

The unique chemical properties of aryl trifluoromethyl sulfides (ArSCF3) have been known for over 60 years.[1] The capacity of SCF3 to act as a lipophilic electron-withdrawing group has resulted in the incorporation of ArSCF3 components into a number of pharmaceutical and agrochemical agents.[2] Unfortunately, direct access to this important class of compounds is complicated by a lack of efficient, safe and general methods.[1a, 3]
Significant advances in Pd-catalyzed cross-coupling processes have allowed for efficient access to a diverse array of functionalized aromatic products, including aryl sulfides.[4] While the coupling of many aromatic or aliphatic thiols with aryl halides has been achieved with very high efficiency,[5] the analogous transformation to form aryl trifluoromethyl sulfides has not been reported. As gaseous CF3SH (b.p. = -36 °C)[6] can be difficult to handle in a laboratory setting, several SCF3 salts have been developed, however, most of these decompose under standard cross-coupling conditions.[3c]
It has been postulated that reductive elimination of Ar–SR from a palladium center is initiated via a nucleophilic attack on the electrophilic hydrocarbyl group by the metal-bound thiolate.[7] Thus, metal-catalyzed Ar–SCF3 coupling might be complicated by the reduced nucleophilicity of the SCF3 anion[2b] as compared to a standard thiolate.
Recent reports from our group regarding novel ligands including BrettPhos (1), t-BuBrettPhos (2), XPhos (3) and 3,4,5,6-tetramethyl(t-Bu)XPhos (4) (Scheme 1), have allowed for the successful coupling of weak nucleophiles traditionally thought to be reluctant participants in the transmetalation or reductive elimination steps of a typical Pd(0)/Pd(II) catalytic cycle. Specifically, using these catalyst systems has allowed for the direct formation of diaryl ether,[8] aryl fluoride,[9] aryl trifluoromethyl,[10] and aryl nitro compounds[11] from their corresponding aryl halides or pseudo halides. In light of these results, we hypothesized that a similar Pd-based system might allow for the formation of an aromatic C–SCF3 bond.
Scheme 1.
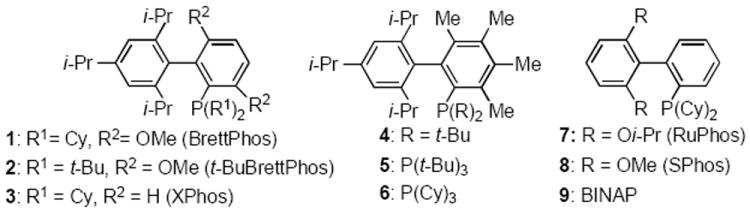
Various ligands used in Pd-catalyzed cross-coupling reactions.
As we suspected that reductive elimination from putative intermediate 11 would be rate limiting in any catalytic process, we began our investigation by attempting its preparation from oxidative addition complex 10 via treatment with AgSCF3 (Scheme 2). We were surprised when this procedure did not provide the expected transmetalation complex but instead led directly to the Ar–SCF3 product 12 (presumably via 11).
Scheme 2.
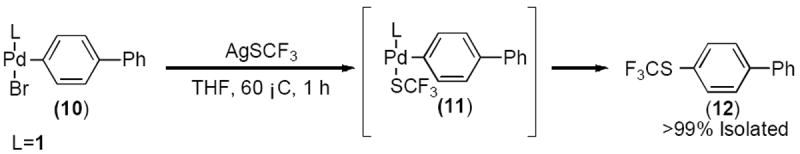
Formation of ArSCF3 via transmetalation and reductive elimination from an isolated LPdAr(Br) complex.
Given this finding, we attempted to convert 4-(4-bromophenyl)morpholine to the corresponding trifluoromethyl sulfide using AgSCF3 and a catalytic quantity of 1 and (COD)Pd(CH2TMS)2 (Table 1). However, under these conditions, none of 13 was observed. We surmised that failure to observe the coupled product might be due to the inefficient transfer of ⊖SCF3 to 10 under catalytic conditions. Thus, we elected to examine the use of a number of alternative previously reported ⊖SCF3 sources (Table 1).[3c, e]
Table 1.
