

In this issue of PNAS, Stoler and colleagues report that typical sporadic colorectal cancers on average contain at least 11,000 genomic alterations per cell (1). Furthermore, they report that the genomic instability responsible for generating this number of mutations starts very early in the neoplastic process and can be found in adenomatous polyps, which are known to be the precursors of cancer in the colon and rectum. Should this conclusion be emblazoned on the front page of the evening news, or does this serve to confirm and extend concepts that we already accept? To grasp the implications of this work, it will be helpful to briefly review the historical background of genomic instability and place this submission in that context. To do this, a series of questions must be posed. How does one measure mutations, and how can one quantitate heterogeneous alterations? Is it possible that tumors simply generate a very large number of alterations at genomic sequences that are irrelevant to issues of tumor development? If there are many mutations and only a few are biologically relevant, how does one determine whether mutations are important? Let's see.
The Historical Context of Genomic Instability.
It has been known—for most of this century—that cancer is often associated with visible derangements in the nucleus of the cell. Solid tumors commonly have duplications, deletions, and rearrangements that occur at the chromosomal level. These were difficult to categorize, particularly before the organization of the human nucleus was understood.
Within 25 years of the discovery of the structure of DNA, oncogenes were isolated, which were frequently mutant versions of normal cellular genes in which an activating point mutation or genetic amplification provided a gain of function for that gene product, and a growth advantage for that cell. While the list of oncogenes was growing, other groups began to realize that tumor growth was also associated with loss of function at tumor suppressor genes. These genetic loci were often inactivated by their deletion from the nucleus, and the phrase “loss of heterozygosity” (LOH) was applied to genetic loci in which both alleles were present in normal tissues (and identified by virtue of their “heterozygosity”), and one copy was lost in the tumor tissue. In many instances, tumor suppressor genes were identified by virtue of germ-line mutations that were present at a high frequency in a rare tumor, such as retinoblastoma. However, within a short period of time it became clear that many of these genes were also associated with a variety of different tumors.
There are no oncogenes or tumor suppressor genes that are activated or deleted from all cancers. Even tumors of a single organ rarely have uniform genetic alterations, although tumor types from one specific organ have a tendency to share mutations in certain genes or in different genes within a single growth-regulatory pathway.
What is not clear as of this writing is how many critical mutations are required to convert a single normal cell into a malignant one. It has recently been reported by Hahn et al. that three defined genetic events were sufficient to convert cultured embryonic human cells into those capable of forming tumors in nude mice (2). However, there is no human tumor for which the story is known to be this simple. Human cells have been notoriously difficult to transform in vitro, and it is not entirely clear whether the human genome is more resistant to the types of mutagenesis that have been attempted, or whether there are more cellular guards against transformation that must be dismantled for this to occur.
The simplest model of tumorigenesis is as follows. Human cells experience a certain number of mutations each day as a consequence of exposure to carcinogens or ordinary “wear and tear,” which alters nucleotide sequences; errors will also occur during new DNA synthesis and during the process of disentangling the chromosomes during mitosis. Most of these errors would be either irrelevant to the life of the cell or deleterious because of the loss of a gene critical for cellular viability. By chance, an occasional genomic misadventure might create a growth advantage for a cell, permitting increased net cellular growth (because of increased proliferation or a reduced cell death) and result in clonal expansion of that lineage. A second genomic alteration might then occur within this expanding clone, again by chance, providing an additional growth advantage for that cell and its progeny. By virtue of these two advantages, the cells of this clone would eventually overgrow neighboring cells, creating yet another expanding clone. This scenario would repeat as a consequence of each new mutation that provided an additional growth advantage.
The accumulation of these growth-promoting mutations is the basis of multistep carcinogenesis. By virtue of the availability of neoplastic tumors in various stages of growth in the human colon, Vogelstein proposed a model of progression in 1988 (3). The genetic alterations in this model included point mutations and allelic deletions. This model addressed neither the rate at which mutations accumulated nor the mechanisms by which this would occur.
For over two decades, Loeb has offered the hypothesis that cancer is characterized by a “mutator phenotype” (4). He has speculated that the spontaneous rate of mutation in normal cells is not sufficient to account for the number of mutations found in human cancers. Implicit in this formulation, a derangement in some cellular process would be required before the development of the first growth-promoting mutation, which would then increase the number of cells and mutational events and facilitate multistep carcinogenesis. When this theory was first proposed, however, there was no mechanism of hypermutability to accommodate it.
The initial experiments that detected the large number of LOH events in colorectal cancer required the extraction of relatively large amounts of DNA from tumors and normal tissue for Southern blot analysis. To detect a deleted tumor suppressor gene, one hoped either that the chromosomal deletion would be large, or that the probe was serendipitously close to the missing tumor suppressor gene. In the classic report of the “allelotype” of colorectal cancer, it was observed that some tumors experienced chromosomal losses at none of the probed loci, whereas others lost more than half of their probed loci (5). How would one make sense of this apparent chaos?
