neurology. 2010;225(1):74–84. doi: 10.1016/j.expneurol.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 47.Gao J, Wang WY, Mao YW, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466(7310):1105–1109. doi: 10.1038/nature09271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donmez G, Arun A, Chung CY, et al. SIRT1 protects against α-synuclein aggregation by activating molecular chaperones. The Journal of Neuroscience. 2012;32(1):124–132. doi: 10.1523/JNEUROSCI.3442-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.Jiang M, Wang J, Fu J, et al. Neuroprotective role of Sirt1 in mammalian models of Huntington’s disease through activation of multiple Sirt1 targets. Nature Medicine. 2012;18:153–158. doi: 10.1038/nm.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, Nguyen MD, Dobbin MM, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. The EMBO Journal. 2007;26(13):3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qin W, Yang T, Ho L, et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of alzheimer disease amyloid neuropathology by calorie restriction. The Journal of Biological Chemistry. 2006;281(31):21745–21754. doi: 10.1074/jbc.M602909200. [DOI] [PubMed] [Google Scholar]
- 52.Bonda DJ, Lee HG, Camins A, et al. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. The Lancet Neurology. 2011;10(3):275–279. doi: 10.1016/S1474-4422(11)70013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wareski P, Vaarmann A, Choubey V, et al. PGC-1α and PGC-1β regulate mitochondrial density in neurons. The Journal of Biological Chemistry. 2009;284(32):21379–21385. doi: 10.1074/jbc.M109.018911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson WT, Johnson LAK, Lukaski HC. Serum superoxide dismutase 3 (extracellular superoxide dismutase) activity is a sensitive indicator of Cu status in rats. Journal of Nutritional Biochemistry. 2005;16(11):682–692. doi: 10.1016/j.jnutbio.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 55.McCord JM. Iron- and manganese-containing superoxide dismutases: structure, distribution, and evolutionary relationships. Advances in Experimental Medicine and Biology. 1976;74:540–550. doi: 10.1007/978-1-4684-3270-1_45. [DOI] [PubMed] [Google Scholar]
- 56.McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein) The Journal of Biological Chemistry. 1969;244(22):6049–6055. [PubMed] [Google Scholar]
- 57.Marklund SL. Human copper-containing superoxide dismutase of high molecular weight. Proceedings of the National Academy of Sciences of the United States of America. 1982;79(24 I):7634–7638. doi: 10.1073/pnas.79.24.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines. Journal of Clinical Investigation. 1984;74(4):1398–1403. doi: 10.1172/JCI111550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Celsi F, Ferri A, Casciati A, et al. Overexpression of superoxide dismutase 1 protects against β-amyloid peptide toxicity: effect of estrogen and copper chelators. Neurochemistry International. 2004;44(1):25–33. doi: 10.1016/s0197-0186(03)00101-3. [DOI] [PubMed] [Google Scholar]
- 60.Iadecola C, Zhang F, Niwa K, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nature Neuroscience. 1999;2(2):157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- 61.Blanchard V, Moussaoui S, Czech C, et al. Time sequence of maturation of dystrophic neurites associated with Aβ deposits in APP/PS1 transgenic mice. Experimental Neurology. 2003;184(1):247–263. doi: 10.1016/s0014-4886(03)00252-8. [DOI] [PubMed] [Google Scholar]
- 62.Aucoin JS, Jiang P, Aznavour N, et al. Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer’s disease. Neuroscience. 2005;132(1):73–86. doi: 10.1016/j.neuroscience.2004.11.047. [DOI] [PubMed] [Google Scholar]
