

Abstract
This G-rich region of the c-MYC promoter has been shown to form a G-quadruplex structure, acting as a silencer element for c-MYC transcriptional control. In the present work, we have synthesized a series of 11-substituted quindoline analogs as c-MYC G-quadruplex–stabilizing compounds, and the cell-free and in vitro activity of these compounds were evaluated. Two lead compounds (4 and 12) demonstrated good cell-free profiles, and compound 4 (2-(4-(10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethylethanamine) significantly downregulated c-MYC expression. However, despite the good cell-free activity and the effect of these compounds on c-MYC gene expression, we have demonstrated, using a cellular assay in a Burkitt’s lymphoma cell line (CA46-specific), that these effects were not mediated through targeting the c-MYC G-quadruplex. Thus, caution should be used in assigning the effects of G-quadruplex-interactive compounds that lower c-MYC to direct targeting of these promoter elements unless this assay, or similar ones, demonstrates direct targeting of the G-quadruplex in cells.
INTRODUCTION
The MYC family of proteins [c-, N-, L-] is involved in promoting cell growth, proliferation, and apoptotic processes under cell stress. c-MYC performs these functions through the regulation of gene expression.1–3 Overexpression of the c-MYC oncogene is linked with cellular proliferation and inhibition of differentiation, leading to its association with a wide range of human cancers, including colon, breast, small-cell lung, osteosarcomas, glioblastomas, lymphoma, and myeloid leukemia.4–6 The transcriptional regulation of c-MYC expression involves multiple promoters and transcriptional start sites, with P1 and P2 being the predominant promoters. The nuclease hypersensitive element (NHE) III1 of the P1 promoter controls 85–90% of c-MYC transcription, and the guanine-rich strand of the promoter element has been shown to form an intramolecular G-quadruplex structure that appears to act as a silencer element when stabilized by the G-quadruplex-interactive compound TMPyP4.7,8
Recently, a variety of small molecules have been synthesized and tested for their ability to interact with c-MYC G-quadruplex. Some of these compounds include quindolines, berberine, methylene blue, and bisaryl diketene analogs.9–12 Quindoline compounds were first reported as telomeric G-quadruplex-interactive agents by Guyen and co-workers.13 Ou et al. synthesized and tested a series of 11-substituted quindoline analogs for G-quadruplex stabilization and c-MYC downregulation.11 Quindoline-i (N1, N1-diethyl-N2-(10H-indolo [3,2-b]quinolin-11-yl)ethane-1,2-diamine) exhibited the best c-MYC G-quadruplex stabilization, and this correlates with c-MYC downregulation and effects on cell proliferation in HepG2 cell lines. Targeted effects of the quindoline analogs were supported by the differential effects in Ramos and CA46 Burkitt’s lymphoma cell lines.
While quindoline-i was an effective downregulator of c-MYC expression, recently published evidence does not support intracellular stabilization of the NHE III1 promoter G-quadruplex as its mechanism of action.14 In comparison, there is much stronger evidence to show that an ellipticine compound exerts its intracellular effects through the c-MYC G-quadruplex, confirming the in vitro existence and targetability of the G-quadruplex structure.14 Here we describe the design, synthesis, and screening of 11-piperazinyl quindolines as c-MYC G-quadruplex–stabilizing compounds in an effort to gain target specificity, with activity in cancer cell lines, and to confirm the mechanism of action. We propose that the addition of a piperazine ring would provide steric bulk to the planar quindoline ring, resulting in increased G-quadruplex binding and reduced duplex DNA binding, which would lead to better selectivity and provide an opportunity to add substituents with varying properties and linker lengths. We synthesized a series of disubstituted analogs with the addition of a second side chain at two different positions on the quindoline ring, which generally increased G-quadruplex stabilization while maintaining cellular potency. A number of these compounds demonstrated good thermal stabilization of the c-MYC G-quadruplex, with minimal effects on other promoters. We also examined the mechanism of action of these compounds using the newly described “CA46 exon-specific” assay, which is presently the only cellular assay that can discriminate between lowering of c-MYC through direct binding to the G-quadruplex in the NHE versus a secondary effect.
MATERIALS AND METHODS
Chemical Synthesis
Polyphosphoric acid, phosphorus oxychloride and 3-(piperazin-1-yl)propan-1-ol were purchased from Alfa Aesar. 1-(3-Methoxypropyl)piperazine and 1-(2-(piperidin-1-yl)ethyl)piperazine were bought from Oakwood Chemicals and 4-(2-(piperazin-1-yl)ethyl)morpholine was purchased from AK Scientific. (4-(2-morpholinoethyl)phenyl)boronic acid was obtained from CombiBlocks. All other chemicals were purchased from Sigma-Aldrich. All the solvents were obtained from Fischer Scientific. Flash chromatography was performed with silica gel (230/400 mesh, Fisher Scientific). All anhydrous reactions were carried out under positive pressure of nitrogen or argon. HPLC-MS analyses were performed on an Agilent 1100 series instrument with a Zorbax C18 reverse-phase column. HRMS results were obtained on an apex-Qe instrument. All 1H-NMR and 13C-NMR spectra were recorded on a Bruker 300 MHz or DRX 500 MHz NMR spectrometer, using deuterated solvents. The spectra are reported in ppm and referenced to deuterated DMSO (2.49 ppm for 1H, 39.5 ppm for 13C) or deuterated chloroform (7.26 ppm for 1H, 77 ppm for 13C). All compounds were analyzed for purity by HPLC using either MS or UV absorbance detectors. All final compounds showed ≥ 95 % purity.
