

Abstract
Breast cancers often have deregulated hepatocyte growth factor (HGF) and c-Met signaling that results in increased tumor growth and invasion. Elucidating the mechanism responsible for HGF/c-Met action in breast cancer progression has been difficult as c-Met communicates with a number of secondary receptors that can lead to various pathological outcomes. Understanding how these secondary receptors facilitate HGF/c-Met cellular responses will aid in the development of better therapeutic treatment options for breast cancer patients with elevated HGF signaling. In the present study it was shown that the epidermal growth factor receptor (EGFR) plays a significant role in HGF/c-Met mediated biological activities indicative of advanced tumor pathology, including enhanced proliferation and invasion. The clinically relevant EGFR inhibitor gefitinib was used to determine the role of EGFR in HGF-induced proliferation and motility in several mammary carcinoma cells including PyVmT, MDA-MB-231 and 4T1. Our analyses indicated that EGFR inhibition significantly blocked HGF activation of c-Met and EGFR and that inhibition of these pathways mitigated HGF induced proliferation and motility. The data indicate that this inhibition was not through a direct effect of gefitinib on c-Met, but that EGFR is necessary for c-Met activation in the assays performed. These results provide a novel mechanism of action for EGFR as a mediator of HGF signaling thereby linking EGFR to the oncogenic potential of c-Met in mammary carcinomas cells.
Keywords: Epidermal growth factor receptor, c-Met, Hepatocyte growth factor, therapy, cross-talk, signaling, motility, growth and breast cancer
INTRODUCTION
Hepatocyte growth factor (HGF) signaling is responsible for promoting cell proliferation, morphogenesis and motility, including cell scattering.1,2 HGF is primarily secreted by stromal fibroblasts that act in a paracrine manner on the receptor tyrosine kinase (RTK), c-Met, which is expressed in epithelial cells.3,4 Under normal physiological conditions HGF mediated signaling is involved in embryonic tissue development, tissue regeneration and wound healing.5 However, increased HGF/c-Met signaling can lead to increased tumor cell growth and invasion.6,7
HGF and c-Met are both critical mediators of breast cancer progression. HGF and c-Met expression dramatically correlate with tumor pathology, showing lowest expression levels in normal tissue and benign hyperplasias while increasing in ductal carcinoma in situ and showing highest expression in invasive breast carcinomas.8 Additionally, high HGF and c-Met expression levels are independently considered as prognostic indicators for poor patient survival.9,10 In addition to predictive expression in human tumors, HGF is a potent tumor inducer in mice. Targeted expression of HGF in mouse mammary epithelium leads to metastatic adenosquamous carcinomas.11 Together these data support a role for c-Met signaling as a direct mediator in breast cancer progression, thus making c-Met a good target for therapeutic intervention. However, elucidating the mechanism responsible for HGF/c-Met action in breast cancer progression has been difficult as c-Met communicates with a number of secondary receptors that can regulate downstream signaling pathways contributing to enhanced growth and motility.12,13 Understanding how these secondary receptors facilitate HGF/c-Met cellular responses will clarify how HGF and c-Met promote breast cancer progression and aid in better therapeutic treatment options for breast cancer patients with elevated HGF signaling.
EGFR might be a likely candidate for receptor cross-talk with c-Met in breast cancers as elevated EGFR expression levels are also correlated with metastatic breast cancers.14,15 In addition EGFR can cross-talk with c-Met in a number of other cancer cell types.16-19 For example, in some tumor cells c-Met communicates through the Ras/MAPK signaling pathway with EGFR, which results in HGF-independent activation of c-Met signaling.16 Furthermore, activation c-Met and EGFR stimulate parallel pathways that elicit coordinated cellular responses including growth and motility.16,19 However, these studies only support a role for EGFR in HGF-independent c-Met signaling. The function of EGFR in HGF-dependent signaling in tumor cells is not clearly understood, though one study has suggested that EGFR plays a significant role in HGF-induced hepatocyte proliferation.20 In breast cancers HGF is clearly a significant signaling molecule that promotes tumor progression, thus understanding HGF-dependent cross-talk is important. Evidence for cooperation between EGFR and c-Met signaling pathways have previously been reported to mediate cell motility and invasion as well as proliferation in other cell types, thus we investigated whether EGFR may play a more global role in HGF-dependent signaling in mammary carcinoma cells.17,18,21
To elucidate EGFR dependence in HGF signaling we analyzed HGF-mediated proliferation, motility and invasion using EGFR tyrosine kinase inhibitors (TKIs) to evaluate HGF-induced proliferation, motility, invasion and cell scattering in several mammary carcinoma cell lines. Our analyses indicated that EGFR inhibition significantly blocked HGF activation of c-Met and EGFR, which mitigated HGF induced biological responses. The requirement of EGFR for HGF-induced biological effects may indicate that in mammary carcinoma cells HGF employs EGFR to enhance c-Met signaling, which results in increased cell proliferation and invasion. These data suggest that patients with breast cancers that have elevated HGF expression may uniquely be suited for EGFR TKI treatment as these cancers could have amplified c-Met signaling due to concurrent EGFR activation.
MATERIALS AND METHODS
Antibodies and reagents
Human recombinant TGF-a (239-A) and HGF (294-HG) were purchased from R&D Systems Inc. (Minnneapolis, MN). Gefitinib (Iressa) was provided by Alan Wakeling (AstraZeneca Pharmaceuticals, Alderley Park, UK). Erlotinib (Tarceva) was a gift from Mark Sliwkowski (Genentech, South San Francisco, CA). [3H]thymidine was purchased from Perkin Elmer Life Science (Wellesley, MA). Antibodies for murine c-Met (07-283) and murine EGFR (06-847) proteins were purchased from Upstate Biotechnology (Lake Placid, NY). The human c-Met (14571-50) antibody was purchased from Abcam Inc. (Cambridge, MA). Phosphotyrosine antibodies recognizing c-Met at Y1234/1235 (3126) and EGFR at Y1068 (2234) were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Anti-rabbit (31462) and anti-mouse (31432) horseradish peroxidase (HRP) conjugated antibodies were purchased from Pierce Biotechnology, Inc. (Rockford, IL). Enhanced chemiluminescence (ECL) kit was purchased from Amersham Pharmacia Biotechnology (Arlington Heights, IL). BioRad protein quantification kit was purchased from BioRad Laboratories (Hercules, CA). Cell Tracker live dye CMTMR (C2927) was purchased from Molecular Probes (Eugene, OR). Matrigel (356231) and 8.0 micron Boyden Chamber Transwells (354578) were purchased from BD Biosciences (Bedford, MA). Mitomycin C (M-0503), Fibronectin (F-4759) and Mayers Hematoxyalin (MHS32) were purchased from Sigma-Aldrich (St. Louis, MO). Aqua Polymount (18606) was purchased from Polyscience, Inc. (Minneapolis, MN).