A number of experimental approaches were developed or modified to tackle this problem. One of these approaches provided an estimate of the number of allelic losses and gains in tumors. Perhaps more importantly, the approach illuminated another unexpected type of mutation, provided an estimate that 105 such mutations were present in some tumors, and predicted the presence of a novel pathway to carcinogenesis (6).
How Does One Measure Mutations in a Tumor?
The most obvious way to determine the number of point mutations in a cancer would be to sequence the entire normal genome, sequence the DNA from a cancer, and compare them. Because of the large size of the human genome, not to mention the variety of alterations in cancer that are not simple nucleotide substitutions, this is impractical. Furthermore, direct sequencing strategies might not detect many of the hemizygous chromosomal deletions (i.e., LOH events) and would probably miss many of the rearrangements. The identification of a small number of unique cytogenetic rearrangements in specific tumors (such as the Philadelphia chromosome in chronic myelogenous leukemia) led to the hope that a systematic cytogenetic characterization of tumors might permit one to understand the basis of tumor formation. However, the tools of cytogenetics were not sufficiently powerful to tackle the problem.
In 1990, Welsh and McClelland used an arbitrarily primed PCR (AP-PCR) to generate genomic “fingerprints” in bacteria and rice (7). Two years later, Peinado et al. used AP-PCR to characterize genetic alterations in colorectal cancers by comparing the fingerprints of normal tissue with that obtained from the tumors. The arbitrary primer sequences yielded reproducible and unbiased fingerprint patterns from template DNA; they literally threw dice to generate the primer sequences. When the PCR products of normal tissue and cancer were directly compared, they were able to estimate the global extent of genetic gains and losses in a single step. Moreover, this approach permitted the direct cloning of the deleted sequences by using the PCR product of the normal tissue to map the appropriate regions (8).
This group, led by Manuel Perucho, made another critical observation (6) that was observed and confirmed in the same general time frame by two other groups (9, 10). In addition to seeing and quantitating chromosomal losses and gains by using AP-PCR, this technique detected subtle length variations in the PCR products in a subset of 12% of cancers that did not have chromosomal gains and losses. Instead, Perucho noted that these tumors had clonal somatic mutations in simple repeated sequences such as mononucleotide (always deletions) and dinucleotide (deletions and insertions) repeats. The unbiased nature of the technique permitted an estimate that tumors carried more than 100,000 such mutations, an astounding conclusion! These tumors did not have the widespread LOH events seen in most other cancers and displayed other distinctive phenotypic and genotypic features. By this observation, a novel pathway to tumorigenesis in 12–20% of cancers was detected, and within a year, the molecular basis of this pathway was definitively traced to inactivation of the human DNA mismatch repair system (11–13).
Tumors with this phenotype are now said to have “microsatellite instability” (MSI), and these tumors are characterized by a very large number of single point mutations and in particular the accumulation of length alterations in simple repeated sequences, which are ubiquitous throughout our genome. The discovery of this alternate pathway for tumor development in the cancer fulfilled Loeb's prediction of a mutator phenotype, provided a mechanism to account for this, and showed that some tumors could have an extremely large number of mutations.
AP-PCR was initially developed to improve on Southern analysis for LOH events. Continuing along this line, Perucho's group also published a study in 1998 using AP-PCR to compare primary and metastatic colorectal cancers (14). They deduced a molecular karyotype by assigning monochromosomal identities to the DNA fingerprints. They observed that gains of sequences were as frequent as losses in cancer. Gains of sequences from chromosomes 8 and 13 occurred in over 75% of tumors. Moreover, they found that losses of sequences from chromosome 4 were associated with metastasis, predicting the presence of a tumor suppressor gene that influences metastasis on that chromosome.
What Mutations Are Important in Tumorigenesis?
Most microsatellite sequences occur in introns or between genes; length variations at these loci in the progeny of a cell with microsatellite instability would be biologically irrelevant in most instances. It has subsequently been shown that a critical small number of genes contain simple repetitive sequences in critical coding regions, that insertion/deletion mutations occur in cancers, and that these length variations create frameshifts that inactivate those critical gene products. This last finding provided a biological story. Mutations occur in a large number of sequences throughout the genome, which will alter the products of a much smaller number of genes involved in regulating cell growth, which then accumulate in a stepwise manner in a developing neoplasm. In an interesting tangent, germ-line mutations in the DNA mismatch repair genes were found to be responsible for the hereditary cancer predisposition known as Lynch Syndrome (15).