Synthesis of Quindoline-i (N1,N1-diethyl-N2-(10H-indolo[3,2-b]quinolin-11-yl)ethane-1,2-diamine)
Quindoline-i was synthesized from 11-chloroquindoline and N,N-diethylethane-1,2-diamine. Detailed procedures and spectral data have been reported earlier.14
General Procedure for the Synthesis of Compounds 2–10
A mixture of 11-chloro-10H-indolo[3,2-b]quinoline 1 and the corresponding amine was heated at an appropriate temperature for a specific time. After cooling, the reaction mixture was poured into ice-water. The precipitated solid was filtered, washed with water, and then dried. The crude product was purified using 2–5 % methanol in chloroform as a solvent by silica gel column chromatography.
11-(piperazin-1-yl)-10H-indolo[3,2-b]quinoline (2)
Compound 1 (100 mg, 0.39 mmol) was reacted with piperazine (1 g) at 100 °C for 24 h to obtain 97 mg (80%) of compound 2 as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 10.91 (s, 1H, NH), 8.43–8.24 (m, 2H, ArH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 7.70–7.48 (m, 4H, ArH), 7.25 (t, J = 7.5 Hz, 1H, ArH), 3.64–3.45 (m, 4H), 3.14–2.97 (m, 4H), 2.75 (br s,1H, NH). 13C NMR (75 MHz, DMSO-d6): δ 161.68, 148.14, 145.87, 144.78, 136.70, 130.32, 129.97, 127.23, 127.01, 125.12, 124.21, 122.12, 121.94, 120.21, 112.89, 51.25, 46.23. MS (ESI): m/z = 303.2 [100%, (M+H)+]. HRMS calcd for C19H19N4 [M+H]+ 303.1604, found 303.1603. HPLC MS purity 99.02%.
11-(4-methylpiperazin-1-yl)-10H-indolo[3,2-b]quinoline (3)
Compound 3 was obtained from 1 (500 mg, 1.98 mmol) and 1-methyl piperazine (5 mL) heated at 100 °C for 12 h. Yield of compound 3 = 530 mg (84%). 1H NMR (300 MHz, DMSO-d6): δ 10.88 (s, 1H, NH), 8.35–8.27 (m, 2H, ArH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 7.68–7.48 (m, 4H, ArH), 7.25 (t, J = 7.1Hz, 1H, ArH), 3.62–3.38 (m, 4H), 2.85–2.56 (m, 4H), 2.35 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 148.16, 145.83, 144.78, 136.78, 130.30, 129.90, 127.26, 127.02, 125.10, 124.28, 124.12, 122.11, 121.94, 120.20, 112.89, 56.31, 51.36, 46.99. MS (ESI): m/z = 317.2 [100%, (M+H)+]. HRMS calcd for C20H21N4 [M+H]+ 317.1761, found 317.1757. HPLC MS purity 98.05%.
2-(4-(10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethylethanamine (4)
Compound 4 was synthesized following the general procedure using 1 (1 g, 3.96 mmol) and 1-[2-(dimethylamino)ethyl] piperazine (5 mL) heated at 100 °C for 16 h to give 1.1 g (79%) of 4 as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 10.88 (s, 1H, NH), 8.38–8.27 (m, 2H, ArH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 7.69–7.49 (m, 4H, ArH), 7.25 (t, J = 7.8 Hz, 1H, ArH), 3.58-3.44 (m, 4H), 2.90–2.65 (m, 4H), 2.62–2.53 (m, 2H), 2.48–2.40 (m, 2H), 2.19 (s, 6H). 13C NMR (75 MHz, DMSO-d6): δ 148.17, 145.80, 144.83, 136.74, 130.28, 129.90, 127.25, 127.00, 125.09, 124.30, 124.13, 122.08, 121.93, 120.19, 112.92, 56.85, 56.11, 54.61, 51.49, 45.88. MS (ESI): m/z = 374.4 [100%, (M+H)+]. HRMS calcd for C23H28N5 [M+H]+ 374.2339, found 374.2334. HPLC MS purity 99.11%.