Cell culture
PyVmT were generated by isolating epithelial cells from mammary tumors in PyVmT transgenic mice in our laboratory,22 human breast cancer MDA-MB-231 cells, human breast cancer MDA-MB-435S and invasive mouse mammary carcinoma 4T1 cells were purchased from American Type Culture Collection (ATCC) (Rockville, MD). Mammary carcinoma cells PyVmT, MDA-MB-231, MDA-MB-435S and 4T1 cells were maintained in Dulbeccos’s Modified Essential Media (DMEM) (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (Gemini Biosciences, Woodland Hills, CA). NMuMG (ATCC) maintained in DMEM supplemented with 10% FBS and 10 mg/ml insulin. The hematopoietic 32D cells and 32D cells stably expressing human-cMet (32D-cMet) were obtained from Dr. Bottaro, National Institute of Health (Bethesda, MD)23 and were maintained in RPMI-1640 medium (HyClone) supplemented with 15% FBS and IL-3 from WEHI-3B conditioned media, also described in.24
Cell proliferation and viability assays
PyVmT and MDA-MB-231 cells were assayed for proliferation by measuring [3H]thymidine incorporation. Cells were plated at 20,000 cells/well in 24-well plates and allowed to recover for 24 h. Cells were washed with phosphate buffered saline (PBS) three times, starved for 20 h in serum free DMEM (SF-DMEM), then incubated in SF-DMEM in the presence or absence of 20 ng/ml or 40 ng/ml HGF or 20 ng/ml TGFα plus inhibitor as indicated for 24 h. Treated cells were pulsed for 2 h with 4 mCi/well [3H]thymidine. Cell were fixed with 1 ml of 10% trichloroacetic acid (TCA) for 30 m at 25°C then washed two times with 10% TCA. DNA was solubilized by incubation in 600 ml 0.2 M NaOH for 30 m. Radioactivity of incorporated [3H]thymindine was counted using 200 μl of solubilized DNA and 4 ml scintillation fluid. To measure cell viability cells were plated at 300,000 cells/well in 6-well plates and allowed to recover for 24 h. Cells were treated with increasing doses of inhibitor as indicated for 24 h, then trypsinized and stained with Trypan Blue. Non-stained cells were then counted using a hemocytometer and measurements represent the number of live cells.
Wound closure assay
NMuMG cells were plated at a high density in 24-well plates and incubated until a confluent monolayer was achieved. A scratch was made in each well and cultures were washed with PBS twice to remove any cell debris. Cells were incubated in DMEM supplemented with 10% FBS and 10 μg/ml insulin and 5 μg/ml Mitomycin C in the presence or absence of 10 ng/ml HGF plus inhibitor as indicated for 20 h. Images were taken at 0 h and 20 h, using an Olympus CK40 inverted microscope and an Olympus DP10 digital camera. Measurements represent the area difference of the wound closure at 0 h and 20 h.
Transwell migration assay
Boyden chamber transwells were coated with fibronectin for 1 h at 37°C and blocked overnight (O/N) in 0.1% BSA/SF-DMEM at 4°C before use. PyVmT and MDA-MB-231 cells were serum starved in SF-DMEM for 24 h before plated in the upper chamber of the transwell at 30,000 cells/well. The lower chamber contained SF-DMEM in the presence or absence of HGF plus inhibitor as indicated. After 8 h, cells that attached to the upper chamber were removed with a cotton swab, and the cells that migrated to the underside were fixed in 10% neutral buffered formalin and stained with Mayer’s hematoxylin for 30 m. Stained cells were photographed using Olympus SZH10 microscope and Olympus DP10 digital camera. Cells were counted from four randomly selected fields per well and averaged.
Matrigel invasion assay
Boyden chamber transwells were coated with Matrigel for 1 h at 37°C prior to use. PyVmT, MDA-MB-231 and NMuMG cells were labeled with a red fluorochrome CMTMR (1.5 μM) for 15 m at 37°C in PBS. Labeled cells recovered O/N in 10% DMEM, then serum starved in SF-DMEM for 24 h before being plated in the upper chamber of the transwell at 30,000 cells/well. The lower chamber contained SF-DMEM in the presence or absence of HGF plus inhibitor as indicated. After 8 h (PyVmT and MDA-MB-231) or 12 h (NMuMG) cells that attached to the upper chamber were removed with a cotton swab and the cells that migrated to the underside were fixed in 10% neutral buffered formalin. Filters containing the migrated cells were then removed and mounted on 25 × 75 mm microslides (Fisher Scientific, Pittsburgh, PA) using Aqua Polymount. At least four randomly selected fields of cells were then photographed using a Zeiss Axioplan 2 inverted fluorescent microscope and Hamamatsu Orca ER camera with Improvision Openlab software. Average pixel density of labeled migrated cells was analyzed using Scion Image and Adobe photoshop software.
Scatter assay
4T1 cells were plated at 500 cells/well in a 6 well plate. Cells were maintained in DMEM supplemented with 10% FBS until single cohesive colonies were present. Cells were then washed in PBS three times to remove any cell debris and serum starved in SF-DMEM for 48 h. Cells were then incubated in SF-DMEM in the presence or absence of 60 ng/ml HGF plus inhibitor as indicated for 24 h. Cells were fixed in 10% neutral buffered formalin for 10 m at 20°C and stained with Mayer’s Hematoxylin for 30 m. Stained cells were photographed using Olympus SZH10 microscope and Olympus DP10 digital camera. Scattered cells were counted from eight colonies per well starting with colonies that had little or no scattered cells as a baseline.
Immunoblot analyses
Sub-confluent PyVmT, MDA-MB-231 and MDA-MB-435S were washed with PBS three times and serum starved in SF-DMEM O/N. 32D and 32D-cMet cells were washed in PBS three times then serum starved for 4 h. Cells were then pre-treated with inhibitor for 2 h, unless indicated otherwise, and treated with 20 ng/ml HGF. Cells were harvested in RIPA (150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris-Cl pH 8.0, 5 mM EDTA) supplemented with protease (50 μg/ml PMSF, 10 μg/ml antipain, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml chymostatin) and phosphatase (4 nM NaF, 0.1 mM Na3VO4) inhibitors. Protein concentrations in cell lysates were determined using the BioRad protein quantification kit. Equal amount of protein lysate (20–40 μg/lane) were separated by 10% SDS-PAGE and transferred to Nitrocellulose membranes. Blots were blocked in 5% Blotto solution (TBST-100 mM Tris, 0.9% NaCl, 0.1% Tween 20 + dry milk) for 2 h. Primary antibody was incubated O/N at 4°C following manufactures instructions, wash 3 × 15 m in TBST and incubated with the appropriate secondary HRP-conjugated antibody for 1 h in 2.5% Blotto solution, then washed 5 × 15 m. Immunoblot detection was visualized using ECL kit. When necessary membranes were stripped as described.23
RESULTS
Inhibition of EGFR blocked HGF-induced proliferation
Previous studies in our laboratory show that the highly invasive mouse mammary carcinoma cell line PyVmT is sensitive to HGF and becomes even more invasive with increased HGF signaling.22 Therefore, we used PyMvT cells to determine if EGFR plays a role in HGF-mediated signaling. Blocking EGFR activation with the small molecule TKI, gefitinib, was used to elucidate the dependence of EGFR in HGF signaling. This small molecule is a competitive inhibitor of ATP binding to the EGFR tyrosine kinase with an in vitro IC50 of 20 nM.25 In intact cells, concentrations of ≤1 μM are considered to be relatively specific to the EGFR.26 Pre-treatment of PyVmT cells with ≤1 μM gefitinib blocked phosphorylation of EGFR at Y1173 and Y1068, both autophosphorylation sites indicative of EGFR kinase activation27 (Fig. 1A), thus establishing that PyVmT cells are sensitive to gefitinib at doses similar to what has been reported for many other responsive cells lines.28-31
Figure 1.

