

Abstract
The intrarenal renin-angiotensin system (RAS) has several pathophysiologic functions not only in blood pressure regulation but also in the development of glomerulonephritis (GN). Angiotensin II (Ang II) is the biologically active product of the RAS. Locally produced Ang II induces inflammation, renal cell growth, mitogenesis, apoptosis, migration, and differentiation, regulates the gene expression of bioactive substances, and activates multiple intracellular signaling pathways, leading to tissue damage. Activation of the Ang II type 1 (AT1) receptor pathway results in the production of proinflammatory mediators, cell proliferation, and extracellular matrix synthesis, which facilitates glomerular injury. Previous studies have shown that angiotensin-converting enzyme inhibitors and/or AT1 receptor blockers have beneficial effects in experimental GN models and humans with various types of GN, and that these effects are more significant than their suppressive effects on blood pressure. In this paper, we focus on intrarenal RAS activation in the pathophysiology of experimental models of GN.
1. Introduction
The role of the renin-angiotensin system (RAS) in blood pressure regulation and sodium and fluid homeostasis is well recognized [1, 2]. The biologically active peptides that are formed from angiotensinogen (AGT) include angiotensin II (Ang II) and Ang 1-7. The balance between the vasoconstricting actions of Ang II, which are mediated by the Ang II type 1 (AT1) receptor, are countered by the vasodilating actions of Ang II, which are mediated by the AT2 receptor [3] and the actions of Ang 1-7 on the G protein-coupled receptor Mas [4]. Formation of Ang II is dependent upon the substrate availability of AGT and Ang I and the activities of renin, angiotensin-converting enzyme (ACE), ACE2, and ACE-dependent enzymatic pathways, including serine proteases, tonin, cathepsin G, trypsin, and kallikrein. The actions of Ang II are determined by signaling via AT1 and AT2 receptors and the putative Ang 1-7 receptor Mas [5].
Local/tissue RAS in specific tissues has become the focus of much recent interest [6]. Emerging evidence has demonstrated the importance of tissue-specific RAS in the brain [7], heart [8], adrenal glands [9], vasculature [10, 11], and the kidneys [12]. Renal RAS in particular is unique, because all of the components necessary to generate intrarenal Ang II are present along the nephron in both interstitial and intratubular compartments [2, 5]. AGT is the only known substrate for renin that is a rate-limiting enzyme of the RAS. Because the level of AGT is close to the Michaelis-Menten constant for renin, not only renin levels but also AGT levels can control RAS activity, and AGT upregulation may lead to elevated angiotensin protein levels and increased blood pressure [13]. Recent studies have shown that AGT plays an important role in the development and progression of hypertension and kidney disease [2, 12]. Renin mRNA and renin-like activity have been observed in cultured proximal tubular cells [14]. The brush border membrane of proximal human kidney tubules also expresses abundant levels of ACE, mRNA [15], and protein [16]. ACE has been measured in proximal and distal tubular fluid, with greater concentrations observed in proximal tubule fluid [17]. Therefore, all the major components required to generate Ang II are expressed within the kidneys [2, 12].
Chronic glomerulonephritis (GN) resulting in substantial renal damage is frequently characterized by relentless progression to end-stage renal disease. Renal Ang II, the production of which is enhanced in chronic GN, can elevate intraglomerular pressure, increase glomerular cell hypertrophy, and augment extracellular matrix (ECM) accumulation [18, 19] (Figure 1). ACE inhibitors and/or AT1 receptor blockers (ARBs) are often administered to patients with proteinuric nephropathies [20, 21]. The efficacy of these agents in this indication suggests that factors other than Ang II play an important role in the progression of renal disease. This paper explores recent findings concerning the involvement of intrarenal RAS activation in experimental models of GN.
Figure 1.