4-(2-(4-(10H-indolo [3,2-b]quinolin-11-yl)piperazin-1-yl)ethyl)morpholine (5)
Compound 1 (200 mg, 0.79 mmol) was heated with 1.5 mL of 4-(2-(piperazin-1-yl) ethyl)morpholine at 110 °C for 24 h to obtain 294 mg (89%) of compound 5 as a solid. 1H NMR (300 MHz, DMSO-d6): δ 10.86 (s, 1H, NH), 8.38–8.25 (m, 2H, ArH), 8.14 (d, J = 8.1 Hz, 1H, ArH), 7.68–7.50 (m, 4H, ArH), 7.25 (t, J = 7.8 Hz, 1H, ArH), 3.65–3.56 (m, 4H), 3.54–3.42 (m, 4H), 2.91–2.68 (m, 4H), 2.68–2.58 (m, 2H), 2.57–2.52 (m, 2H), 2.48–2.38 (m, 4H). 13C NMR (75 MHz, DMSO-d6): δ 148.19, 145.83, 144.82, 136.73, 130.28, 129.91, 127.25, 126.99, 125.08, 124.29, 124.13, 122.12, 121.93, 120.19, 112.90, 67.10, 56.78, 56.16, 54.80, 54.61, 51.56. MS (ESI): m/z = 416.3 [100%, (M+H)+]. HRMS calcd for C25H29N5O [M+H]+ 416.2445, found 416.2448. HPLC MS purity 100%.
11-(4-(2-(piperidin-1-yl)ethyl)piperazin-1-yl)-10H-indolo[3,2-b]quinoline (6)
Compound 6 was obtained from compound 1 (350 mg, 1.38 mmol) and 3 mL of 1-(2-(piperidin-1-yl)ethyl)piperazine heated at 120 °C for 12 h. Yield = 330 mg (57%). 1H NMR (300 MHz, DMSO-d6): δ 10.87 (s, 1H, NH), 8.38–8.25 (m, 2H, ArH), 8.13 (d, J = 8.1 Hz, 1H, ArH), 7.68–7.50 (m, 4H, ArH), 7.25 (t, J = 6.6 Hz, 1H, ArH), 3.60–3.43 (m, 4H), 2.90–2.67 (m, 4H), 2.65–2.53 (m, 2H), 2.50–2.45 (m, 2H), 2.44–2.30 (m, 4H), 1.58–1.45 (m, 4H), 1.45–1.35 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 148.18, 145.83, 144.82, 136.74, 130.27, 129.90, 127.24, 126.99, 125.07, 124.29, 124.13, 122.11, 121.92, 120.18, 112.90, 57.03, 56.39, 55.26, 54.79, 51.55, 26.34, 24.82. MS (ESI): m/z = 414.5 [100%, (M+H)+]. HRMS calcd for C26H31N5 [M+H]+ 414.2652, found 414.2658. HPLC MS purity 100%.
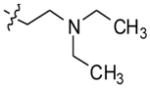
2-(4-(10H-indolo [3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-diethylethanamine (7)
Compound 7 was synthesized in 92% yield (735 mg) from compound 1 (500 mg, 1.98 mmol) and 3 mL of N,N-diethyl-2-(piperazin-1-yl)ethanamine heated at 100 °C for 48 h. 1H NMR (500 MHz, DMSO-d6): δ 10.90 (s, 1H, NH), 8.33 (d, J = 8.5 Hz, 1H, ArH), 8.29 (d, J = 7.5 Hz, 1H, ArH), 8.14 (d, J = 8.5 Hz, 1H, ArH), 7.68–7.52 (m, 4H, ArH), 7.25 (t, J = 8.0 Hz, 1H, ArH), 3.60–3.40 (m, 4H), 2.90–2.70 (m, 4H), 2.67–2.60 (m, 2H), 2.58–2.48 (m, 6H), 1.00 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, DMSO-d6): δ 148.18, 145.83, 144.82, 136.75, 130.27, 129.90, 127.25, 126.98, 125.07, 124.30, 124.13, 122.12, 121.92, 120.18, 112.90, 57.12, 54.86, 51.57, 50.93, 47.64, 12.68. MS (ESI): m/z = 402.3 [65%, (M+H)+], 201.6 (100%). HRMS calcd for C25H31N5 [M+H]+ 402.2652, found 402.2645. HPLC MS purity 100%.
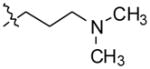
3-(4-(10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethylpropan1-amine (8)
Compound 8 was synthesized from 1 (500 mg, 1.98 mmol) and N,N-dimethyl-3-(piperazin-1-yl)propan-1-amine (2 mL) heated at 110 °C for 24 h to obtain 633 mg (82%) of 8 as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 10.86 (s, 1H, NH), 8.40–8.25 (m, 2H, ArH), 8.14 (d, J = 8.4 Hz, 1H, ArH), 7.68–7.50 (m, 4H, ArH), 7.25 (t, J = 7.4 Hz, 1H, ArH), 3.60–3.45 (m, 4H), 2.85–2.60 (m, 4H), 2.47(t, J = 7.2 Hz, 2H), 2.29 (t, J = 7.2 Hz, 2H), 2.16 (s, 6H), 1.73–1.58 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 148.18, 145.82, 144.82, 136.76, 130.28, 129.90, 127.24, 127.00, 125.08, 124.29, 124.14, 122.11, 121.93, 120.19, 112.90, 58.20, 57.11, 54.51, 51.59, 46.09, 25.41. MS (ESI): m/z = 388.3 [100%, (M+H)+]. HRMS calcd for C24H29N5 [M+H]+ 388.2496, found 388.2496. HPLC MS purity 99.08%.