- 63.Tong XK, Nicolakakis N, Kocharyan A, Hamel E. Vascular remodeling versus amyloid β-induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer’s disease. The Journal of Neuroscience. 2005;25(48):11165–11174. doi: 10.1523/JNEUROSCI.4031-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Omar RA, Chyan YJ, Andorn AC, Poeggeler B, Robakis NK, Pappolla MA. Increased expression but reduced activity of antioxidant enzymes in Alzheimer’s disease. Journal of Alzheimer’s Disease. 1999;1(3):139–145. doi: 10.3233/jad-1999-1301. [DOI] [PubMed] [Google Scholar]
- 65.Esposito L, Raber J, Kekonius L, et al. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. The Journal of Neuroscience. 2006;26(19):5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(32):13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fujita K, Nakabeppu Y, Noda M. Therapeutic effects of hydrogen in animal models of Parkinson’s disease. Parkinson’s Disease. 2011;2011:9 pages. doi: 10.4061/2011/307875.307875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Noda M, Fujita K, C. H. Lee, Yoshioka T. The principle and the potential approach to ROS-dependent cytotoxicity by non-pharmaceutical therapies: optimal use of medical gases with antioxidant properties. Current Pharmaceutical Design. 2011;17(22):2253–2263. doi: 10.2174/138161211797052600. [DOI] [PubMed] [Google Scholar]
- 69.Von Burg R. Carbon monoxide. Journal of Applied Toxicology. 1999;19(5):379–386. doi: 10.1002/(sici)1099-1263(199909/10)19:5<379::aid-jat563>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 70.Sjostrand T. Early studies of CO production. Annals of the New York Academy of Sciences. 1970;174(1):5–10. doi: 10.1111/j.1749-6632.1970.tb49767.x. [DOI] [PubMed] [Google Scholar]
- 71.Maines MD. The heme oxygenase system: update 2005. Antioxidants and Redox Signaling. 2005;7(11-12):1761–1766. doi: 10.1089/ars.2005.7.1761. [DOI] [PubMed] [Google Scholar]
- 72.Piantadosi CA. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radical Biology and Medicine. 2008;45(5):562–569. doi: 10.1016/j.freeradbiomed.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brancho D, Tanaka N, Jaeschke A, et al. Mechanism of p38 MAP kinase activation in vivo. Genes and Development. 2003;17(16):1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim HP, Wang X, Zhang J, et al. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38β MAPK and heat shock factor-1. Journal of Immunology. 2005;175:2622–2629. doi: 10.4049/jimmunol.175.4.2622. [DOI] [PubMed] [Google Scholar]
- 75.Wang B, Cao W, Biswal S, Dore S. Carbon monoxide-activated Nrf2 pathway leads to protection against permanent focal cerebral ischemia. Stroke. 2011;42(9):2605–2610. doi: 10.1161/STROKEAHA.110.607101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thom SR, Fisher D, Xu YA, Notarfrancesco K, Lschiropoulos H. Adaptive responses and apoptosis in endothelial cells exposed to carbon monoxide. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(3):1305–1310. doi: 10.1073/pnas.97.3.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carballal S, Trujillo M, Cuevasanta E, et al. Reactivity of hydrogen sulfide with peroxynitrite and other oxidants of biological interest. Free Radical Biology and Medicine. 2011;50(1):196–205. doi: 10.1016/j.freeradbiomed.2010.10.705. [DOI] [PubMed] [Google Scholar]
- 78.Whiteman M, Armstrong JS, Chu SH, et al. The novel neuromodulator hydrogen sulfide: an endogenous peroxynitrite “scavenger”? Journal of Neurochemistry. 2004;90(3):765–768. doi: 10.1111/j.1471-4159.2004.02617.x. [DOI] [PubMed] [Google Scholar]
- 79.Qingyou Z, Junbao D, Weijin Z, Hui Y, Chaoshu T, Chunyu Z. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochemical and Biophysical Research Communications. 2004;317(1):30–37. doi: 10.1016/j.bbrc.2004.02.176. [DOI] [PubMed] [Google Scholar]