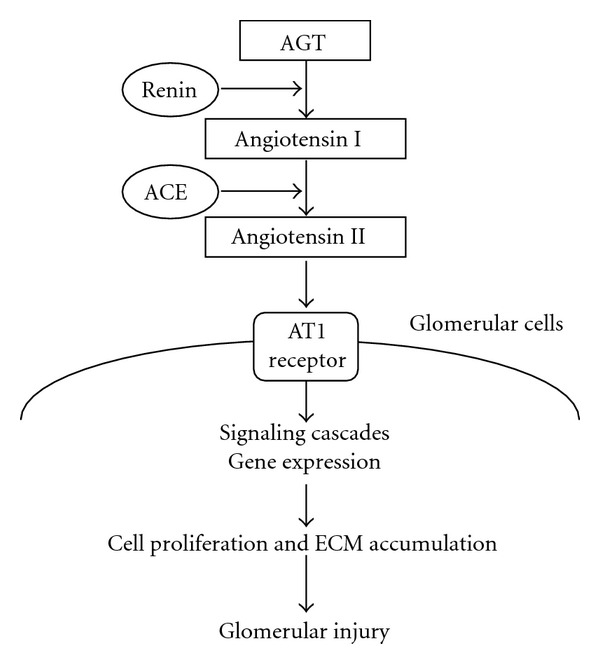
Working schematic of the renin-angiotensin system in glomerulonephritis. AGT: angiotensinogen, ACE: angiotensin-converting enzyme, AT1 receptor: angiotensin II type 1 receptor, and ECM: extracellular matrix.
2. RAS Activation in a Model of Progressive Mesangioproliferative GN
Anti-Thy-1 antibody-induced GN is the most common model of experimental nephritis [22, 23], because selective damage to mesangial cells (MCs) allows for the study of mesangial function and pathophysiology. This antibody-antigen reaction initiates complement activation to form a membrane attack complex. Repeated anti-Thy-1 antibody injections may produce progressive glomerular lesions ending in sclerosis, resembling human progressive GN [24]. Glomerulosclerosis is characterized by a continuative accumulation of the ECM, due to increased synthesis and decreased degeneration of the ECM, and overproduction of transforming growth factor (TGF)-β1 in the glomerulus. Furthermore, uninephrectomized rats treated with a single injection of the anti-Thy-1 antibody develop hypertension, massive proteinuria, and severe glomerular injury, finally resulting in chronic mesangioproliferative glomerulosclerosis [25].
To elucidate the involvement of intrarenal RAS activation in the development of glomerulosclerosis during the course of anti-Thy-1 GN with uninephrectomy, we performed an interventional study using the ARB candesartan in a rat model of progressive mesangioproliferative GN [26]. Reactive oxygen species (ROS) produced by NADPH oxidase have been implicated in the development and progression of GN [27, 28]. Ang II induces the activation of NADPH oxidase and the development of oxidative stress in GN [29]. We therefore also examined whether the interaction between the RAS and ROS is important for the development of progressive GN. Nephritis was induced in rats by a single intravenous injection of 2 mg of the nephritogenic anti-Thy-1 antibody 1 week after uninephrectomy. Rats were divided into 4 groups and administered daily oral doses of (1) vehicle, (2) 1% probucol, a free radical scavenger, (3) 70 mg/L candesartan in drinking water, or (4) probucol plus candesartan. Rats in each group were killed at 56 days after the injection of anti-Thy1-1 antibody. As controls, rats were injected with phosphate-buffered saline 1 week after unilateral nephrectomy and were killed on day 56. The ARB candesartan was found to considerably reduce proteinuria, the level of TGF-β1, and ECM accumulation, finally inhibiting the progression of glomerulosclerosis. The combination of probucol and candesartan not only completely eliminated NADPH oxidase components and superoxide production, but also normalized urinary protein excretion and TGF-β1 expression and prevented ECM accumulation, resulting in full prevention of the progression of GN. It seems likely that the beneficial effect of such combined treatment is due to the synergistic action of Ang II inhibition with a receptor antagonist and the elimination of ROS with a radical scavenger. These findings suggest that RAS activation and NADPH oxidase-associated ROS production may play a pivotal role in the progression of GN.
Hydrogen peroxide-inducible clone-5 (Hic-5) was originally discovered as a gene that is induced by TGF-β1 and hydrogen peroxide (H2O2), in a study on the growth-inhibitory functions of TGF-β1 in cellular senescence [30]. Recently, Hic-5 has been shown to function as an adaptor protein in focal adhesions and to be involved in integrin-mediated signal transduction, remodeling of the actin cytoskeleton, and regulation of the cellular phenotype [31–33]. To investigate the significance of Hic-5 expression in GN, we analyzed the changes in Hic-5 expression in a rat model of progressive mesangial proliferative GN and examined the combined effects of an ARB and probucol on Hic-5 expression in GN [34]. Glomerular Hic-5 expression increased in parallel with α-smooth muscle actin (SMA) expression in progressive mesangial proliferative GN. Combined therapy with an ARB and probucol in this model improved the histology and expression of Hic-5 and α-SMA. These results suggest that Hic-5 is involved in changes in the MC phenotype, which produce abnormal ECM remolding in GN.