11-(4-(3-methoxypropyl)piperazin-1-yl)-10H-indolo[3,2-b]quinoline (9)
Compound 9 was prepared from 1 (200 mg, 0.79 mmol) and 2 mL of 1-(3-methoxypropyl)-piperazine heated at 110 °C for overnight to obtain 264 mg (88%) of compound 9. 1H NMR (500 MHz, DMSO-d6): δ 10.87 (s, 1H, NH), 8.33 (d, J = 8.5 Hz, 1H, ArH), 8.30 (d, J = 7.5 Hz, 1H, ArH), 8.14 (d, J = 8.5 Hz, 1H, ArH), 7.65–7.58 (m, 3H, ArH), 7.55 (t, J = 7.5 Hz, 1H, ArH), 7.26 (t, J = 8.0 Hz, 1H, ArH), 3.60–3.46 (m, 4H), 3.43 (t, J = 6.5 Hz, 2H), 3.27 (s, 3H), 2.90–2.60 (m, 4H), 2.56–2.50 (m, 2H), 1.90–1.74 (m, 2H). 13C NMR (125 MHz, DMSO-d6): δ 147.81, 145.44, 144.44, 136.36, 129.90, 129.52, 126.87, 126.62, 124.71, 123.92, 123.75, 121.73, 121.54, 119.81, 112.52, 70.78, 58.39, 55.57, 54.11, 51.20, 27.11. MS (ESI): m/z = 375.2 [40 %, (M+H)+]. HRMS calcd for C23H26N4O [M+H]+ 375.2179, found 375.2178. HPLC MS purity 99.4%.
3-(4-(10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)propan-1-ol (10)
Compound 10 was prepared from 1 (500 mg, 1.98 mmol) and 1-(3-hydroxy propyl)piperazine (1.5 mL) by heating at 100 °C for 24 h to obtain 620 mg (86%) of 10 as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 10.88 (br s, 1H, NH), 8.42–8.25 (m, 2H, ArH), 8.14 (d, J = 8.1 Hz, 1H, ArH), 7.76–7.50 (m, 4H, ArH), 7.25 (t, J = 6.4 Hz,1H, ArH), 5.10–4.30 (m, OH), 3.70–3.40 (m, 6H), 2.92–2.63 (m, 4H), 2.58–2.50 (m, 2H), 1.80–1.58 (m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 148.17, 145.80, 144.81, 136.73, 130.30, 129.89, 127.23, 127.02, 125.10, 124.27, 124.14, 122.09, 121.94, 120.20, 112.89, 60.29, 56.25, 54.53, 51.55, 30.54. MS (ESI): m/z = 361.2 [100%, (M+H)+]. HRMS calcd for C22H25N4O [M+H]+ 361.2023, found 361.2022. HPLC MS purity 100%.
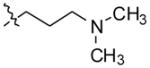
N,N-dimethyl-3-(11-(4-methylpiperazin-1-yl)-10H-indolo[3,2-b]quinolin-10-yl)propan-1-amine (11)
To a solution of compound 3 (100 mg, 0.31 mmol) in DMF at 0 °C, sodium hydride (60% in mineral oil) (22 mg, 0.93 mmol) was added. After stirring the reaction mixture for 1 h, 3-chloro-N,N-dimethylpropan-1-amine hydrochloride (98 mg, 0.62 mmol) was added. The stirring was continued for three days at room temperature. After completion of the reaction, the mixture was poured in ice-cold water. The reaction was quenched with saturated ammonium chloride solution and extracted with CHCl3 (2 × 50 mL). The organic layer was dried over anhydrous MgSO4 and concentrated to give the product. This crude product was purified by silica gel column chromatography, eluting with chloroform to give 93 mg (73%) of 11 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 8.42-8.31 (m, 2H, ArH), 8.23 (d, J = 8.4 Hz, 1H, ArH), 7.75–7.54 (m, 4H, ArH), 7.36–7.28 (m, 1H, ArH), 4.84 (t, J = 6.9 Hz, 2H), 3.68–3.18 (m, 4H), 3.00–2.52 (m, 4H), 2.46–2.30 (m, 5H), 2.19 (s, 6H), 2.03–1.87(m, 2H). 13C NMR (75 MHz, DMSO-d6): δ 148.27, 145.92, 145.59, 136.49, 130.90, 130.83, 129.94, 127.56, 126.64, 125.22, 124.79, 122.00, 121.91, 120.61, 110.51, 57.11, 55.90, 51.36, 47.22, 46.03, 42.95, 28.16. MS (ESI): m/z = 402.4 [100 %, (M+H)+]. HRMS calcd for C25H32N5 [M+H]+ 402.2652, found 402.2647. HPLC MS purity 100%.