Examination of different SCF3 sources.[a]
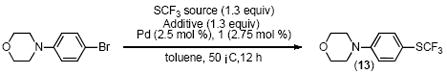
| |||
|---|---|---|---|
| Entry | SCF3 Source | Additive | Yield [%] |
| 1 | CsSCF3 | none | 10 |
| 2 | AgSCF3 | none | 0 |
| 3 | (Me)4NSCF3 | None | 20 |
| 4 | AgSCF3 | (Me)4NI | 0 |
| 5 | AgSCF3 | (Bu)4NCl | 0 |
| 6 | AgSCF3 | (Bu)4NBr | 56 |
| 7 | AgSCF3 | (Bu)4NI | 55 |
| 8 | AgSCF3 | Ph(Me)3NI | 80 |
| 9 | AgSCF3 | Ph(Et)3NI | >99 |
(COD)Pd(CH2TMS)2 (2.5 mol %), PhMe (4 mL); all reactions are run on 0.2 mmol scale and all reported yields are based on GC data
Clark’s[3d] work on the use of (Bu)4NI and AgSCF3 for SNAr reactions with aryl halides indicated to us that the addition of a quaternary ammonium salt might be beneficial. Consistent with this hypothesis, the addition of 1 equivalent of (Bu)4NI to the reaction mixture increased the yield of 13 from 0 % to 55 % (Table 1). Further examination of different ammonium salts revealed that Ph(Me)3NI was more effective than (Bu)4NI and that switching to a more soluble ammonium salt, Ph(Et)3NI, provided a nearly quantitative yield of the desired product (Table 1). Based on work done by Clark, it is presumed that the iodide anion binds to AgSCF3 in order generate an anionic “ate” complex. We hypothesize that a large diffuse cation further aids in the solubility of this complex. It is worth noting that while the use of quaternary ammonium iodides and bromides allowed for catalytic turnover, the corresponding chloride analogs were ineffective.
With the optimal combination of Ph(Et)3NI and AgSCF3 realized, we re-examined various other previously reported ligands, which have enjoyed a measure of success in Pd-catalyzed cross-coupling reactions.[12] Our survey revealed that only dialkylbiarylphosphine based ligands were successful carrying out this transformation, while other ligands such as 5 or 6 did not perform well even with higher catalyst loadings.
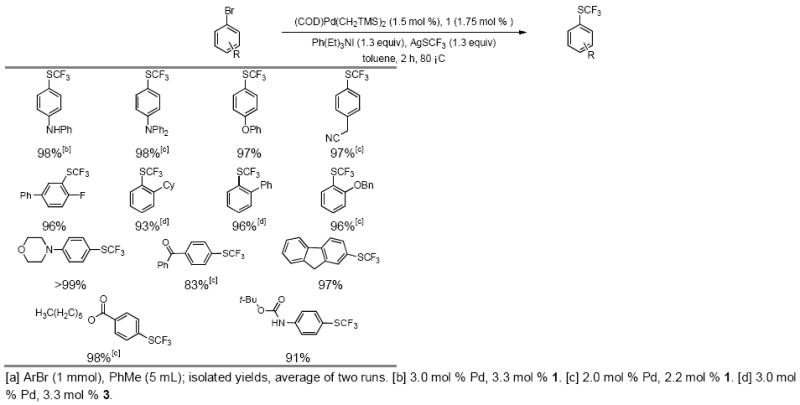
Accordingly, we were successful in converting electron-rich, -neutral and -deficient aryl bromides to their respective aryl trifluoromethyl sulfides in 2 hours at 80 °C using 1.5 - 3.5 mol % of Pd and 1.65 – 3.85 mol % of 1. Electron-neutral and electron-rich substrates were coupled more efficiently than their electron-poor analogs. This effect has previously been noted in the coupling of aryl halides with NaNO2.[11] Substrates containing acid-sensitive functional groups, such as BOC-protected anilines and nitriles, were tolerated and coupled in high yield along with substrates containing ketones, esters, and free NH groups of anilines (Table 3). Aryl bromides containing bulky ortho-groups, e.g., o-cyclohexyl and o-phenyl groups, could also be coupled successfully, although they required the use of the smaller ligand XPhos (3) (Table 3).
Table 3.