The mitogen-activated protein kinase (MAPK) signaling pathway is a highly conserved module involved in various cellular functions, including cell proliferation, cell survival, differentiation, and migration [35]. Extracellular stimuli, such as growth factors and environmental stress, induce sequential activation of the MAPK cascade. At least 4 members of the MAPK family have been identified, namely, extracellular signal-regulated kinase 1/2 (ERK1/2), c-Jun-amino-terminal kinase, p38, and ERK5 [35]. It was recently reported that ERK1/2 activation occurs in the rat Thy-1 model of mesangioproliferative nephritis, and that blocking of the ERK1/2 pathway results in a significant reduction in MC proliferation in this model [36]. In addition, ERK1/2 activation in human glomerulopathies is associated with cell proliferation, histologic lesions, and renal dysfunction [37]. ERK5-mediated MC growth has been reported to be involved in the pathogenesis of diabetic nephropathy [38]. To further elucidate whether ERK signaling is involved in the pathogenesis of GN, we examined the expression and phosphorylation levels of glomerular ERK signals in progressive models of rat mesangioproliferative GN characterized by MC proliferation and ECM accumulation. In addition, the potential role of ERK signaling in MC-mediated pathologic mesangial remodeling was investigated in cultured MCs [39]. Glomerular alterations in progressive models of rat mesangial proliferative GN were examined on days 3, 7, 14, 28, and 56 after anti-Thy1 antibody injection. Light microscopy revealed that almost all glomeruli exhibited faint MC proliferation on day 3, followed by mesangial matrix expansion on day 7. Thereafter, massive accumulation of mesangial ECM and MC proliferation were the prominent features of nephritic glomeruli on days 28 and 56. Immunostaining of kidneys obtained at different time points revealed that phospho-ERK1/2 expression increased on day 7 during the phase of enhanced MC proliferation and decreased thereafter. On the other hand, phospho-ERK5 was weakly expressed in control glomeruli but dramatically increased in a typical mesangial pattern after 28 and 56 days of GN. Semi-quantitative assessment indicated that glomerular phospho-ERK5 expression closely paralleled the accumulation of ECM and collagen type 1, as well as glomerular expression of ROS and Ang II. The in vitro study revealed that H2O2 and Ang II each induced ERK5 phosphorylation by cultured MCs. Costimulation with both H2O2 and Ang II synergistically increased ERK5 phosphorylation in MCs. Cultured MCs transfected with ERK5-specific small interference RNA showed a significant decrease in H2O2 or Ang II-induced cell viability and soluble collagen secretion compared with control cells. Finally, treatment of GN rats with an ARB resulted in a significant decrease in phospho-ERK5 expression and collagen accumulation, accompanied by remarkable histologic improvement. Taken together, these results suggest that MC ERK5 phosphorylation by Ang II or H2O2 enhances cell viability and ECM accumulation in chronic GN.
Several studies demonstrated that the MAPK signaling controls cell behaviors via under RAS activation in other experimental GN models [40–42]. In diabetic conditions, high glucose generates ROS as a result of glucose auto-oxidation, metabolism, and formation of advanced glycation end production [43]. All these signaling molecules are involved into MAPK signaling pathways in glomerular cells [44]. High-glucose-induced diabetic complications have been implicated, in part, to the activation of MAPK [44]. Interestingly, the stimulation of Ang II by hyperglycemia or oxidative stress activates the MAPK cascade [45]. These results suggest that high-glucose-induced ROS/MAPK pathway and intrarenal RAS activation play key roles in diabetic nephropathy.