- 80.Szabo C. Hydrogen sulphide and its therapeutic potential. Nature Reviews Drug Discovery. 2007;6(11):917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 81.Ohsawa I, Ishikawa M, Takahashi K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nature Medicine. 2007;13(6):688–694. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 82.Nagata K, Nakashima-Kamimura N, Mikami T, Ohsawa I, Ohta S. Consumption of molecular hydrogen prevents the stress-induced impairments in hippocampus-dependent learning tasks during chronic physical restraint in mice. Neuropsychopharmacology. 2009;34(2):501–508. doi: 10.1038/npp.2008.95. [DOI] [PubMed] [Google Scholar]
- 83.Gu Y, Huang CS, Inoue T, et al. Drinking hydrogen water ameliorated cognitive impairment in senescence-accelerated mice. Journal of Clinical Biochemistry and Nutrition. 2010;46(3):269–276. doi: 10.3164/jcbn.10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fukuda KI, Asoh S, Ishikawa M, Yamamoto Y, Ohsawa I, Ohta S. Inhalation of hydrogen gas suppresses hepatic injury caused by ischemia/reperfusion through reducing oxidative stress. Biochemical and Biophysical Research Communications. 2007;361(3):670–674. doi: 10.1016/j.bbrc.2007.07.088. [DOI] [PubMed] [Google Scholar]
- 85.Buchholz BM, Kaczorowski DJ, Sugimoto R, et al. Hydrogen inhalation ameliorates oxidative stress in transplantation induced intestinal graft injury. American Journal of Transplantation. 2008;8(10):2015–2024. doi: 10.1111/j.1600-6143.2008.02359.x. [DOI] [PubMed] [Google Scholar]
- 86.Nakao A, Kaczorowski DJ, Wang Y, et al. Amelioration of rat cardiac cold ischemia/reperfusion injury with inhaled hydrogen or carbon monoxide, or both. Journal of Heart and Lung Transplantation. 2010;29(5):544–553. doi: 10.1016/j.healun.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 87.Hayashida K, Sano M, Ohsawa I, et al. Inhalation of hydrogen gas reduces infarct size in the rat model of myocardial ischemia-reperfusion injury. Biochemical and Biophysical Research Communications. 2008;373(1):30–35. doi: 10.1016/j.bbrc.2008.05.165. [DOI] [PubMed] [Google Scholar]
- 88.Ohsawa I, Nishimaki K, Yamagata K, Ishikawa M, Ohta S. Consumption of hydrogen water prevents atherosclerosis in apolipoprotein E knockout mice. Biochemical and Biophysical Research Communications. 2008;377(4):1195–1198. doi: 10.1016/j.bbrc.2008.10.156. [DOI] [PubMed] [Google Scholar]
- 89.Fujita K, Seike T, Yutsudo N, et al. Hydrogen in drinking water reduces dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. PLoS ONE. 2009;4(9) doi: 10.1371/journal.pone.0007247.e7247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fu Y, Ito M, Fujita Y, et al. Molecular hydrogen is protective against 6-hydroxydopamine-induced nigrostriatal degeneration in a rat model of Parkinson’s disease. Neuroscience Letters. 2009;453(2):81–85. doi: 10.1016/j.neulet.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 91.Fornai F, Schlüter OM, Lenzi P, et al. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and α-synuclein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(9):3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Setsukinai KI, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. The Journal of Biological Chemistry. 2003;278(5):3170–3175. doi: 10.1074/jbc.M209264200. [DOI] [PubMed] [Google Scholar]
- 93.Liang LP, Huang J, Fulton R, Day BJ, Patel M. An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in vivo. The Journal of Neuroscience. 2007;27(16):4326–4333. doi: 10.1523/JNEUROSCI.0019-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sato Y, Kajiyama S, Amano A, et al. Hydrogen-rich pure water prevents superoxide formation in brain slices of vitamin C-depleted SMP30/GNL knockout mice. Biochemical and Biophysical Research Communications. 2008;375(3):346–350. doi: 10.1016/j.bbrc.2008.08.020. [DOI] [PubMed] [Google Scholar]
- 95.Nakao A, Toyoda Y, Sharma P, Evans M, Guthrie N. Effectiveness of hydrogen rich water on antioxidant status of subjects with potential metabolic syndrome—an open label pilot study. Journal of Clinical Biochemistry and Nutrition. 2010;46(2):140–149. doi: 10.3164/jcbn.09-100. [DOI] [PMC free article] [PubMed] [Google Scholar]