

Cytoplasmic calcium serves as a ubiquitous signal for acute cellular activation and for regulation of important cellular processes such as cell growth, division, differentiation, and even cell death and apoptosis. Increases in cytoplasmic calcium or calcium signals can be generated either by release of calcium from intracellular stores or by influx of calcium across the plasma membrane, but commonly by both means. The release of intracellular calcium comes from the endoplasmic reticulum or its specialized counterpart in muscle, the sarcoplasmic reticulum, and is generally signaled by the formation or influx of small mediators such as inositol 1,4,5-trisphosphate (IP3) (1), cyclic ADP ribose (2), and even calcium itself (3). Mechanisms for controlling calcium influx are somewhat more varied (4). Calcium influx can be more directly controlled by the membrane potential (5) or by the binding of extracellular neurotransmitters directly to the channels (6). Calcium influx can be signaled by second messengers such as cyclic nucleotides (7); however, in the case of receptors coupled to the phospholipase C pathway, the classical messengers of this pathway, IP3 and diacylglycerol, do not appear to be the primary mediators of activated calcium entry. Rather, in most instances, the signal for entry is somehow derived from the IP3-mediated depletion of calcium from intracellular stores, a process called “capacitative calcium entry” or “store-operated calcium entry” (8, 9).
Two key issues regarding the mechanism of capacitative calcium entry have been (i) the nature of the signal from the endoplasmic reticulum that activates the channel, and (ii) the molecular identity of the channels themselves. It has been generally thought that the answer to the second question, the molecular identity of the channels, would likely lead to knowledge of the mechanism of signaling. Recent findings suggest that this may indeed be the case. In a report appearing in this issue of PNAS, Boulay et al. (10) provide compelling evidence for the involvement of a member of the TRP family of channel proteins in this signaling pathway, and for signaling activation of the TRP channels through interaction with IP3 receptors.
Mammalian TRP proteins are homologues of the Drosophila photoreceptor mutants TRP and TRPL. There are at least seven mammalian genes, designated TRP1 through TRP7 (11, 12). Expression of these genes in various cell lines has resulted in varying patterns of response, including constitutive activity, augmentation of capacitative calcium entry, apparent direct activation by receptors, activation by IP3, and activation by diacylglycerol (13). Thus, despite considerable speculation regarding the function of these proteins in capacitative calcium entry, none of the available evidence directly implicates one or more of these gene products in this pathway in nontransfected cells.
A number of mechanisms of activation of capacitative calcium entry have been proposed. The two major categories of mechanisms include those involving a diffusible messenger (14, 15) and a more direct communication with the endoplasmic reticulum involving interaction of the plasma membrane channels with closely underlying IP3 receptors (16, 17). The latter, conformational coupling model was originally proposed by Irvine (16) based on a suggested homology with the coupling that occurs in skeletal muscle between the ryanodine receptor calcium release channels in the sarcoplasmic reticulum and surface membrane (t-tubule) dihydropyridine receptor calcium channels.
In support of the conformational coupling model, recent evidence indicates that the activation of capacitative calcium entry channels requires close physical interactions between endoplasmic reticulum and plasma membrane constituents (18, 19). There is also recent evidence that TRP3, when exogenously expressed in cells, can be activated by an IP3 receptor-dependent conformational coupling mechanism. Kiselyov et al. (20) demonstrated that depletion of intracellular Ca2+ stores activated single channels in the plasma membrane of cells stably transfected with the gene for human TRP3. On excision, channel activity was lost but could be restored by IP3. With more extensive washing, restoration of channel activity required addition of both IP3 and the IP3 receptor. These results suggest that expressed TRP3 is gated by IP3-liganded IP3 receptors, consistent with the conformation coupling model. In a more recent study, Kiselyov et al. (21) demonstrated that TRP3 activation depended on a limited region of the IP3 receptor, spanning the IP3 binding domain.
In the study by Boulay et al. (10), evidence was first obtained for direct protein–protein interaction of TRP3 and IP3 receptors. In cells stably expressing epitope-tagged TRP3 or its homolog TRP6, IP3 receptor could be detected in immunoprecipitates of TRP, and, reciprocally, TRPs could be detected in immunoprecipitates of the IP3 receptor. So next, Boulay et al. sought to determine the sites of interaction of these proteins and to determine the functional significance of these interactions. To accomplish this, they generated glutathione S-transferase fusions of peptides coded by cDNA fragments of the human type 3 IP3 receptor and human TRP3 and used a series of glutathione S-transferase pull-down experiments to map minimal interacting domains on the two proteins. A sequence in the C-terminal domain of TRP3 consisting of 54 amino acids (designated C7, from position 742 to position 795) was identified, as well as a shorter sequence of 21 amino acids (C8, 777 to 797), which strongly bound specific sequences within the IP3 receptor. A 289-aa sequence just downstream from the IP3 binding domain of the IP3 receptor, designated F2r (positions 638 through 926), strongly bound TRP3, and within this sequence, a number of smaller interacting sequences were identified [see Boulay et al. (10)].