3. RAS Activation in a Model of Crescentic GN
Crescentic GN, also known as antiglomerular basement membrane (anti-GBM) disease or Goodpasture's syndrome, is characterized by the formation and deposition of antibodies on the basement membranes of glomeruli and alveoli [46]. The disease progresses rapidly, and patients present with renal failure, dyspnea, hemoptysis, a sudden decrease in hemoglobin level, pallor, and circulatory disturbances. Understanding the underlying proinflammatory responses may help to facilitate the identification of therapeutic targets for arresting the progression of anti-GBM disease. In Wistar-Kyoto (WKY) rats, the administration of a minute dose of anti-GBM antibodies induces severe proliferative and necrotizing GN with crescent formation [47, 48]. In rat models of anti-GBM disease, glomerular infiltration by T lymphocytes, monocytes/macrophages, and some neutrophils is the earliest and most prominent pathologic change [48]. To investigate whether local RAS activation occurs in nephritic glomeruli with crescent formation and whether the final effector molecule Ang II contributes to the induction of ROS and inflammation, as well as glomerular pathologic alterations, we studied the effects of an ARB on rat anti-GBM antibody-induced GN by evaluating indexes of glomerular RAS activation, oxidative stress, inflammation, and TGF-β1 expression in GN rats [49]. Progressive anti-GBM GN was induced in 7-week-old male WKY rats by a single injection of anti-GBM antiserum. Vehicle-treated nephritic rats showed severe proteinuria and developed crescentic GN accompanied by marked macrophage infiltration and enhanced expression of glomerular α-SMA, AGT, Ang II, AT1 receptor, and NADPH oxidase on day 28. Treatment with an ARB improved proteinuria and pathologic alterations such as crescent formation and glomerulosclerosis, and had a significant inhibitory effect on these parameters on day 28 of GN. Enhanced superoxide production in nephritic glomeruli was also decreased by the ARB. Moreover, Ang II and TGF-β1 in the supernatant of cultured glomeruli was increased significantly in vehicle-treated nephritic rats, whereas the production of these compounds was significantly inhibited in ARB-treated rats on day 28. These findings indicate that increased glomerular RAS activity and the resulting increase in Ang II production plays an important role in progressive glomerular injury by inducing oxidative stress and TGF-β1 expression.
Recent studies have revealed that monocyte chemoattractant protein-1 (MCP-1) is involved in the pathogenesis of crescentic GN [50]. MCP-1 is presumed to be a key mediator of chemotaxis and the activation of macrophages [51]. Chronic Ang II infusion in rats activates MCP-1 and TGF-β1, which in turn induces macrophage infiltration in renal tissues [52]. CC chemokine receptor 2 (CCR2), a cognate receptor of MCP-1 expressed mainly on monocytes, has been reported to be involved in human crescentic GN [53]. Based on these principles, we hypothesized that therapeutic management of anti-GBM disease may be achieved by blocking the MCP-1/CCR2 signaling pathway and RAS [54]. Whereas treatment with a CCR2 antagonist (CA) or ARB alone only moderately ameliorated kidney injury in a rat model of crescentic GN, combination treatment with a CA and an ARB dramatically prevented proteinuria and markedly reduced glomerular crescent formation. Further, combination treatment suppressed macrophage infiltration, reduced MCP-1, AGT, Ang II, and TGF-β1 expression, and reversed the fibrotic change in glomeruli. Primary cultured glomerular MCs stimulated by Ang II showed significant increases in MCP-1 and TGF-β1 expression. Furthermore, a coculture model consisting of MCs, parietal epithelial cells, and macrophages showed an increase in Ang II-induced cell proliferation and collagen secretion. ARB treatment attenuated these effects. These data suggest that Ang II enhances glomerular crescent formation, and inhibition of the MCP-1/CCR2 pathway with a CA/ARB combination effectively reduces renal injury in anti-GBM nephritis.
Recent reports indicated roles of RAS activation and MCP-1/CCR2 pathway in other nephritis models [55–58]. Most studies concluded that activation of RAS is an important determinant of local MCP-1 expression, either directly or indirectly through glomerular hemodynamic effects. However, whether the manipulation of MCP-1/CCR2 pathway and RAS is beneficial with respect to the progression of human and experimental GN remains to be investigated.
4. Conclusion
The present studies reveal that intrarenal RAS activation has important pathophysiologic functions in the development of progressive mesangioproliferative and crescentic GN. Additional studies are needed to determine the relationship between RAS activation and glomerular injury, such as cell proliferation, sclerosis, and crescent formation, and to clarify the mechanisms underlying Ang II-induced pathologic glomerular changes. Furthermore, it is necessary to determine whether RAS inhibition may provide a clinically significant pharmacologic strategy for the therapeutic treatment of progressive GN.