So, How Many Mutations in a Tumor?
Returning to the work from Anderson's group, in 1997, Basik and colleagues used an intersimple sequence repeat PCR (INTER-SSR-PCR) to assess the degree of genomic instability among 57 colon cancers (16). These investigators used a single PCR primer homologous to dinucleotide repeats, anchored at the 3′ end by two nonrepetitive nucleotides as illustrated in Fig. 1. They were able to evaluate the degree of genomic instability by observing the appearance (gains) or disappearance (losses) of bands when comparing the amplifiable segments obtained from tumors and matched normal colonic tissues. The use of this primer has an intrinsic priming bias. Nonetheless, it allowed an estimate of the number of events that occurred in each tumor cell and a calculation of a rate of genomic instability, by using the following formula:
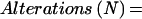 |
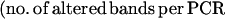 |
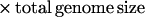 |
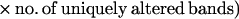 |
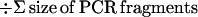 |
In this issue of PNAS (1), Anderson's group has studied 58 colon cancers and 14 polyps with INTER-SSR-PCR. They estimated that >104 such events were detectable per tumor cell. Notably—and this is an issue that bears careful consideration—the authors reported a range of genomic instability indices that were similar between sporadic cancers and polyps, including 11 adenomas (benign neoplasms) and 2 hyperplastic polyps (which are not considered neoplastic), suggesting that genomic instability is an early event in multistep carcinogenesis and occurs in one possibly nonneoplastic lesion.
Figure 1.

Schematic representation of the INTER-SSR-PCR (adapted from ref. 16). The PCR product is obtained by using a single primer homologous to dinucleotide repeats and anchored at 3′ by two nonrepetitive sequences. Genomic instability is deduced by the appearance or disappearance of bands when comparing results from tumors and matched normal tissues. The new bands can be larger versions of the original “normal” amplicon because of an insertion in the inter-SSR site, a smaller band because of a deletion or a rearrangement, creating a novel INTER-SSR site. One cannot determine what alteration has occurred without analyzing the PCR products.
These data raise several important questions. If the same genomic instability index is found in colorectal adenomas, cancers, and hyperplastic polyps, how does this fit with the concept of clonal expansion and multistep carcinogenesis? Would not one expect that the accumulation of mutations is critical in the evolution of neoplasia? Because the number of mutations is quite high, one would expect that many more genomic alterations would be detectable as one traced the process from the early stages to the later stages as the tumor progresses. Colorectal cancer is thought to take several decades to fully develop, on the basis of the slow growth rates of adenomatous polyps and the slow progression of these polyps to cancer. Moreover, what accounts for genomic instability in hyperplastic polyps, which have “ordinary” proliferation indices and are not considered neoplasms? One could make the case that the number of genomic alterations in adenomas (true benign neoplasms) is reasonable, but if hyperplastic polyps are truly not neoplasms, we must revise our thinking either about these polyps or about the implications of the INTER-SSR-PCR data from them. Additional work will be required to evaluate the persistence and evolution of genomic instability throughout these stages to fully comprehend the meaning of these findings.
The authors suggest that INTER-SSR-PCR is an easy method to assess the number of mutational events during tumor development; however, these findings almost surely reflect an overestimation of the alterations necessary for tumorigenesis and the identification of silent sequences aberrations, which will not lead to a growth advantage. To prove their point, Stoler et al. need to provide inter-SSR sequence data to identify the targets of these rearrangements. In the only example provided, the genetic alteration was a 4-bp insertion mutation (of the MSI variety). This is the type of genetic alteration initially described by Perucho's group and does not suggest that INTER-SSR-PCR accurately measures the form of genomic instability characterized by duplications, deletions, and rearrangements of chromosomes. Thus, it is not yet established that INTER-SSR-PCR is a suitable technique for quantitating “chromosomal instability” (also called CIN) rather than MSI.
In a more direct measure of CIN, Lengauer et al. used fluorescence in situ hybridization to demonstrate that gains or losses of chromosomes occur in aneuploid colorectal cancer cell lines but not in diploid cell lines or normal lymphocytes (17). Again, the technique involved provided a quantitative estimate of DNA damage and mapped chromosomal targets.
What Mechanisms Might Account for Genomic Instability in Non-MSI Cancers?
A mechanistic explanation is available to account for MSI in tumors, namely loss of DNA mismatch repair activity. But how does one account for gains and losses (not to mention rearrangements) of large chromosomal segments that are demonstrated by LOH at Southern analysis and have been proposed by results with INTER-SSR-PCR? Several theories have been proposed to account for this common phenomenon. The p53 gene, as “guardian of the genome,” was thought to be a candidate for this, but LOH of the wild-type p53 allele occurs as the adenoma becomes malignant in the colon (3, 18). Thus, it occurs too late to also account for CIN in benign neoplasms. Altered mitotic spindle checkpoint genes, such as hBub-1, have been proposed and are mechanistically attractive candidates (19), but the evidence to date indicates that this is a relatively rare event in colorectal neoplasia, and their functional significance remains to be established. Several other genetic alterations have been proposed, but the evidence supporting their role in human cancers remains thin.