Pd-catalyzed coupling of aryl bromides.[a]
|
Heteroaryl bromides such as those containing indoles, pyridines, quinolines, thiophenes and furans, were also viable substrates (Table 4). Unfortunately, attempts to extend this methodology to the coupling of aryl chlorides or aryl triflates were unsuccessful. We are currently working to understand and overcome these limitations.
Table 4.
Pd-catalyzed formation of heteroaryl–SCF3 compounds.[a]
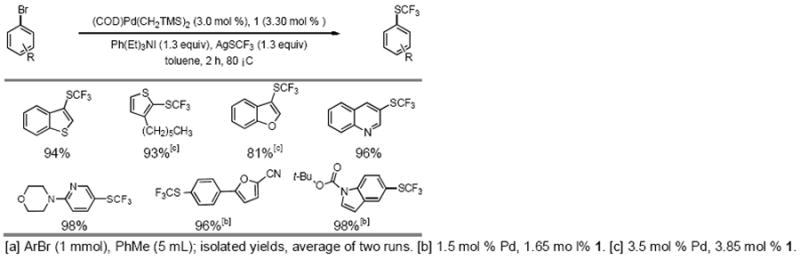
|
Finally, to demonstrate the utility of this method, we prepared an intermediate in the reported synthesis of Toltrazuril,[13] an antiprotozoal agent. Intermediate 14 can be assembled from readily available starting materials in an overall yield of 88%. The key C–SCF3 bond-forming process proceeded in 95% yield (Scheme 3).
Scheme 3.
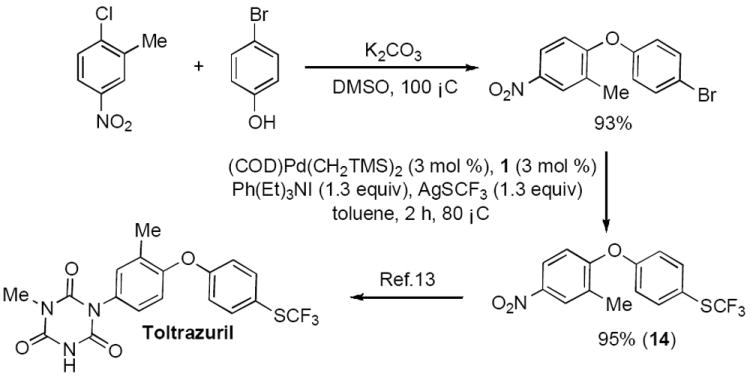
Synthesis of Toltrazuril intermediate.
In summary, we have developed a general method for the Pd-catalyzed Ar–SCF3 bond-forming reaction. Using this method, a wide range of aryl bromides were converted into their corresponding aryl trifluoromethyl sulfides. Additionally, we have been successful in generating a variety of heterocyclic aryl trifluoromethyl sulfides from heteroaryl bromide precursors. Due to the utility of Ar–SCF3 compounds as biologically active agents, and the mild reaction conditions employed, we expect this method to be immediately implemented in the discovery of novel compounds with pharmaceutical and agrochemical applications.
Supplementary Material
Table 2.
Examination of various ligands commonly employed in Pd-catalyzed reactions.[a]
| |||
|---|---|---|---|
| Entry | Ligand | Time (h) | Yield (%) |
| 1 | 1 | 1 | >99[b] |
| 2 | 2 | 1.5 | 60[b] |
| 3 | 3 | 1 | 84[b] |
| 4 | 7 | 2 | 36[c] |
| 5 | 8 | 2 | 3[c] |
| 6 | 5 | 2 | 29[d] |
| 7 | 6 | 2 | 0[d] |
| 8 | 9 | 2 | 0[d] |
| 9 | 4 | 2 | <1[c] |
PhMe (4 mL); all reactions are run on 0.2 mmol scale and all reported yields are based on GC data.
1.15 mol % Pd, 1.27 mol % L.
1.5 mol % Pd, 1.65 mol % L.