Acknowledgments
The authors acknowledge the critical discussion and/or excellent technical assistance from Maki Shimizu, Masanori Takamatsu, Kenichi Suga, Sato Matuura, Naomi Okamoto (The University of Tokushima Graduate School), and L. Gabriel Navar, Hiroyuki Kobori (Tulane University). These studies were supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the National Institute of Diabetes and Digestive and Kidney Diseases and the National Center for Research Resources.
References
- 1.Navar LG, Harrison-Bernard LM, Imig JD, Wang CT, Cervenka L, Mitchell KD. Intrarenal angiotensin II generation and renal effects of AT1 receptor blockade. Journal of the American Society of Nephrology. 1999;10(supplement 12):S266–S272. [PubMed] [Google Scholar]
- 2.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacological Reviews. 2007;59(3):251–287. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 3.Carey RM, Siragy HM. The intrarenal renin-angiotensin system and diabetic nephropathy. Trends in Endocrinology and Metabolism. 2003;14(6):274–281. doi: 10.1016/s1043-2760(03)00111-5. [DOI] [PubMed] [Google Scholar]
- 4.Santos RAS, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8258–8263. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navar LG, Prieto-Carrasquero MC, Kobori H. Chapter 1: Molecular Aspects of the Renal Renin-Angiotensin System. 1st edition. Oxfordshine, UK: Taylor & Francis Medical; 2006. [Google Scholar]
- 6.Dzau VJ, Re R. Tissue angiotensin system in cardiovascular medicine: a paradigm shift? Circulation. 1994;89(1):493–498. doi: 10.1161/01.cir.89.1.493. [DOI] [PubMed] [Google Scholar]
- 7.Baltatu O, Silva JA, Jr., Ganten D, Bader M. The brain renin-angiotensin system modulates angiotensin II-induced hypertension and cardiac hypertrophy. Hypertension. 2000;35(1, part 2):409–412. doi: 10.1161/01.hyp.35.1.409. [DOI] [PubMed] [Google Scholar]
- 8.Dell’Italia LJ, Meng QC, Balcells E, et al. Compartmentalization of angiotensin II generation in the dog heart: evidence for independent mechanisms in intravascular and interstitial spaces. Journal of Clinical Investigation. 1997;100(2):253–258. doi: 10.1172/JCI119529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mazzocchi G, Malendowicz LK, Markowska A, Albertin G, Nussdorfer GG. Role of adrenal renin-angiotensin system in the control of aldosterone secretion in sodium-restricted rats. American Journal of Physiology. 2000;278(6):E1027–E1030. doi: 10.1152/ajpendo.2000.278.6.E1027. [DOI] [PubMed] [Google Scholar]
- 10.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circulation Research. 1994;74(6):1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 11.Danser AHJ, Admiraal PJJ, Derkx FHM, Schalekamp MADH. Angiotensin I-to-II conversion in the human renal vascular bed. Journal of Hypertension. 1998;16(12, part 2):2051–2056. doi: 10.1097/00004872-199816121-00029. [DOI] [PubMed] [Google Scholar]
- 12.Navar LG, Harrison-Bernard LM, Nishiyama A, Kobori H. Regulation of intrarenal angiotensin II in hypertension. Hypertension. 2002;39(2, part 2):316–322. doi: 10.1161/hy0202.103821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brasier AR, Li J. Mechanisms for inducible control of angiotensinogen gene transcription. Hypertension. 1996;27(3, part 2):465–475. doi: 10.1161/01.hyp.27.3.465. [DOI] [PubMed] [Google Scholar]
- 14.Henrich WL, McAllister EA, Eskue A, Miller T, Moe OW. Renin regulation in cultured proximal tubular cells. Hypertension. 1996;27(6):1337–1340. doi: 10.1161/01.hyp.27.6.1337. [DOI] [PubMed] [Google Scholar]
- 15.Sibony M, Gasc JM, Soubrier F, Alhenc-Gelas F, Corvol P. Gene expression and tissue localization of the two isoforms of angiotensin I converting enzyme. Hypertension. 1993;21(6, part 1):827–835. doi: 10.1161/01.hyp.21.6.827. [DOI] [PubMed] [Google Scholar]