Boulay et al. next transfected HEK-293T cells with fluorescent protein fusions of three of these peptides and examined the effects of the potential TRP3 and IP3 receptor binding peptides on activation of capacitative calcium entry. The C7 peptide inhibited entry by 25–30%, whether the entry was activated by a phospholipase C-linked agonist or by depletion of intracellular calcium stores with the calcium pump inhibitor, thapsigargin. Likewise, F2l, an interacting subfragment of F2r, inhibited entry by a lesser, albeit significant, degree. Another interacting subfragment of F2r, F2 g, caused slight but significant augmentation of calcium entry. This is potentially interesting given the well documented calcium-dependent negative feedback regulation of capacitative calcium entry (22).
The findings of Boulay et al. are significant because they provide evidence for involvement of a TRP protein in calcium entry in a cell whose TRP and IP3 receptors have not been manipulated. Similarly, they provide evidence for interaction between TRP and IP3 receptors in the endogenous cellular mechanism of activation of entry. The findings complement previous work with cells transfected with exogenous TRP proteins that demonstrated a requirement for both IP3 receptors and IP3 for activation of TRP-containing channels (20, 21).
As pointed out by Boulay et al. (10), the simplest mechanism by which signaling might occur from depleted calcium stores would involve a calcium-dependent conformational change in the IP3 receptor. This was first predicted by Irvine (16), and, subsequently, a number of investigators documented effects of endoplasmic reticulum luminal calcium on IP3 receptor binding and function (23, 24). A conformational change in the receptor as calcium levels in the endoplasmic reticulum fall would be transmitted to a TRP component of the store-operated channel, presumably through interaction of C7 and F2r domains.
A major unresolved problem is the role played by IP3 in this process. Previous studies have clearly demonstrated a requirement for IP3 to gate these channels (20, 21, 25, 26). However, it is equally clear that depletion of calcium stores by means that do not involve activation of phospholipase C can induce maximal capacitative calcium entry (cf. ref. 9). A possible solution to this apparent conundrum involves spatial compartmentalization of steps in the signaling process. By analogy with the situation in the Drosophila photoreceptor, phospholipase C may be associated with TRP through mutual interaction with a scaffolding protein, thus creating a spatially compact signaling complex between phospholipase C, TRP, and an IP3 receptor (27, 28). Within this spatially restricted domain, IP3 provided by the basal activity of phospholipase C may be sufficient to satisfy the IP3 requirement of the receptors that regulate TRP containing channels. In the absence of receptor activation of phospholipase C, IP3-metabolizing enzymes would prevent diffusion of IP3 beyond this spatial restricted compartment, thereby preventing significant depletion of endoplasmic reticulum stores (29). Thus, IP3 may be a required but constitutive component of this signaling pathway, but would not usually function as a regulator or activator of entry. The signal for channel activation is the depletion of endoplasmic reticulum stores, the sine qua non of capacitative calcium entry.
Fig. 1 depicts a current view of the regulation of capacitative calcium entry channels and the involvement of TRP and IP3 receptor proteins. In addition to these two key interacting proteins, the model hypothesizes an unknown scaffolding protein involved in assembling the capacitative calcium entry signaling complex. This could be a mammalian homolog of the PDZ-domain protein that fulfils this function in Drosophila, InaD (27, 28, 30), or possibly SNAP-25 or a similar docking protein (18). It is likely that we still do not have all of the players in this important signaling mechanisms yet, and we can look forward to a continued assembly of the pathway as additional molecular and cellular constituents are discovered.
Figure 1.
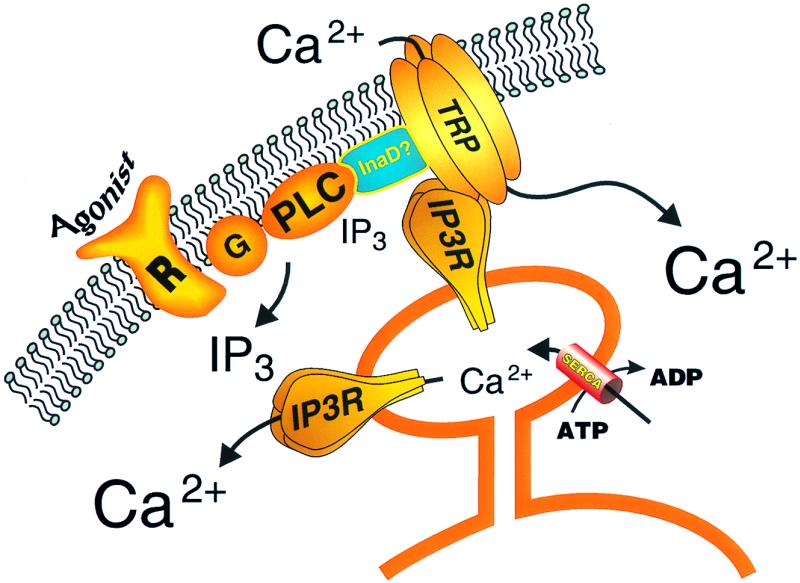
Model for regulation of capacitative calcium entry channels by IP3 receptors. Agonist activation of a surface membrane receptor (R), perhaps through a heterotrimeric G-protein (G), activates phospholipase C (PLC), leading to the production of the calcium-mobilizing messenger, IP3. IP3 releases calcium from a critical endoplasmic reticulum store. The fall in luminal calcium in this store causes a conformational change in an IP3 receptor that interacts with a TRP subunit of the capacitative calcium entry channel, causing it to open. A signaling complex containing the PLC, TRP, and IP3 receptor may require an as yet uncharacterized scaffolding protein, perhaps similar to InaD. The close spatial arrangement of this signaling complex results in a constant supply of IP3 from basal PLC activity for the IP3 receptor, such that IP3 levels are not normally limiting; rather, it is depletion of calcium from the specific intracellular store that provides the critical signal for channel activation.