2.5 mol % Pd, 2.75 mol % L
Footnotes
We thank the National Institutes of Health (NIH) for financial support of this project (GM-58160). We thank BASF for a gift of Pd compounds, FMC Lithium for a gift of tBu2PCl and Nippon Chemical for a gift of Cy2PCl and unrestricted support. G.T. would also like to thank the DoD for the NDSEG Fellowship (32 CFR 168a). The Varian NMR instrument used was supported by the NSF (CHE-980861).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Manteau B, Pazenok S, Vors JP, Leroux FR. J Fluorine Chem. 2010;131:140. [Google Scholar]; b) Jeschke P. Pest Manag Sci. 2010;66:10. doi: 10.1002/ps.1829. [DOI] [PubMed] [Google Scholar]
- 2.a) Leo A, Hansch C, Elkins D. Chem Rev. 1971;71:525. [Google Scholar]; b) Yagupolskii LM, Il’chenko AY, Kondratenko NV. Russ Chem Rev. 1974;43:32. [Google Scholar]; c) Leroux F, Jeschke P, Schlosser M. Chem Rev. 2005;105:827. doi: 10.1021/cr040075b. [DOI] [PubMed] [Google Scholar]
- 3.a) Kieltsch I, Eisenberger P, Togni A. Angew Chem Int Ed. 2007;46:754. doi: 10.1002/anie.200603497. [DOI] [PubMed] [Google Scholar]; b) Pooput C, Medebielle M, Dolbier WR. Org Lett. 2004;6:301. doi: 10.1021/ol036303q. [DOI] [PubMed] [Google Scholar]; c) Tyrra W, Naumann D, Hoge B, Yagupolskii YL. J Fluorine Chem. 2003;119:101. [Google Scholar]; d) Adams DJ, Clark JH. J Org Chem. 2000;65:1456. doi: 10.1021/jo9915933. [DOI] [PubMed] [Google Scholar]; e) Clark JH, Jones CW, Kybett AP, Mcclinton MA, Miller JM, Bishop D, Blade RJ. J Fluorine Chem. 1990;48:249. [Google Scholar]; f) Wakselman C, Tordeux M. J Chem Soc Chem Comm. 1984:793. [Google Scholar]; g) Yagupolskii LM, Kondratenko NV, Sambur VP. Synthesis. 1975:721. [Google Scholar]; h) Adams DJ, Goddard A, Clark JH, Macquarrie DJ. Chem Commun. 2000:987. [Google Scholar]; i) Boiko VN, Shchupak GM, Yagupolskii LM. Zh Org Khim. 1977;13:1057. [Google Scholar]
- 4.a) Surry DS, Buchwald SL. Chemical Science. 2011;2:27. doi: 10.1039/C0SC00331J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cacchi S, Fabrizi G. Chem Rev. 2005;105:2873. doi: 10.1021/cr040639b. [DOI] [PubMed] [Google Scholar]; c) Miyaura N. In: Metal-Catalyzed Cross-Coupling Reactions. Second ed. de Meijere A, Diedrech F, editors. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- 5.Fernández-Rodríguez MA, Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:2180. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]
- 6.Zack NR, Shreeve JM. Synthetic Commun. 1974;4:233. [Google Scholar]
- 7.Mann G, Baranano D, Hartwig JF, Rheingold AL, Guzei IA. J Am Chem Soc. 1998;120:9205. [Google Scholar]
- 8.Burgos CH, Barder TE, Huang X, Buchwald SL. Angew Chem Int Ed. 2006;45:4321. doi: 10.1002/anie.200601253. [DOI] [PubMed] [Google Scholar]
- 9.Watson DA, Su MJ, Teverovskiy G, Zhang G, Garcia-Fortanet J, Kinzel T, Buchwald SL. Science. 2009;325:1661. doi: 10.1126/science.1178239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679. doi: 10.1126/science.1190524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fors BP, Buchwald SL. J Am Chem Soc. 2009;131:12898. doi: 10.1021/ja905768k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Barder TE, Walker SD, Martinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]; b) Milne JE, Buchwald SL. J Am Chem Soc. 2004;126:13028. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]; c) Wolfe JP, Buchwald SL. J Am Chem Soc. 1996;118:7215. [Google Scholar]; d) Hartwig JF, Kawatsura M, Hauck SI, Shaughnessay KH, Alcazar-Roman LM. J Org Chem. 1999;64:5575. doi: 10.1021/jo990408i. [DOI] [PubMed] [Google Scholar]; e) Reddy NP, Tanaka M. Tetrahedron Lett. 1997;38:4807. [Google Scholar]
- 13.Günther A, Mohrmann K-H, Stubbe M, Ziemann H, Bayer AG. 1986 DE3516630. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.