- 16.Vío CP, Jeanneret VA. Local induction of angiotensin-converting enzyme in the kidney as a mechanism of progressive renal diseases. Kidney International, Supplement. 2003;64(86):S57–S63. doi: 10.1046/j.1523-1755.64.s86.11.x. [DOI] [PubMed] [Google Scholar]
- 17.Casarini DE, Boim MA, Stella RCR, Krieger-Azzolini MH, Krieger JE, Schor N. Angiotensin I-converting enzyme activity in tubular fluid along the rat nephron. American Journal of Physiology. 1997;272(3, part 2):F405–F409. doi: 10.1152/ajprenal.1997.272.3.F405. [DOI] [PubMed] [Google Scholar]
- 18.Kohan DE. Angiotensin II and endothelin in chronic glomerulonephritis. Kidney International. 1998;54(2):646–647. doi: 10.1046/j.1523-1755.1998.00038.x. [DOI] [PubMed] [Google Scholar]
- 19.Urushihara M, Kobori H. Angiotensinogen expression is enhanced in the progression of glomerular disease. International Journal of Clinical Medicine. 2011;2(4):378–387. doi: 10.4236/ijcm.2011.24064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horita Y, Tadokoro M, Taura K, et al. Low-dose combination therapy with temocapril and losartan reduces proteinuria in normotensive patients with immunoglobulin A nephropathy. Hypertension Research. 2004;27(12):963–970. doi: 10.1291/hypres.27.963. [DOI] [PubMed] [Google Scholar]
- 21.Ruggenenti P, Perna A, Gherardi G, Gaspari F, Benini R, Remuzzi G. Renal function and requirement for dialysis in chronic nephropathy patients on long-term ramipril: REIN follow-up trial. The Lancet. 1998;352(9136):1252–1256. doi: 10.1016/s0140-6736(98)04433-x. [DOI] [PubMed] [Google Scholar]
- 22.Ishizaki M, Masuda Y, Fukuda Y. Experimental mesangioproliferative glomerulonephritis in rats induced by intravenous administration of anti-thymocyte serum. Acta Pathologica Japonica. 1986;36(8):1191–1203. doi: 10.1111/j.1440-1827.1986.tb02839.x. [DOI] [PubMed] [Google Scholar]
- 23.Sakai N, Iseki K, Suzuki S, et al. Uninephrectomy induces progressive glomerulosclerosis and apoptosis in anti-Thy1 glomerulonephritis. Pathology International. 2005;55(1):19–26. doi: 10.1111/j.1440-1827.2005.01781.x. [DOI] [PubMed] [Google Scholar]
- 24.Kawachi H, Iwanaga T, Toyabe S, Oite T, Shimizu F. Mesangial sclerotic change with persistent proteinuria in rats after two consecutive injections of monoclonal antibody 1-22-3. Clinical and Experimental Immunology. 1992;90(1):129–134. doi: 10.1111/j.1365-2249.1992.tb05844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura T, Obata JE, Kimura H, et al. Blocking angiotensin II ameliorates proteinuria and glomerular lesions in progressive mesangioproliferative glomerulonephritis. Kidney International. 1999;55(3):877–889. doi: 10.1046/j.1523-1755.1999.055003877.x. [DOI] [PubMed] [Google Scholar]
- 26.Kondo S, Shimizu M, Urushihara M, et al. Addition of the antioxidant probucol to angiotensin II type I receptor antagonist arrests progressive mesangioproliferative glomerulonephritis in the rat. Journal of the American Society of Nephrology. 2006;17(3):783–794. doi: 10.1681/ASN.2005050519. [DOI] [PubMed] [Google Scholar]
- 27.Gaertner SA, Janssen U, Ostendorf T, Koch KM, Floege J, Gwinner W. Glomerular oxidative and antioxidative systems in experimental mesangioproliferative glomerulonephritis. Journal of the American Society of Nephrology. 2002;13(12):2930–2937. doi: 10.1097/01.asn.0000034908.43113.5d. [DOI] [PubMed] [Google Scholar]
- 28.Budisavljevic MN, Hodge L, Barber K, et al. Oxidative stress in the pathogenesis of experimental mesangial proliferative glomerulonephritis. American Journal of Physiology. 2003;285(6):F1138–F1148. doi: 10.1152/ajprenal.00397.2002. [DOI] [PubMed] [Google Scholar]