Footnotes
See companion article on page 14955.
References
- 1.Berridge M J. Nature (London) 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 2.Lee H C. Physiol Rev. 1997;77:1133–1164. doi: 10.1152/physrev.1997.77.4.1133. [DOI] [PubMed] [Google Scholar]
- 3.Fabiato A. Am J Physiol. 1983;245:C1–C4. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 4.Barritt G J. Biochem J. 1999;337:153–169. [PMC free article] [PubMed] [Google Scholar]
- 5.Miller R J. J Biol Chem. 1992;267:1403–1406. [PubMed] [Google Scholar]
- 6.Burnashev N. Cell Calcium. 1998;24:325–332. doi: 10.1016/s0143-4160(98)90056-2. [DOI] [PubMed] [Google Scholar]
- 7.Zagotta W N, Siegelbaum S A. Annu Rev Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- 8.Putney J W., Jr Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 9.Putney J W., Jr . Capacitative Calcium Entry. Austin, TX: Landes Biomedical Publishing; 1997. [Google Scholar]
- 10.Boulay G, Brown D M, Qin N, Jiang M, Dietrich A, Zhu M X, Chen Z, Birnbaumer M, Mikoshiba K, Birnbaumer L. Proc Natl Acad Sci USA. 1999;96:14955–14960. doi: 10.1073/pnas.96.26.14955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M. Proc Natl Acad Sci USA. 1996;93:15195–15202. doi: 10.1073/pnas.93.26.15195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- 13.Putney J W, Jr, McKay R R. BioEssays. 1999;21:38–46. doi: 10.1002/(SICI)1521-1878(199901)21:1<38::AID-BIES5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 14.Putney J W., Jr Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 15.Randriamampita C, Tsien R Y. Nature (London) 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 16.Irvine R F. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- 17.Berridge M J. Biochem J. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yao Y, Ferrer-Montiel A V, Montal M, Tsien R Y. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- 19.Patterson R L, van Rossum D B, Gill D L. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- 20.Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Nature (London) 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 21.Kiselyov K, Mignery G A, Zhu M X, Muallem S. Mol Cell. 1999;4:423–429. doi: 10.1016/s1097-2765(00)80344-5. [DOI] [PubMed] [Google Scholar]
- 22.Parekh A B, Penner R. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 23.Missiaen L, Parys J B, DeSmedt H, Oike M, Casteels R. Mol Cell Endocrinol. 1994;98:147–156. doi: 10.1016/0303-7207(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 24.Taylor C W. Biochim Biophys Acta. 1998;1436:19–33. doi: 10.1016/s0005-2760(98)00122-2. [DOI] [PubMed] [Google Scholar]
- 25.Vaca L, Kunze D L. Am J Physiol. 1994;267:C920–C925. doi: 10.1152/ajpcell.1994.267.4.C920. [DOI] [PubMed] [Google Scholar]
- 26.Zubov A I, Kaznacheeva E V, Alexeeno V A, Kiselyov K, Muallem S, Mozhayeva G. J Biol Chem. 1999;274:25983–25985. doi: 10.1074/jbc.274.37.25983. [DOI] [PubMed] [Google Scholar]
- 27.Huber A, Sander P, Gobert A, Bähner M, Hermann R, Paulsen R. EMBO J. 1996;15:7036–7045. [PMC free article] [PubMed] [Google Scholar]
- 28.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker C S. Nature (London) 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 29.Putney J W., Jr Cell. 1999;99:5–8. doi: 10.1016/s0092-8674(00)80056-2. [DOI] [PubMed] [Google Scholar]
- 30.Philipp S, Flockerzi V. FEBS Lett. 1997;413:243–248. doi: 10.1016/s0014-5793(97)00877-6. [DOI] [PubMed] [Google Scholar]