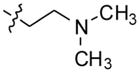
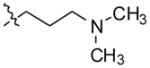
3-(11-(4-(2-(dimethylamino)ethyl)piperazin-1-yl)-10H-indolo[3,2-b]quinolin-10 -yl)-N,N-dimethylpropan-1-amine (12)
Compound 4 (500 mg, 1.3 mmol), was reacted with sodium hydride (60% in mineral oil) (288 mg, 12 mmol) and 3-chloro-N,N-dimethylpropan-1-amine hydrochloride (612 mg, 3.9 mmol) to give compound 12 (523 mg, 85%) as a solid. 1H NMR (300 MHz, CDCl3): δ 8.49 (d, J = 7.8 Hz, 1H, ArH), 8.42–8.28 (m, 2H, ArH), 7.68–7.55 (m, 2H, ArH), 7.52–7.41 (m, 2H, ArH), 7.29 (t, J = 7.5 Hz, 1H, ArH), 4.82 (t, J = 6.3 Hz, 2H), 4.45–3.65 (m, 2H), 3.60–2.80 (m, 4H), 2.75–2.50 (m, 4H), 2.50–2.29 (m, 10H), 2.28–2.10 (m, 6H), 2.06–1.90 (m, 2H). 13C NMR (75 MHz, CDCl3): δ 148.71, 146.01, 145.46, 135.92, 130.76, 130.13, 129.97, 127.49, 126.10, 124.47, 124.44, 122.18, 120.12, 109.43, 57.51, 57.40, 57.24, 54.63, 51.45, 46.32, 46.03, 42.82, 28.40. MS (ESI): m/z = 459.2 [100%, (M+H)+]. HRMS calcd for C28H39N6 [M+H]+ 459.3231, found 459.3231. HPLC MS purity 100%.
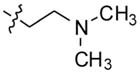
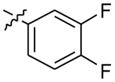
2-(4-(2-(3,4-difluorophenyl)-10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethylethanamine (13)
A mixture of 2-(4-(2-bromo-10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethylethanamine (200 mg, 0.44 mmol), 3,4-difluorophenylboronic acid (104 mg, 0.66 mmol), sodium carbonate (141 mg, 1.32 mmol), and Pd(dppf)2Cl2 (10 mol %) in 1,2-dimethoxyethane:water (4:1) was heated at 90 °C for 6 h. After completion of the reaction, the reaction mixture was cooled to room temperature, poured into ice-cold water, and extracted with ethyl acetate. The organic layer was separated, dried over anhydrous MgSO4, and evaporated under vacuum to give the product. The crude product was purified by silica gel column chromatography using 5% MeOH in CHCl3 as an eluent to afford 186 mg (86%) of pure product 13 as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 11.00 (s, 1H, NH), 8.51 (d, J = 2.1 Hz, 1H, ArH), 8.30 (d, J = 7.5 Hz, 1H, ArH), 8.22 (d, J = 8.7 Hz, 1H, ArH), 7.98–7.84 (m, 2H, ArH), 7.70–7.56 (m, 4H, ArH), 7.27 (t, J = 7.2 Hz, 1H, ArH), 3.63–3.48 (m, 4H), 2.93–2.65 (m, 8H), 2.44 (br s, 6H). 13C NMR (75 MHz, DMSO-d6): δ 148.44, 148.27, 145.35, 144.93, 139.12, 137.07, 134.52, 130.75, 130. 47, 127.55, 126.15, 124.58, 124.22, 122.08, 121.99, 121.93, 120.35, 119.16, 118.94, 116.93, 116.70, 112.99, 56.35, 55.46, 54.58, 54.18, 51.54, 45.44. MS (ESI): m/z = 486.2 [100%, (M+H)+] HRMS calcd for C29H29F2N5 [M+H]+ 486.2463, found 486.2465. HPLC MS purity 100%.
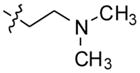
N,N-dimethyl-2-(4-(2-(4-(2-morpholinoethyl)phenyl)-10H-indolo[3,2-b]quinolin-11-yl) piperazin-1-yl)ethanamine (14)
Compound 14 was prepared following a procedure similar to that for compound 13, using 2-(4-(2-bromo-10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethyl ethanamine (200 mg, 0.44 mmol), 4-(2-morpholinoethyl)phenylboronic acid (156 mg, 0.66 mmol), sodium carbonate (141 mg, 1.32 mmol), and Pd(dppf)2Cl2 (10 mol %) to give 14 (217 mg, 87%) as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 10.89 (s, 1H, NH), 8.51 (d, J = 1.2 Hz, 1H, ArH), 8.29 (d, J = 7.8 Hz, 1H, ArH), 8.20 (d, J = 8.7 Hz, 1H, ArH), 7.92 (dd, J = 8.7 Hz, 1H, ArH), 7.72 (d, J = 8.1 Hz, 2H, ArH), 7.68–7.56 (m, 2H, ArH), 7.40 (d, J = 8.1 Hz, 2H, ArH), 7.26 (t, J = 7.5 Hz, 1H, ArH), 3.66–3.58 (m, 4H), 3.57-3.46 (m, 4H), 2.87–2.74 (m, 4H), 2.66–2.56 (m, 4H), 2.55–2.51 (m, 4H), 2.48–2.40 (m, 4H), 2.24 (br s, 6H). 13C NMR (75 MHz, DMSO-d6): δ 148.17, 145.20, 144.85, 140.58, 138.97, 136.87, 136.57, 130.58, 130.36, 127.65, 127.50, 126.26, 124.36, 122.17, 121.92, 121.18, 120.26, 112.93, 67.07, 60.85, 57.43, 56.67, 54.86, 54.15, 51.61, 46.27, 32.87. MS (ESI): m/z = 563.4 [100%, (M+H)+]. HRMS calcd for C35H42N6O [M+H]+ 563.3492, found 563.3491. HPLC MS purity 98.17%.