- 29.Gorin Y, Ricono JM, Wagner B, et al. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochemical Journal. 2004;381, part 1:231–239. doi: 10.1042/BJ20031614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibanuma M, Mashimo JI, Kuroki T, Nose K. Characterization of the TGFβ1-inducible hic-5 gene that encodes a putative novel zinc finger protein and its possible involvement in cellular senescence. Journal of Biological Chemistry. 1994;269(43):26767–26774. [PubMed] [Google Scholar]
- 31.Matsuya M, Sasaki H, Aoto H, et al. Cell adhesion kinase β forms a complex with a new member, Hic-5, of proteins localized at focal adhesions. Journal of Biological Chemistry. 1998;273(2):1003–1014. doi: 10.1074/jbc.273.2.1003. [DOI] [PubMed] [Google Scholar]
- 32.Tumbarello DA, Turner CE. Hic-5 contributes to epithelial-mesenchymal transformation through a RhoA/ROCK-dependent pathway. Journal of Cellular Physiology. 2007;211(3):736–747. doi: 10.1002/jcp.20991. [DOI] [PubMed] [Google Scholar]
- 33.Dabiri G, Tumbarello DA, Turner CE, Van de Water L. Hic-5 promotes the hypertrophic scar myofibroblast phenotype by regulating the TGF-β1 autocrine loop. Journal of Investigative Dermatology. 2008;128(10):2518–2525. doi: 10.1038/jid.2008.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suga K, Kondo S, Matsuura S, et al. Glomerular expression of hydrogen peroxide inducible clone-5 in human and rat progressive mesagnial proliferative glomerulonephritis. Nephron Experimental Nephrology. 2012;120(2):e59–e68. doi: 10.1159/000335780. [DOI] [PubMed] [Google Scholar]
- 35.Nishimoto S, Nishida E. MAPK signalling: ERK5 versus ERK1/2. EMBO Reports. 2006;7(8):782–786. doi: 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bokemeyer D, Panek D, Kramer HJ, et al. In vivo identification of the mitogen-activated protein kinase cascade as a central pathogenic pathway in experimental mesangioproliferative glomerulonephritis. Journal of the American Society of Nephrology. 2002;13(6):1473–1480. doi: 10.1097/01.asn.0000017576.50319.ac. [DOI] [PubMed] [Google Scholar]
- 37.Masaki T, Stambe C, Hill PA, Dowling J, Atkins RC, Nikolic-Paterson DJ. Activation of the extracellular-signal regulated protein kinase pathway in human glomerulopathies. Journal of the American Society of Nephrology. 2004;15(7):1835–1843. doi: 10.1097/01.asn.0000130623.66271.67. [DOI] [PubMed] [Google Scholar]
- 38.Suzaki Y, Yoshizumi M, Kagami S, et al. BMK1 is activated in glomeruli of diabetic rats and in mesangial cells by high glucose conditions. Kidney International. 2004;65(5):1749–1760. doi: 10.1111/j.1523-1755.2004.00576.x. [DOI] [PubMed] [Google Scholar]
- 39.Urushihara M, Takamatsu M, Shimizu M, et al. ERK5 activation enhances mesangial cell viability and collagen matrix accumulation in rat progressive glomerulonephritis. American Journal of Physiology Renal Physiology. 2009;298(1):F167–F176. doi: 10.1152/ajprenal.00124.2009. [DOI] [PubMed] [Google Scholar]
- 40.Okada H, Inoue T, Kikuta T, et al. A possible anti-inflammatory role of angiotensin II type 2 receptor in immune-mediated glomerulonephritis during type 1 receptor blockade. American Journal of Pathology. 2006;169(5):1577–1589. doi: 10.2353/ajpath.2006.060178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohashi N, Urushihara M, Satou R, Kobori H. Glomerular angiotensinogen is induced in mesangial cells in diabetic rats via reactive oxygen speciesERK/JNK pathways. Hypertension Research. 2010;33(11):1174–1181. doi: 10.1038/hr.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lakshmanan AP, Thandavarayan RA, Watanabe K, et al. Modulation of AT-1R/MAPK cascade by an olmesartan treatment attenuates diabetic nephropathy in streptozotocin-induced diabetic mice. Molecular and cellular endocrinology. 2012;348(1):104–111. doi: 10.1016/j.mce.2011.07.041. [DOI] [PubMed] [Google Scholar]
- 43.Ha H, Hi Bahl Lee Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney International, Supplement. 2000;58(77):S19–S25. doi: 10.1046/j.1523-1755.2000.07704.x. [DOI] [PubMed] [Google Scholar]