A recent candidate with the ability to create chromosomal instability in human epithelial cells is the T antigen, which is a complex viral transforming gene found in the SV40 virus and in two human viruses: JC virus and BK virus. The work of Hahn et al. (2) indicates that T antigen is a candidate-transforming gene in a human cell model, and work from our group has recently demonstrated that JC viral sequences are present in normal human colons, in colon cancers, and in one colon cancer cell line with CIN (20). Understanding all of the varieties of genomic instability remains an unsolved problem and is likely to occupy the attention of cancer researchers for some time to come.
Acknowledgments
The authors are supported, in part, by the Research Service of the Veteran's Administration Medical Center, by the California Cancer Research Program, and by National Institutes of Health grant CA72851.
Footnotes
See companion article on page 15121.
References
- 1.Stoler DL, Chen N, Basik M, Kahlenberg M S, Rodriguez-Bigas M A, Petrelli N J, Anderson G R. Proc Natl Acad Sci USA. 1999;96:15121–15126. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hahn W C, Counter C M, Lundberg A S, Beijersbergen R L, Brooks M W, Weinberg R A. Nature (London) 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Fearon E R, Hamilton S R, Kern S E, Preisinger A C, Leppert M, Nakamura Y, White R, Smits A M, Bos J L. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 4.Loeb L A. Cancer Res. 1994;54:5059–5063. [PubMed] [Google Scholar]
- 5.Vogelstein B, Fearon E R, Kern S E, Hamilton S R, Preisinger A C, Nakamura Y, White R. Science. 1989;244:207–211. doi: 10.1126/science.2565047. [DOI] [PubMed] [Google Scholar]
- 6.Ionov Y, Peinado M A, Malkhosyan S, Shibata D, Perucho M. Nature (London) 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 7.Welsh J, McClelland M. Nucleic Acids Res. 1990;25:7123–7128. doi: 10.1093/nar/18.24.7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peinado M A, Malkhosyan S, Velazquez A, Perucho M. Proc Natl Acad Sci USA. 1992;89:10065–10069. doi: 10.1073/pnas.89.21.10065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aaltonen L A, Peltomaki P, Leach F S, Sistonen P, Pylkkanen L, Mecklin J P, Jarvinen H, Powell S M, Jen J, Hamilton S R, et al. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 10.Thibodeau S N, Bren G, Schaid D. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 11.Peltomaki P, Aaltonen L A, Sistonen P, Pylkkanen L, Mecklin J P, Jarvinen H, Green J S, Jass J R, Weber J L, Leach F S, et al. Science. 1993;260:810–812. doi: 10.1126/science.8484120. [DOI] [PubMed] [Google Scholar]
- 12.Fishel R, Lescoe M K, Rao M R, Copeland N G, Jenkins N A, Garber J, Kane M, Kolodner R. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 13.Leach F S, Nicolaides N C, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen L A, Nystrom-Lahti M, et al. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 14.Malkhosyan S, Yasuda J, Soto J L, Sekiya T, Yokota J, Perucho M. Proc Natl Acad Sci USA. 1998;95:10170–10175. doi: 10.1073/pnas.95.17.10170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boland C R. In: The Genetic Basis of Human Cancer. Vogelstein B, Kinzler K W, editors. New York: McGraw–Hill; 1998. pp. 333–346. [Google Scholar]
- 16.Basik M, Stoler D L, Kontzoglou K C, Rodriguez-Bigas M A, Petrelli N J, Anderson G R. Genes Chromosomes Cancer. 1997;18:19–29. [PubMed] [Google Scholar]
- 17.Lengauer C, Kinzler K W, Vogelstein B. Nature (London) 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 18.Boland C R, Sato J, Appelman H D, Bresalier R S, Feinberg A P. Nat Med. 1995;1:902–909. doi: 10.1038/nm0995-902. [DOI] [PubMed] [Google Scholar]
- 19.Cahill D P, Lengauer C, Yu J, Riggins G J, Willson J K, Markowitz S D, Kinzler K W, Vogelstein B. Nature (London) 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 20.Laghi L, Randolph A E, Chauhan D P, Marra G, Major E O, Neel J V, Boland C R. Proc Natl Acad Sci USA. 1999;96:7484–7489. doi: 10.1073/pnas.96.13.7484. [DOI] [PMC free article] [PubMed] [Google Scholar]