N,N-dimethyl-2-(4-(2-(pyridin-4-yl)-10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)ethanamine (15)
Compound 15 was synthesized following a procedure similar to that for compound 13, using 2-(4-(2-bromo-10H-indolo[3,2-b]quinolin-11-yl)piperazin-1-yl)-N,N-dimethyl ethanamine (100 mg, 0.22 mmol), pyridin-4-ylboronic acid (40.5 mg, 0.33 mmol), sodium carbonate (69.9 mg, 0.66 mmol), and Pd(dppf)2Cl2 (10 mol%) to obtain 93 mg (93%) of compound 15 as a yellow solid. 1H NMR (300 MHz, DMSO-d6): δ 11.10 (s, 1H, NH), 8.72 (d, J = 5.1 Hz, 2H, ArH), 8.68 (s, 1H, ArH), 8.31(d, J = 7.5 Hz, 1H, ArH), 8.27 (d, J = 9.0 Hz, 1H, ArH), 8.05 (d, J = 8.7 Hz, 1H, ArH), 7.86 (d, J = 5.1 Hz, 2H, ArH), 7.72–7.58 (m, 2H, ArH), 7.28 (t, J = 6.9 Hz, 1H, ArH), 3.70–3.55 (m, 4H), 3.26–3.13 (m, 2H), 2.98–2.82 (m, 6H), 2.76 (s, 6H). 13C NMR (75 MHz, DMSO-d6): δ 151.28, 148.86, 148.14, 145.93, 145.02, 137.37, 133.47, 131.01, 130.63, 127.47, 125.60, 124.11, 122.55, 122.27, 122.09, 122.04, 120.44, 113.03, 54.53, 54.22, 53.52, 51.52, 44.09. MS (ESI): m/z = 451.3 [70 %, (M+H)+]. HRMS calcd for C28H30N6 [M+H]+ 451.2605, found 451.2602. HPLC MS purity 100%.
4-(4-(10H-indolo[3,2-b]quinolin-11-yl)phenethyl)morpholine (16)
A mixture of 1 (200 mg, 0.79 mmol), 4-(2-morpholino ethyl)phenylboronic acid (279 mg, 1.19 mmol), sodium carbonate (252 mg, 2.38 mmol), and Pd(dppf)2Cl2 (10 mol %) in 1,2-dimethoxyethane:water (3:1) was heated at 90 °C overnight. After completion of the reaction, the reaction mixture was cooled to room temperature, poured in ice-cold water, and extracted with ethyl acetate (2 × 100 mL). The organic layer was separated, dried over anhydrous MgSO4, and evaporated under vacuum to give the crude product. The crude product was purified by silica gel column chromatography using 2% MeOH in CHCl3 as an eluent to afford (235 mg, 72%) of pure product 16. 1H NMR (300 MHz, DMSO-d6): δ 10.97 (s, 1H, NH), 8.37 (d, J = 7.5 Hz, 1H, ArH), 8.25 (d, J = 8.7 Hz, 1H, ArH), 7.80 (d, J = 8.1 Hz, 1H, ArH), 7.67 (t, J = 8.1 Hz, 1H, ArH), 7.63–7.42 (m, 7H, ArH), 7.29 (t, J = 7.2 Hz, 1H, ArH), 3.74–3.50 (m, 4H), 3.02–2.83 (m, 2H), 2.80–2.63 (m, 2H), 2.60–2.50 (m, 4H). 13C NMR (75 MHz, DMSO-d6): δ 146.33, 145.30, 144.66, 141.52, 132.44, 131.26, 131.03, 130.47, 130.27, 130.09, 126.74, 126.16, 125.93, 125.73, 125.23, 122.15, 122.02, 120.30, 112.78, 67.06, 60.80, 54.16, 33.08. MS (ESI): m/z = 408.3 [40%, (M+H)+]. HRMS calcd for C27H25N3O [M+H]+ 408.2070, found 408.2071. HPLC MS purity 98.2%.
Thermal Stabilization
Spectral characteristics and thermal stability of the c-MYC G-quadruplex (Table 1) in the absence and presence of compounds were recorded on a J-810 spectropolarimeter (Jasco, Easton, MD). Briefly, the c-MYC G-quadruplex was diluted to 5 μM in a 50 mM Tris-HCl solution (pH 7.4), incubated at 25 °C for 10 min, heated to 95 °C for 10 min, and allowed to cool in the air over ~30 minutes before the addition of compound at 1 equivalence. Positive molecular ellipticity at the parallel G-quadruplex peak (262 nm) was confirmed by spectral examination before each mixture was heated from 5 to 95 °C at 2 °C/min. Molecular ellipticity as a function of temperature was used to calculate a Tm (the temperature at which 50% of the formed higher order DNA structure was melted) for each condition using GraphPad Prism software and a nonlinear regression model with variable slope. ΔTm were calculated as Tm-compound–Tm-control.
Table 1.