- 44.Tomlinson DR. Mitogen-activated protein kinases as glucose transducers for diabetic complications. Diabetologia. 1999;42(11):1271–1281. doi: 10.1007/s001250051439. [DOI] [PubMed] [Google Scholar]
- 45.Tsiani E, Lekas P, Fantus IG, et al. High glucose-enhanced activation of mesangial cell p38 MAPK by ET-1, ANG II, and platelet-derived growth factor. American Journal of Physiology. 2002;282(1):E161–E169. doi: 10.1152/ajpendo.2002.282.1.E161. [DOI] [PubMed] [Google Scholar]
- 46.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. The New England Journal of Medicine. 2003;348(25):2543–2556. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 47.Shimizu M, Kondo S, Urushihara M, et al. Role of integrin-linked kinase in epithelial-mesenchymal transition in crescent formation of experimental glomerulonephritis. Nephrology Dialysis Transplantation. 2006;21(9):2380–2390. doi: 10.1093/ndt/gfl243. [DOI] [PubMed] [Google Scholar]
- 48.Wada T, Yokoyama H, Furuichi K, et al. Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (MCAF/MCP-1) The FASEB Journal. 1996;10(12):1418–1425. [PubMed] [Google Scholar]
- 49.Kinoshita Y, Kondo S, Urushihara M, et al. Angiotensin II type I receptor blockade suppresses glomerular renin-angiotensin system activation, oxidative stress, and progressive glomerular injury in rat anti-glomerular basement membrane glomerulonephritis. Translational Research. 2011;158(4):235–248. doi: 10.1016/j.trsl.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Wada T, Furuichi K, Sakai N, et al. A new anti-inflammatory compound, FR167653, ameliorates crescentic glomerulonephritis in Wistar-Kyoto rats. Journal of the American Society of Nephrology. 2000;11(8):1534–1541. doi: 10.1681/ASN.V1181534. [DOI] [PubMed] [Google Scholar]
- 51.Matsushima K, Larsen CG, DuBois GC, Oppenheim JJ. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. Journal of Experimental Medicine. 1989;169(4):1485–1490. doi: 10.1084/jem.169.4.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozawa Y, Kobori H, Suzaki Y, Navar LG. Sustained renal interstitial macrophage infiltration following chronic angiotensin II infusions. The American Journal of Physiology. 2007;292(1):F330–F339. doi: 10.1152/ajprenal.00059.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Segerer S, Cui Y, Hudkins KL, et al. Expression of the chemokine monocyte chemoattractant protein-1 and its receptor chemokine receptor 2 in human crescentic glomerulonephritis. Journal of the American Society of Nephrology. 2000;11(12):2231–2242. doi: 10.1681/ASN.V11122231. [DOI] [PubMed] [Google Scholar]
- 54.Urushihara M, Ohashi N, Miyata K, Satou R, Acres OW, Kobori H. Addition of angiotensin II Type 1 receptor blocker to CCR2 antagonist markedly attenuates crescentic glomerulonephritis. Hypertension. 2011;57(3):586–593. doi: 10.1161/HYPERTENSIONAHA.110.165704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wada T, Matsushima K, Kobayashi KI. Monocyte chemoattractant protein-1: does it play a role in diabetic nephropathy? Nephrology Dialysis Transplantation. 2003;18(3):457–459. doi: 10.1093/ndt/18.3.457. [DOI] [PubMed] [Google Scholar]
- 56.Kato S, Luyckx VA, Ots M, et al. Renin-angiotensin blockade lowers MCP-1 expression in diabetic rats. Kidney International. 1999;56(3):1037–1048. doi: 10.1046/j.1523-1755.1999.00643.x. [DOI] [PubMed] [Google Scholar]
- 57.Zoja C, Corna D, Rottoli D, et al. Effect of combining ACE inhibitor and statin in severe experimental nephropathy. Kidney International. 2002;61(5):1635–1645. doi: 10.1046/j.1523-1755.2002.00332.x. [DOI] [PubMed] [Google Scholar]
- 58.Chen YM, Hu-Tsai MI, Lin SL, Tsai TJ, Hsieh BS. Expression of CX3CL1/fractalkine by mesangial cells in vitro and in acute anti-Thy1 glomerulonephritis in rats. Nephrology Dialysis Transplantation. 2003;18(12):2505–2514. doi: 10.1093/ndt/gfg457. [DOI] [PubMed] [Google Scholar]