EGFR inhibition by gefitinib blocked HGF-mediated proliferation. (A) PyVmT cells were serum starved for 20 h, pre-treated with gefitinib (0.25 μM and 1.0 μM) for 2 h, and then treated with 20 ng HGF for 15 m. Extracts from each treatment were prepared and subjected to immunoblot analyses. Immunoblots were analyzed for EGFR autophosphorylation using Y1173 and Y1068 phosphotyrosine antibodies. Immunoblots were then stripped and re-probed for total EGFR levels. Immunoblots are representatives of three separate experiments. PyVmT cells were serum starved for 20 h then treated with (B). HGF at the doses indicated or with (C). Gefitinib (0.25 μM, 0.5 μM, 1.0 μM) and either 20 ng TGFα, HGF (+) or 40 ng HGF (++) for 24 h or with (D). gefitinib (0.25 μM, 0.5 μM, 1.0 μM) and either 10% Fetal Bovine Serum (FBS) or 20 ng HGF for 24 h. DNA synthesis was measured by labeling the cells with 3H-Thymidine for the last 2 h of treatment. Results are presented as counts per minute (CPM) 3H-Thymidine incorporation ± standard error of six replicates.
Next, we examined HGF-induced proliferation (DNA synthesis) in PyVmT cells. Initial analysis of PyVmT cells showed that incubation with increasing doses of HGF yielded a linear proliferative response (Fig. 1B). To examine the role of EGFR in HGF-induced proliferation, we used pharmacologically relevant doses of gefitinib, ranging from 0.25 μM to 1.0 μM. The EGFR ligand TGFα was used as an internal control for gefitinib function (Fig. 1C) and consistent with previous results,28 TGFα induced-PyMvT cell proliferation was inhibited in dose-dependent manner to gefitinib treatment. Our experimental data showed that HGF-mediated proliferation was also significantly inhibited by gefitinib at both 20 and 40 ng/ml HGF treatments (Fig. 1C). Additionally, gefitinib preferentially inhibited HGF-induced proliferation, as treatment with gefitinib on 10% serum-induced proliferation was significantly less inhibited (Fig. 1D). To verify that we were not observing a specific drug effect we used a related EGFR small molecule TKI, erlotinib. Erlotinib, like gefitinib, is a synthetic anilinoquinazoline agent that binds to the intracellular domain of EGFR thus, inhibiting kinase activation.32 Analysis of both gefitinib and erlotinib treatment showed a similar decrease in HGF-mediated proliferation (Fig. 2A).
Figure 2.

Erlotinib and gefitinib functioned similarly in PyMvT cells. (A) PyVmT cells were serum starved for 20 h and treated with either gefitinib or erlotinib (0.25 μM, 0.5 μM, 1.0 μM) and 20 ng HGF for 24 h. DNA synthesis was measured by labeling the cells with 3H-Thymidine for the last 2 h of treatment. Results are presented as counts per minute (CPM) 3H-Thymidine incorporation ± standard error of six replicates. (B) PyVmT cells were serum starved for 20 h, treated with either gefitinib or erlotinib at the indicated doses for another 24 h and trypsinized and stained with trypan blue. The number of non-blue (viable) cells were counted. Results are presented as number of average viable cells ± standard error of the mean of three replicates.
To determine whether the inhibitory effects of gefitinib and erlotinib were due to mitogenic effects or cell toxicity effects, we examined PyVmT cell toxicity in response to gefitinib and erlotinib. Cells were treated with either gefitinib or erlotinib for 24 h to mimic the [3H]thymindine incorporation assays, cells were stained with Trypan Blue then viable, non-stained, cells were counted. Analysis of cell viability indicated that cell numbers did not appreciably decline until inhibitor doses were well above 1 μM (Fig. 2B). As doses >1 μM were not used in our studies, these data suggest that the effects observed on HGF-induced proliferation when using either gefitinib or erlotinib were direct.
Inhibition of EGFR blocked HGF-induced cell motility
To address the role of EGFR in HGF-induced cell motility we used a wound closure assay that measures the distance of cells migrating to fill a cell free area. As gefitinib is known to inhibit EGFR activation and EGF induced proliferation in NMuMG cells,33 we analyzed the effect of gefitinib on HGF-induced motility. Inhibition of EGFR with treatment of either gefitinib (Fig. 3A) or erlotinib (Fig. 3B) significantly decreased HGF induced cell motility at both 0.25 μM and 1.0 μM inhibitor concentrations. An increase in NMuMG DNA synthesis was not detected upon HGF treatment using [3H]thymindine incorporation assay (data not shown), indicating that gefitinib and erlotinib blocked HGF induced motility without affecting basal proliferation. A comparison of the wound closure data between gefitinib and erlotinib inhibition indicated that both inhibitors equally blocked NMuMG cell migration. As gefitinib and erlotinib seem to function similarly in HGF-mediated proliferation and motility we continued to investigate the role of EGFR in HGF signaling using only gefitinib.
Figure 3.
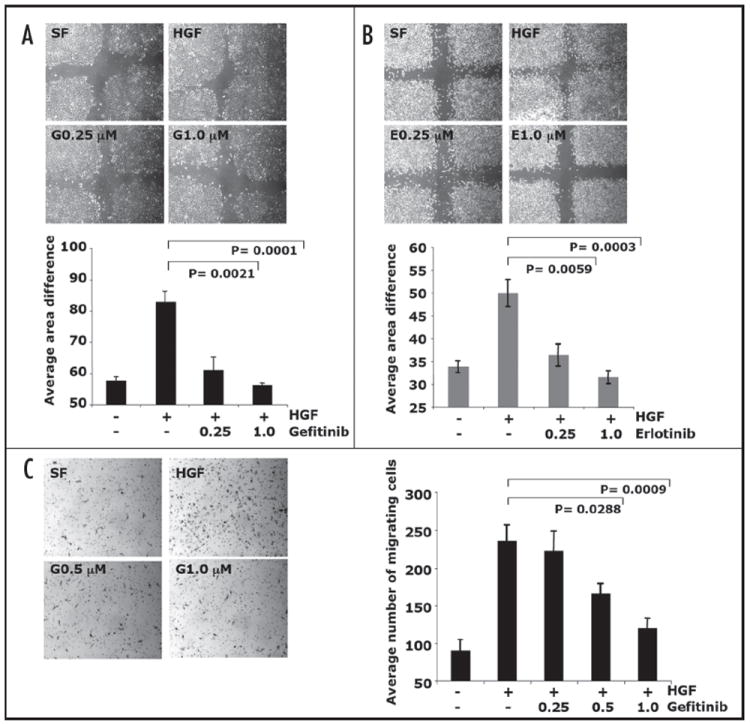
Inhibition of EGFR blocked HGF-mediated cell motility. Confluent NMuMG cells were wounded then treated with (A) gefitinib or with (B) erlotinib (0.25 μM or 1.0 μM) and 10 ng HGF for 20 h. Analysis of the area of the wound at 0 h to the area of wound closure at 20 h were done. Results are presented as average of area difference (0 h-20 h) ± standard error of the mean of six replicates. (C) PyVmT cells were serum starved for 24 h and plated in the upper chamber, the lower chamber contained either serum free media, 40 ng/ml HGF, or 40 ng/ml HGF + gefitinib (0.25 μM, 0.5 μM or 1.0 μM) for 8 h. Cells that migrated to the underside of the fibronectin coated filter were fixed and stained with hematoxylin. Images of four fields per filter were taken at 10 X and the number of migrated cells counted. Results are presented as average of number of cells of four fields ± standard error of the mean of at least four replicates.