Oligonucleotide sequences (5′–3′).
| d(AT) | ATATATATATATATATATATATATATATATATAT |
| d(GC) | GCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGCGC |
| c-MYC G4 | TGGGGAGGGTGGGGAGGGTGGGGAAGG |
| Bcl-2 G4 | AGGGGCGGGCGCGGGAGGAAGGGGGCGGGAGCGGGGC |
| PDGFA G4 | GGAGGCGGGGGGGGGGGGGCGGGGGCGGGGGCGGGGGAGGGGCGCGGC |
| PDGFR-β G4 | GCTGGGAGAAGGGGGGGCGGCGGGGCAGGGAGGGTGGA |
| hTERT G4 | GGGGAGGGGCTGGGAGGGCCCGGAGGGGGCTGGGCCGGGGACCCGGGAGGGGTCGGGACGGGGCGGGG |
| HIF-1α G4 | GCGCGGGGAGGGGAGAGGGGGCGGGAGCGCG |
Competition Dialysis
All DNA (75 μM) were prepared for the experiment in 50 mM Tris-HCl and 25 mM KCl solution. Duplex DNA sequences were annealed by incubating complementary strands together for 10 min at RT, heating at 95 °C for 10 min, and slowly (1 °C/min) decreasing temperature to the Tm, holding at that temperature (°C) for 10 min, and slowly (1 °C/min) decreasing temperature to RT. G-quadruplexes were formed by heating to 95 °C for 10 min then rapidly cooling (in air) to RT. All topologies were confirmed by circular dichroism before continuing with the assay. 250 μL of DNA, in their formed and confirmed topologies, were added to pre-wet mini-dialysis units in a flotation device, placed in a beaker containing 250 mL BPES buffer (6 mM Na2HPO4, 2 mM NaH2PO4,1 mM disodium EDTA) with 25 mM KCl plus 2 μM of compound 4 or 12, and placed in a cold room on a stirring device for 48 h. Following the published protocol,15 the concentration of compound bound to DNA was determined by measuring UV absorbance and using a standard absorbance curve. Binding per putative site value was calculated based on two binding sites per G-quadruplex, and intercalation between every other base pair in dsDNA. The experiment was performed in duplicate.
Cellular Viability
Cells were maintained in a 37 °C, 5% CO2 incubator, in exponential growth, for the duration of experimentation in either DMEM (HCT-116) or RMPI-1640 (Raji and CA46) media supplemented with 10% fetal bovine serum and 1× penicillin/streptomycin. Cytotoxicity of compounds in cells (ATCC, Manassas, VA) was determined using the MTS assay16 with compound diluted over a 5–6 log range in 0.5 log steps. IC50s were determined with GraphPad Prism software using non-linear regression modeling. The experiments were performed with biological triplicates.
Transcriptional Regulation
CA46 or HCT-116 cells were seeded in a T-25 flask or 6-well plates respectively at a density of 0.5–1.0 × 106 cells/mL in 1 mL overnight before incubation with the appropriate IC50 concentration of 4, 12, or quindoline-i for the prescribed times. Cells were washed with PBS (2×) and lysed, and RNA was isolated with an RNeasy Mini kit (Qiagen, Valencia, CA) using the Qiacube automated system. 200–500 ng of cDNA was reverse transcribed with the Quantitect cDNA synthesis kit (Qiagen) prior to quantitative real-time PCR detection on the BioRad MyIQ thermocycler. FAM-labeled TaqMan primers were obtained from Applied Biosystems (Carlsbad, CA) for GAPDH, HIF-1α, VEGFA, PDGFRβ, hTERT, and c-MYC (exon 1 and exon 2).14 The ΔΔCt method was used to calculate changes in expression, normalized to the appropriate time-matched DMSO vehicle-treated control cells. Experiments were performed in at least duplicates, with duplicate measurements within each qPCR reaction; two-tailed unpaired student t-tests were utilized to determine statistical significance.
RESULTS AND DISCUSSION
Chemistry
Quindoline analogs (Table 2) were synthesized from 11-chloroquindoline 1 (Schemes 1 and 4) or substituted 11-chloroquindoline (Schemes 2 and 3). Anthranilic acid and aniline were used in a multistep procedure to synthesize 11-chloroquindoline as reported in literature.17,18
Table 2.
Structures for substituted quindoline analogs.
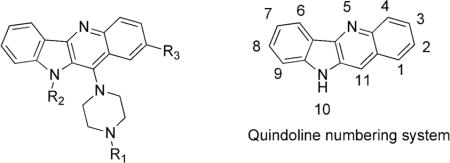
| |||
|---|---|---|---|
| Compound | R1 | R2 | R3 |
| 2 | –H | –H | –H |
| 3 | –CH3 | –H | –H |
| 4 |
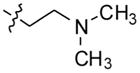
|
–H | –H |
| 5 |
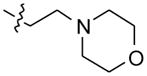
|
–H | –H |
| 6 |
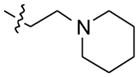
|
–H | –H |
| 7 |
|
–H | –H |
| 8 |
|
–H | –H |
| 9 |
|
–H | –H |
| 10 |
|
–H | –H |
| 11 | –CH3 |
|
–H |
| 12 |
|
|
–H |
| 13 |
|
–H |
|
| 14 |
|
–H |
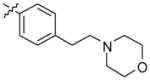
|
| 15 |
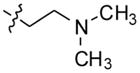
|
–H |
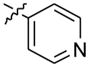
|
| 16 |
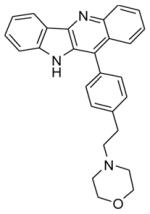
|
||
| Quindoline-i |
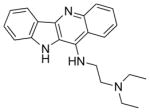
|
||
Scheme 1.