To determine if gefitinib affects HGF-induced motility in other cell lines we analyzed PyVmT cells. PyVmT cell morphology is more mesenchymal and thus not as suitable to wound closure analysis, plus we wanted to determine if migration occurred on other matrixes besides plastic. Therefore, a Boyden chamber transwell assay was used to measure PyVmT cell migration through a fibronectin matrix. Similar to NMuMG cells, gefitinib treatment significantly inhibited HGF-induced PyVmT cell migration (Fig. 3C). PyVmT cells were observed to be less sensitive to gefitinib compared to NMuMG cells. Gefitinib was found to inhibit HGF-induced cell motility of PyVmT cells at doses of 0.5 μM and 1.0 μM, while gefitinib inhibited migration of NMuMG cells at 0.25 μM, suggesting that gefitinib sensitivity may be cell type specific. Taken together these data showed that EGFR is involved in HGF-mediated cell migration in mammary epithelial cells.
Gefitinib inhibition of EGFR mitigates HGF-induced cell invasion
Clinical data from breast cancer patients show a strong association between elevated HGF expression and increased invasiveness.8,34 Thus, we investigated the possible role of EGFR in HGF-mediated invasion through Matrigel using a boyden chamber transwell assay. NMuMG mammary epithelial cells, PyVmT mammary carcinoma cells and human MDA-MB-231 breast cancer cells were labeled using a fluorescent tracking dye and their ability to invade the Matrigel matrix was assessed. HGF-dependent invasion was detected in all cell lines tested NMuMG (Fig. 4A), PyVmT (Fig. 4B) and MDA-MB-231 (Fig. 4C). The addition of gefitinib significantly inhibited HGF-induced cell invasion in NMuMG and MDA-MB-231 cells at concentrations ranging from 0.25 μM–1.0 μM (Fig. 4A and C). Gefitinib inhibited HGF-induced PyVmT cell invasion at doses of 0.5 μM and 1.0 μM (Fig. 4B), indicating differences of gefitinib sensitivity among cell lines. Overall, gefitinib negatively effects HGF-mediated invasion in several mammary carcinoma cells.
Figure 4.
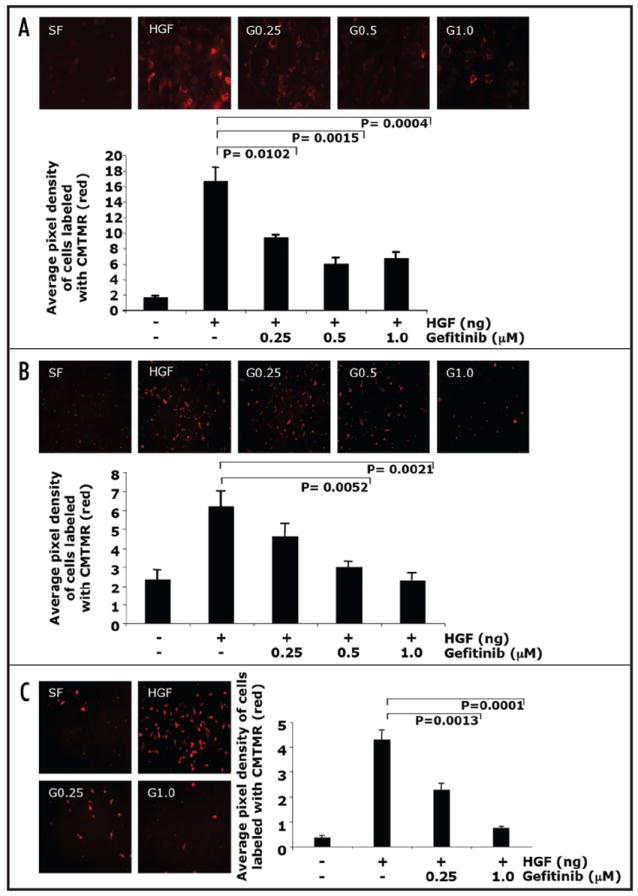
Inhibition of EGFR by gefitinib blocked HGF-mediated cell invasion. (A) NMuMG cells, (B) PyVmT and (C) MDA-MB-231 cells were labeled with a red fluorochrome and plated in the upper chamber, the lower chamber contained either serum free media, HGF or HGF + gefitinib (0.25 μM, 0.5 μM or 1.0 μM) for 8–12 h. Cells that migrated to the underside of the matrigel coated filter were fixed and mounted on slides. Images of at least four fields per filter were taken at 20X at constant exposure. The number of migrated cells were quantified by assaying the pixel density of the labeled migrated cells using software from Scion Image and Adobe Photoshop. Results are presented as average of pixel density of at least four fields ± standard error of the mean of at least four replicates.
Gefitinib inhibition of EGFR blocked HGF-mediated cell scattering
HGF-mediated cell scattering is a hallmark feature of this growth factor, which causes cohesive epithelial cell colonies to spread out, separate into individual cells, and assume a fibroblastic morphology. The role of EGFR signaling in this biological effect of HGF was investigated using the malignant mammary epithelial 4T1 cells, as they have the unique ability to form small colonies of cells when plated sparsely, unlike NMuMG, PyVmT or MDA-MB-231 cells. Once suitable individual colonies were apparent, cells were serum starved and then treated with 60 ng/ml HGF in the presence or absence gefitinib at doses of 0.25 μM and 1.0 μM for 24 h. Analysis of the number of scattered cells indicate that HGF induced cell scattering was inhibited in a dose-dependent manner by gefitinib (Fig. 5).
Figure 5.
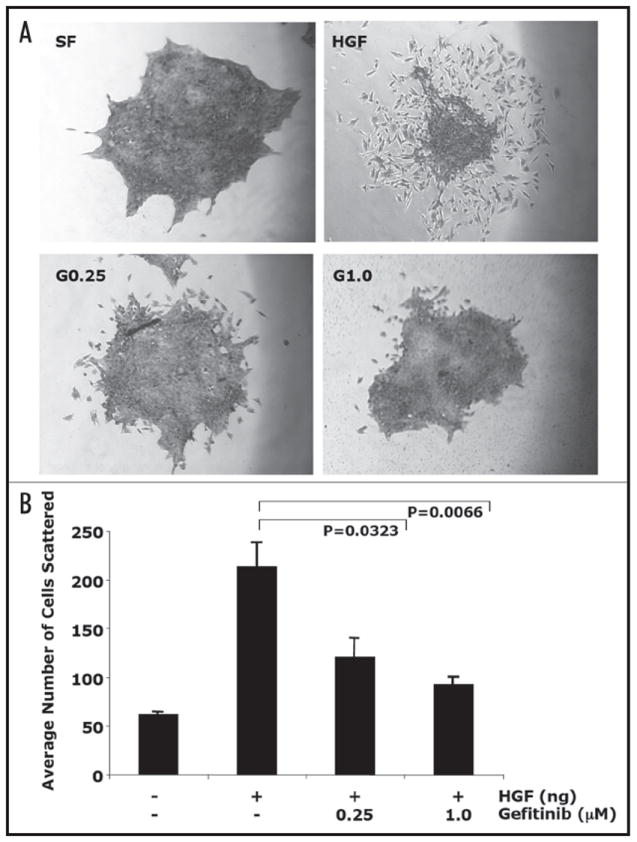
EGFR inhibition by gefitinib blocked HGF-induced cell scattering. 4T1 cells were plated at low density and maintained until single cohesive colonies were present. Cells were serum starved for 48 h and treated with gefitinib (0.25 μM or 1.0 μM) and 60 ng/ml HGF for and additional 24 h. (A) Cells were fixed, stained with hematoxylin, and photographed at 10X. Images of eight colonies per well were taken starting with colonies that had little on no scattered cells. (B) Results are presented as average of number of scattered cells of eight colonies ± standard error of the mean of at least four replicates.
HGF transactivated EGFR
To elucidate a mechanism of gefitinib action in inhibiting HGF-induced biological responses, we investigated whether EGFR and c-Met cross talk. One mechanism of cross talk is transactivation, thus we examined if HGF could activate EGFR in PyMvT cells. Analysis of Y1068 and Y1234/Y1235, autophosphorylation sites indicative of EGFR and c-Met, kinase activation, respectively,27,35 showed that at 0 h c-Met had no activation while EGFR has some basal activation (Fig. 6A and B). HGF dramatically induced c-Met phosphorylation with maximal activation at 5 m while EGFR was fully activated at 15 m. At 1 h both receptors returned to basal levels of activation (Fig. 6A). These data showed that HGF transactivates EGFR in a time-dependent manner.
Figure 6.
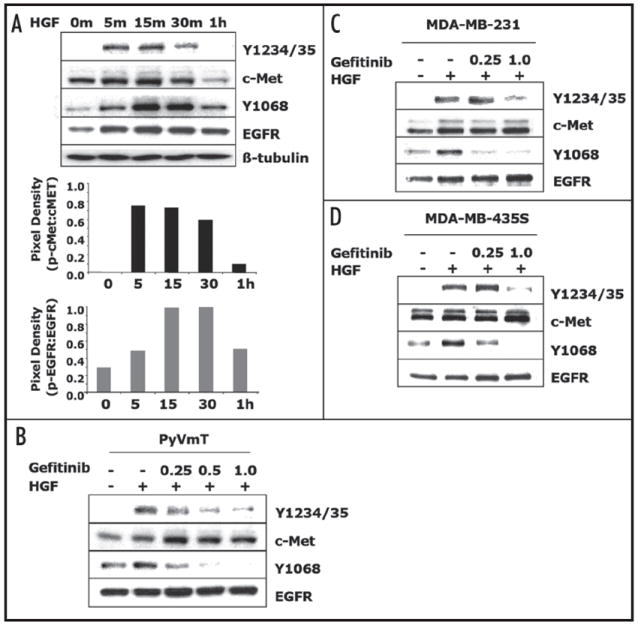
Gefitinib inhibited HGF activation c-Met and EGFR. (A) PyVmT cells were serum starved for 20 h treated with 20 ng/ml HGF for the indicated time points. Extracts from each time point were prepared and subjected to immunoblot analyses using phosphotyrosine antibodies for c-Met-Y1234/Y1235 and EGFR-Y1068. Immunoblots were stripped and re-probed for total c-Met and EGFR levels. Pixel density ratios of phosphorylated receptor to total receptor levels are depicted below the immunoblot data. Immunoblots are representatives of two separate experiments. (B) PyVmT cells, (C) MDA-MB-231 cells and (D) MDA-MB-435S cells were serum starved for 20 h, pre-treated with gefitinib (0.25–1.0 μM) for 2 h and treated with 20 ng/ml HGF for 15–30 m. Extracts were prepared and subjected to immunoblot analyses using phosphotyrosine antibodies for c-Met-Y1234/Y1235 and EGFR-Y1068. Immunoblots were stripped and re-probed for total c-Met and EGFR levels. Immunoblots are representatives of three separate experiments.
To further elucidate the mechanism of EGFR dependence in HGF-mediated biological responses we used the EGFR inhibitor gefitinib to examine the role of EGFR in HGF activation of c-Met. PyVmT cells were serum starved for 20 h to deplete the receptors of ligand, then pre-treated with gefitinib for 2 h. Our analysis of pre-treatment times for PyVmT sensitivity to gefitinib as well as previously published pre-incubation time points show that 2–20 hrs is an adequate time to block EGFR activation (data not shown).36,37 PyVmT cells were then stimulated for 15 m with HGF as that was the time point that both c-Met and EGFR were maximally stimulated by HGF (Fig. 6A). Gefitinib inhibited HGF-dependent EGFR activation at its auto-phosphorylation site at Y1068 (Fig. 6B). Activation of c-Met at its autophosphorylation sites at Y1234/Y1235 was also inhibited by gefitinib (Fig. 6B). To determine if this was cell type specific we looked at MDA-MB-231 and MDA-MB-435S cells. We showed that HGF activated both EGFR and c-Met in the human breast cancer cells MDA-MB-231 (Fig. 6C) and MDA-MB-435S (Fig. 6D) and that treatment with gefitinib blocked HGF-mediated activation of EGFR at concentrations of 0.25 μM and 1.0 μM and blocked c-Met activation at 1.0 μM in both MDA-MB-231 (Fig. 6C) and MDA-MB-435S (Fig. 6D) cells. These data suggest that gefitinib may inhibit HGF-mediated biological activities through blocking activation of its cognate receptor.
Inhibition of c-Met activation by gefitinib was EGFR-dependent
To determine the necessity of EGFR in the observed gefitinib-induced effects on c-Met signaling, we used the 32D myeloid cell line shown to be devoid of the erbB family of receptors and ligands.38,39 The 32D cells also lack c-Met, thus we used a 32D cell line stably expressing human c-Met (32D-cMet) for our analysis.23 Previous studies using the 32D-cMet cells showed c-Met receptor activation upon HGF ligand binding and intact downstream signaling pathways.23,24 Through Western blot analysis, we verified that the 32D-cMet cells lacked erbB receptors and expressed high levels c-Met (Fig. 7A). Additional analyses of 32D-cMet cells showed that 20 ng/ml of HGF was sufficient to induce activation of c-Met at autophosphorylation sites, Y1234/Y1235 (Fig. 7B). Treatment of 32D-cMet cells with gefitinib at doses ranging from 0.25 μM to 1.0 μM, which are the same concentrations used to treat PyVmT, MDA-MB-231, MDA-MB-435S cells, failed to block HGF activation of c-Met (Fig. 7C). Previous reports have suggested that gefitinib may target c-Met at higher doses of 3 μM.40 Analysis of gefitinib doses up to 3.0 μM also failed to inhibit HGF activation of c-Met (Fig. 7D). These data strongly suggest that gefitinib acts through an EGFR-dependent mechanism and not through any direct or non-specific affect on c-Met.
Figure 7.
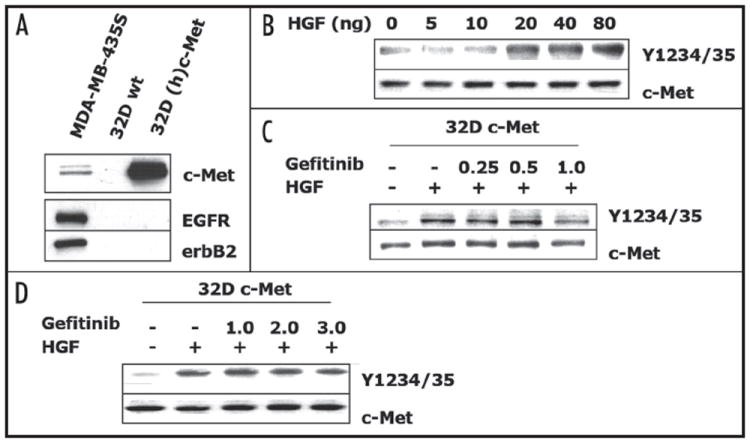
Gefitinib did not inhibit HGF activation of c-Met in 32D cells. (A) Analysis of 32D cells RTK expression levels. 32D c-Met cells were serum starved for 4 h and (B) treated with increasing doses of HGF for 30 m or pre-treated with gefitinib (C) 0.25–1.0 μM or (D) 1.0–3.0 μM, for 2 h and treated with 20 ng/ml HGF for 30 m. Extracts were prepared and subjected to immunoblot analyses using phosphotyrosine antibodies for c-Met-Y1234/Y1235. Immunoblots were then stripped and re-probed for total c-Met. Immunoblots are representatives of three separate experiments.
DISCUSSION
An increasingly recognized mechanism of action during carcinogenesis is the modulation of RTK signaling pathways. The RTK c-Met, and its cognate ligand HGF, are both critical mediators of breast cancer progression. High HGF and c-Met expression levels correlate with metastatic breast cancer and are independently considered as prognostic indicators for patient survival.9,10,34,41 Determining the specific role of HGF/c-Met signaling in breast cancer has been complicated by observations that c-Met communicates with a number of other signaling proteins that can regulate downstream signaling pathways, contributing to cellular responses that enhance growth and motility.12 Our studies focused on c-Met cross talk with EGFR as this receptor is also correlated with metastatic breast cancer14 and has been linked to c-Met signaling in other carcinomas cells.16,18,19
EGFR and c-Met cross talk has previously only been examined in HGF-independent situations, however since breast cancers display elevated HGF expression and c-Met signaling we sought to evaluate EGFR in HGF-dependent c-Met signaling. One report has suggested that EGFR activation is essential for HGF mediated hepatocyte proliferation,20 however little is known about the dependence of EGFR in other biological responses indicative of HGF signaling. Our data showed that EGFR may play a more global role in HGF mediated signaling, specifically in breast carcinoma cells. We evaluated the dependence of EGFR in HGF-induced proliferation, motility, invasion and cell scattering as these biological responses are relevant to breast cancer progression. The data presented here show that HGF-induced proliferation in PyVmT cells as well as HGF-induced motility and invasion in NMuMG and MDA-MB-231 cells was significantly inhibited upon treatment with gefitinib. Additional analysis of HGF-induced cell scattering in 4T1 cells showed a dose-dependent inhibition of cell scattering upon gefitinib treatment. Our analysis of multiple mammary cell lines showed a similar dependence of EGFR in HGF-mediated biological responses suggesting that EGFR-dependent HGF signaling was not cell line specific.
To evaluate a possible mechanism of communication between EGFR and c-Met the ability of HGF to transactivate EGFR was analyzed. Treatment of mammary carcinoma cells with HGF resulted in activation of c-Met and EGFR in a time-dependent manner. Previous reports have shown that EGFR ligands can active c-Met via intracellular mechanisms involving the Ras/MAPK signaling pathway, thus perhaps HGF is using a similar mechanism to transactivate EGFR.16,19 Additionally, a least one report suggests that EGFR and c-Met can directly associate.18 However, our analysis of EGFR and c-Met association showed minimal EGFR associating with c-Met (data not shown). Further analysis of HGF activation of EGFR and c-Met showed that gefitinib treatment blocked HGF activation of both EGFR and c-Met in PyMvT, MDA-MB-231 and MDA-435S cells, suggesting that gefitinib action is not cell line specific. These data showed that gefitinib may block HGF-mediated biological responses by inhibiting HGF activation c-Met and EGFR.
Previous reports show that gefitinib can directly inhibit c-Met with an IC50 of 3.2 ± 1.1μM.40 However, our studies in mammary carcinoma cells showed c-Met inhibition at gefitinib doses of ≤1 mM, well below the IC50 reported for c-Met sensitivity to gefitinib, suggesting that gefitinib may be functioning via EGFR. To further determine if gefitinib was directly targeting c-Met we analyzed the affect of gefitinib treatment on HGF activation of c-Met in 32D-cMet cells that do not express EGFR. These data showed that in 32D-cMet cells, c-Met activation was not affected by gefitinib at concentrations of ≤3.0 μM, suggesting that gefitinib uses an EGFR-dependent mechanism of action to inhibit HGF-induced c-Met activation. Our studies examined HGF activated c-Met phosphorylation while the previous study examined basal c-Met activation.40 Similar results in the 32D-cMet cells indicate that gefitinib can moderately decrease basal c-Met activation after 24 h treatment (data not shown). However, since our data showed gefitinib inhibition of HGF-mediated c-Met activation after only 2 h of treatment, it is unlikely that this is a relevant mechanism of action in our studies in mammary carcinoma cells. In summary our data showed that HGF-mediated activation of c-Met is inhibited by gefitinib in an EGFR-dependent manner that negatively regulates the ability of HGF/c-Met to induced cell proliferation, motility, invasion and cell scattering.
These data suggest that EGFR modulates c-Met activity in mammary carcinoma cells. As with α6β4 integrin,42 EGFR may contribute to c-Met signaling by amplification of common down-stream pathways thereby increasing the potential of c-Met to induce tumor-promoting effects including proliferation and invasion. Both EGFR and c-Met activate similar signaling cascades, including the Ras/MAPK mitogenic pathway and the PI3K motogenic pathway, that lead to similar biological outcomes.43 Furthermore, EGFR and c-Met can cooperate in liver regeneration through an additive effect on growth by concurrent activation of the Ras/MAPK pathway.21 These studies show that activation of both EGFR and c-Met signaling cascades can result in cooperative activation of downstream signaling effectors that enhance biological outcomes, similar to what we reported. We propose that in mammary carcinoma cells HGF-dependent c-Met signaling is amplified by concurrent activation of EGFR that results in increased HGF-induced proliferation and invasion, which is inhibited by gefitinib (Fig. 8).
Figure 8.

Model of HGF-dependent c-Met and EGFR cross talk in mammary carcinoma cells. HGF activates both c-Met and EGFR that can potentially amplify common signaling pathways responsible for tumor progression. The EGFR tyrosine kinase inhibitor gefitinib blocks EGFR activation and thus blocks intracellular downstream signaling. Gefitinib also blocks c-Met activation in an EGFR-dependent manner. Therefore, gefitinib blocks HGF mediated biological responses via blocking EGFR and c-Met activation thus, which also blocks down-stream signaling pathways responsible for tumor progression.
The implications of EGFR-dependent HGF signaling in tumori-genesis are significant. Our studies show a coordinated activation of two critical RTKs involved in breast cancer progression, suggesting that breast cancer patients that have elevated HGF expression and thus increased c-Met and EGFR signaling may benefit from the FDA approved EGFR tyrosine kinase inhibitor gefitinib.44 Currently, there are no FDA approved c-Met small molecule inhibitors but two compounds show promise in animal models, PHA665752 and SU11274.45,46 However, our data suggest that HGF-responsive mammary carcinomas evoke the cooperation between both EGFR and c-Met, thus perhaps a dual inhibitor approach might be more appropriate. This approach is supported by pre-clinical studies evaluating combination treatment of gefitinib and the HGF-antagonist NK4 in gastric cancer models, which show a synergistic inhibitory effect on tumor growth.47 EGFR and c-Met are both frequently misregulated in breast carcinogenesis and we showed that there is extensive signaling cross-talk between these two RTKs, which will allow for greater selectivity in treatment options for patients.
Acknowledgments
Dr. Bottaro kindly provided the 32D and 32D-cMet cells. We thank Dr. Emily Wang and Dr. Marianela Perez-Torres for their helpful discussions. This work was supported by grants CA 085492 and CA 102162 to HLM and the T.J. Martell foundation.
ABBREVIATIONS
- EGFR
epidermal growth factor receptor
- HGF
hepatocyte growth factor
- RTK
receptor tyrosine kinase
- TKIs
tyrosine kinase inhibitors
References
- 1.Gherardi E, Stoker M. Hepatocyte growth factor-scatter factor: Mitogen, motogen, and met. Cancer Cells. 1991;3:227–32. [PubMed] [Google Scholar]
- 2.Jeffers M, Rao MS, Rulong S, Reddy JK, Subbarao V, Hudson E, Vande Woude GF, Resau JH. Hepatocyte growth factor/scatter factor-met signaling induces proliferation, migration, and morphogenesis of pancreatic oval cells. Cell Growth Differ. 1996;7:1805–13. [PubMed] [Google Scholar]
- 3.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251:802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 4.Naldini L, Weidner KM, Vigna E, Gaudino G, Bardelli A, Ponzetto C, Narsimhan RP, Hartmann G, Zarnegar R, Michalopoulos GK. Scatter factor and hepatocyte growth factor are indistinguishable ligands for the met receptor. EMBO J. 1991;10:2867–78. doi: 10.1002/j.1460-2075.1991.tb07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsumoto K, Nakamura T. Emerging multipotent aspects of hepatocyte growth factor. J Biochem (Tokyo) 1996;119:591–600. doi: 10.1093/oxfordjournals.jbchem.a021283. [DOI] [PubMed] [Google Scholar]
- 6.Weidner KM, Hartmann G, Naldini L, Comoglio PM, Sachs M, Fonatsch C, Rieder H, Birchmeier W. Molecular characteristics of hgf-sf and its role in cell motility and invasion. Exs. 1993;65:311–28. [PubMed] [Google Scholar]
- 7.Maulik G, Shrikhande A, Kijima T, Ma PC, Morrison PT, Salgia R. Role of the hepatocyte growth factor receptor, c-met, in oncogenesis and potential for therapeutic inhibition. Cytokine Growth Factor Rev. 2002;13:41–59. doi: 10.1016/s1359-6101(01)00029-6. [DOI] [PubMed] [Google Scholar]
- 8.Jin L, Fuchs A, Schnitt SJ, Yao Y, Joseph A, Lamszus K, Park M, Goldberg ID, Rosen EM. Expression of scatter factor and c-met receptor in benign and malignant breast tissue. Cancer. 1997;79:749–60. doi: 10.1002/(sici)1097-0142(19970215)79:4<749::aid-cncr12>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 9.Yamashita J, Ogawa M, Yamashita S, Nomura K, Kuramoto M, Saishoji T, Shin S. Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res. 1994;54:1630–3. [PubMed] [Google Scholar]
- 10.Edakuni G, Sasatomi E, Satoh T, Tokunaga O, Miyazaki K. Expression of the hepatocyte growth factor/c-met pathway is increased at the cancer front in breast carcinoma. Pathol Int. 2001;51:172–8. doi: 10.1046/j.1440-1827.2001.01182.x. [DOI] [PubMed] [Google Scholar]
- 11.Gallego MI, Bierie B, Hennighausen L. Targeted expression of hgf/sf in mouse mammary epithelium leads to metastatic adenosquamous carcinomas through the activation of multiple signal transduction pathways. Oncogene. 2003;22:8498–508. doi: 10.1038/sj.onc.1207063. [DOI] [PubMed] [Google Scholar]
- 12.Orian-Rousseau V, Chen L, Sleeman JP, Herrlich P, Ponta H. Cd44 is required for two consecutive steps in hgf/c-met signaling. Genes Dev. 2002;16:3074–86. doi: 10.1101/gad.242602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertotti A, Comoglio PM. Tyrosine kinase signal specificity: Lessons from the hgf receptor. Trends Biochem Sci. 2003;28:527–33. doi: 10.1016/j.tibs.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br Med Bull. 1991;47:87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- 15.Wakeling AE. Inhibitors of growth factor signalling. Endocr Relat Cancer. 2005;12:S183–7. doi: 10.1677/erc.1.01014. [DOI] [PubMed] [Google Scholar]
- 16.Bergstrom JD, Westermark B, Heldin NE. Epidermal growth factor receptor signaling activates met in human anaplastic thyroid carcinoma cells. Exp Cell Res. 2000;259:293–9. doi: 10.1006/excr.2000.4967. [DOI] [PubMed] [Google Scholar]
- 17.Pai R, Nakamura T, Moon WS, Tarnawski AS. Prostaglandins promote colon cancer cell invasion; signaling by cross-talk between two distinct growth factor receptors. Faseb J. 2003;17:1640–7. doi: 10.1096/fj.02-1011com. [DOI] [PubMed] [Google Scholar]
- 18.Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-met signal pathways in transformed cells. J Biol Chem. 2000;275:8806–11. doi: 10.1074/jbc.275.12.8806. [DOI] [PubMed] [Google Scholar]
- 19.Fischer OM, Giordano S, Comoglio PM, Ullrich A. Reactive oxygen species mediate met receptor transactivation by g protein-coupled receptors and the epidermal growth factor receptor in human carcinoma cells. J Biol Chem. 2004;279:28970–8. doi: 10.1074/jbc.M402508200. [DOI] [PubMed] [Google Scholar]
- 20.Scheving LA, Stevenson MC, Taylormoore JM, Traxler P, Russell WE. Integral role of the egf receptor in hgf-mediated hepatocyte proliferation. Biochem Biophys Res Commun. 2002;290:197–203. doi: 10.1006/bbrc.2001.6157. [DOI] [PubMed] [Google Scholar]
- 21.Moriuchi A, Hirono S, Ido A, Ochiai T, Nakama T, Uto H, Hori T, Hayashi K, Tsubouchi H. Additive and inhibitory effects of simultaneous treatment with growth factors on DNA synthesis through mapk pathway and g1 cyclins in rat hepatocytes. Biochem Biophys Res Commun. 2001;280:368–73. doi: 10.1006/bbrc.2000.4063. [DOI] [PubMed] [Google Scholar]
- 22.Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. Loss of tgf-beta type ii receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of tgf-alpha-, msp- and hgf-mediated signaling networks. Oncogene. 2005;24:5053–68. doi: 10.1038/sj.onc.1208685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day RM, Soon L, Breckenridge D, Bridges B, Patel BK, Wang LM, Corey SJ, Bottaro DP. Mitogenic synergy through multilevel convergence of hepatocyte growth factor and interleukin-4 signaling pathways. Oncogene. 2002;21:2201–11. doi: 10.1038/sj.onc.1205289. [DOI] [PubMed] [Google Scholar]
- 24.Day RM, Cioce V, Breckenridge D, Castagnino P, Bottaro DP. Differential signaling by alternative hgf isoforms through c-met: Activation of both map kinase and pi 3-kinase pathways is insufficient for mitogenesis. Oncogene. 1999;18:3399–406. doi: 10.1038/sj.onc.1202683. [DOI] [PubMed] [Google Scholar]
- 25.Wakeling AE, Guy SP, Woodburn JR, Ashton SE, Curry BJ, Barker AJ, Gibson KH. Zd1839 (iressa): An orally active inhibitor of epidermal growth factor signaling with potential for cancer therapy. Cancer Res. 2002;62:5749–54. [PubMed] [Google Scholar]
- 26.Arteaga CL, Johnson DH. Tyrosine kinase inhibitors-zd1839 (iressa) Curr Opin Oncol. 2001;13:491–8. doi: 10.1097/00001622-200111000-00012. [DOI] [PubMed] [Google Scholar]
- 27.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: Mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 28.Anderson NG, Ahmad T, Chan K, Dobson R, Bundred NJ. Zd1839 (iressa), a novel epidermal growth factor receptor (egfr) tyrosine kinase inhibitor, potently inhibits the growth of egfr-positive cancer cell lines with or without erbb2 overexpression. Int J Cancer. 2001;94:774–82. doi: 10.1002/ijc.1557. [DOI] [PubMed] [Google Scholar]
- 29.Vicentini C, Festuccia C, Gravina GL, Angelucci A, Marronaro A, Bologna M. Prostate cancer cell proliferation is strongly reduced by the epidermal growth factor receptor tyrosine kinase inhibitor zd1839 in vitro on human cell lines and primary cultures. J Cancer Res Clin Oncol. 2003;129:165–74. doi: 10.1007/s00432-003-0420-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (egfr) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 31.Janmaat ML, Rodriguez JA, Gallegos-Ruiz M, Kruyt FA, Giaccone G. Enhanced cytotoxicity induced by gefitinib and specific inhibitors of the ras or phosphatidyl inositol-3 kinase pathways in non-small cell lung cancer cells. Int J Cancer. 2006;118:209–14. doi: 10.1002/ijc.21290. [DOI] [PubMed] [Google Scholar]
- 32.Albanell J, Gascon P. Small molecules with egfr-tk inhibitor activity. Curr Drug Targets. 2005;6:259–74. doi: 10.2174/1389450053765888. [DOI] [PubMed] [Google Scholar]
- 33.Barnes CJ, Bagheri-Yarmand R, Mandal M, Yang Z, Clayman GL, Hong WK, Kumar R. Suppression of epidermal growth factor receptor, mitogen-activated protein kinase, and pak1 pathways and invasiveness of human cutaneous squamous cancer cells by the tyrosine kinase inhibitor zd1839 (iressa) Mol Cancer Ther. 2003;2:345–51. [PubMed] [Google Scholar]
- 34.Yao Y, Jin L, Fuchs A, Joseph A, Hastings HM, Goldberg ID, Rosen EM. Scatter factor protein levels in human breast cancers: Clinicopathological and biological correlations. Am J Pathol. 1996;149:1707–17. [PMC free article] [PubMed] [Google Scholar]
- 35.Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM. Tyrosines1234-1235 are critical for activation of the tyrosine kinase encoded by the met proto-oncogene (hgf receptor) Oncogene. 1994;9:49–57. [PubMed] [Google Scholar]
- 36.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant egfr mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albitar L, Laidler LL, Abdallah R, Leslie KK. Regulation of signaling phosphoproteins by epidermal growth factor and iressa (zd1839) in human endometrial cancer cells that model type i and ii tumors. Mol Cancer Ther. 2005;4:1891–9. doi: 10.1158/1535-7163.MCT-05-0274. [DOI] [PubMed] [Google Scholar]
- 38.Pierce JH, Ruggiero M, Fleming TP, Di Fiore PP, Greenberger JS, Varticovski L, Schlessinger J, Rovera G, Aaronson SA. Signal transduction through the egf receptor transfected in il-3-dependent hematopoietic cells. Science. 1988;239:628–31. doi: 10.1126/science.3257584. [DOI] [PubMed] [Google Scholar]
- 39.Ewald JA, Wilkinson JC, Guyer CA, Staros JV. Ligand- and kinase activity-independent cell survival mediated by the epidermal growth factor receptor expressed in 32d cells. Exp Cell Res. 2003;282:121–31. doi: 10.1016/s0014-4827(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 40.Brehmer D, Greff Z, Godl K, Blencke S, Kurtenbach A, Weber M, Muller S, Klebl B, Cotten M, Keri G, Wissing J, Daub H. Cellular targets of gefitinib. Cancer Res. 2005;65:379–82. [PubMed] [Google Scholar]
- 41.Lengyel E, Prechtel D, Resau JH, Gauger K, Welk A, Lindemann K, Salanti G, Richter T, Knudsen B, Vande Woude GF, Harbeck N. C-met overexpression in node-positive breast cancer identifies patients with poor clinical outcome independent of her2/neu. Int J Cancer. 2005;113:678–82. doi: 10.1002/ijc.20598. [DOI] [PubMed] [Google Scholar]
- 42.Trusolino L, Bertotti A, Comoglio PM. A signaling adapter function for alpha6beta4 integrin in the control of hgf-dependent invasive growth. Cell. 2001;107:643–54. doi: 10.1016/s0092-8674(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 43.Grant S, Qiao L, Dent P. Roles of erbb family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci. 2002;7:d376–89. doi: 10.2741/grant. [DOI] [PubMed] [Google Scholar]
- 44.Cohen MH, Williams GA, Sridhara R, Chen G, McGuinn WD, Jr, Morse D, Abraham S, Rahman A, Liang C, Lostritto R, Baird A, Pazdur R. United states food and drug administration drug approval summary: Gefitinib (zd1839; iressa) tablets. Clin Cancer Res. 2004;10:1212–8. doi: 10.1158/1078-0432.ccr-03-0564. [DOI] [PubMed] [Google Scholar]
- 45.Christensen JG, Burrows J, Salgia R. C-met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 2005;225:1–26. doi: 10.1016/j.canlet.2004.09.044. [DOI] [PubMed] [Google Scholar]
- 46.Ma PC, Schaefer E, Christensen JG, Salgia RA. Selective small molecule c-met inhibitor, pha665752, cooperates with rapamycin. Clin Cancer Res. 2005;11:2312–9. doi: 10.1158/1078-0432.CCR-04-1708. [DOI] [PubMed] [Google Scholar]
- 47.Namiki Y, Namiki T, Yoshida H, Date M, Yashiro M, Matsumoto K, Nakamura T, Yanagihara K, Tada N, Satoi J, Fujise K. Pre-clinical study of a “Tailor-made” Combination of nk4-expressing gene therapy and gefitinib (zd1839, iressatrade mark) for disseminated peritoneal scirrhous gastric cancer. Int J Cancer. 2006;118:1545–55. doi: 10.1002/ijc.21531. [DOI] [PubMed] [Google Scholar]