Synthesis of compounds 2–10.
Scheme 4.

Synthesis of compound 16.
Scheme 2.

Synthesis of compounds 11 and 12.
Scheme 3.

Synthesis of compounds 13–15.
Synthesis of 11-substituted Quindoline Analogs
11-substituted quindoline analogs 2–10 were synthesized from 11-chloroquindoline and commercially available piperazines (Schemes 1–4). Reactions were performed by heating two reactants together under neat conditions. Products were isolated in 60–80% yields. It was expected that the addition of a piperazine ring would increase the binding to G-quadruplex. The piperazine N4 atom also provides a site for the addition of groups with varying properties.
Disubstituted Quindoline Analogs
A second set of quindoline analogs was prepared using compound 4 as a lead molecule and substituting at two different positions: indole NH and C2 on the quindoline ring (Scheme 2). It was expected that substitution of a second side chain would lead to an increase in the G-quadruplex binding and cellular activity of the analogs. Compound 12 was prepared by alkylation at indole –NH of compound 4. Compounds 13–15 were synthesized using a palladium-catalyzed Suzuki reaction (Scheme 3). A 3-bromo analog was reacted with corresponding boronic acids to obtain compounds 13–15 in ~90% yield.
Cell-Free and Cellular Evaluation
Evaluation of Mono-substituted Quindoline Analogs
Thermal Stabilization
Quindoline analogs substituted at the R1 position were tested for their ability to thermally stabilize the c-MYC G-quadruplex, resulting in increase in melting temperature (ΔTm) (Table 3). This increase in Tm value (ΔTm) for each compound can be directly correlated with the strength of binding with the G-quadruplex structure.19,20 ΔTm values ranged from 1.9 to 11.4 °C. The lead compound, quindoline-i, shows a ΔTm of 5.2 °C, while compound 2 (11-piperazinylquindoline) shows a moderate ΔTm at 6.7 °C. Compounds 4, 5, and 9 demonstrated comparable thermal stabilization of the c-MYC G-quadruplex, whereas compounds 3, 10, 11 and 16 were less efficacious, and compounds 6–8 demonstrated higher ΔTms in the range of 8.8–11.4 °C.
Table 3.
Melting temperature and cytotoxicity of single-substituted compounds against HCT-116 (colon) and Raji (lymphoma) cell lines.
| Compound | ΔTm in °C (1 equiv.)a | HCT-116 IC50 (μM) 96 h | Raji IC50 (μM) 96 h |
|---|---|---|---|
| Quindoline-i | 5.2 | 0.11 | 0.70 |
| 2 | 6.7 | 4.3 | 33.9 |
| 3 | 3.0 | 3.5 | 5.44 |
| 4 | 6.7 | 0.97 | 2.33 |
| 5 | 5.9 | 9.1 | 52.9 |
| 6 | 8.8 | 0.5 | 56.1 |
| 7 | 10.3 | 3.4 | 26 |
| 8 | 11.4 | 0.9 | 11.8 |
| 9 | 6.3 | 6.3 | 21.3 |
| 10 | 1.3 | 2.4 | >100 |
| 11 | 2.8 | 2.98 | 7.03 |
| 16 | 1.9 | >100 | >100 |
Increase in melting temperature of c-MYC G-quadruplex in the presence of one equivalent of ligand (DNA:Ligand = 1:1).
Cellular Cytotoxicity
Both the colon cancer cell line HCT-116 and the Burkitt’s lymphoma cell line Raji exhibit high levels of c-MYC expression. As an initial indication of both intracellular accumulation of the compound and potential c-MYC downregulation, cytotoxicity in each of these two cell lines was measured after 96 h exposure (Table 3). With the exception of compound 16, all compounds demonstrated greater than 10 μM potency in the HCT-116 cell line, with 96 h IC50 values ranging from 0.5 to 9.1 μM and compounds 4, 6, and 8 showing high nanomolar efficacy. Raji cells, on the other hand, generally have higher IC50 values, ranging from 2.33 to 100 μM. The most potent analog tested in these cells is compound 4. Thus, considering the moderate thermal stabilization by this compound, and the cellular potency, second-generation molecules were derived from compound 4.
Evaluation of Disubstituted Compounds
Thermal Stabilization
In comparison to compound 4, all disubstituted quindoline analogs demonstrated comparable or improved thermal stabilization of the parallel c-MYC G-quadruplex structure (Table 4). For compounds 12–15, ΔTm values ranged from 6.5 to 16.5 °C, with compound 12 showing the greatest stabilization (ΔTm = 16.5 °C). This compound is better when compared to both compounds 4 and 11, since it is a combination of the two side-chains, and there is apparent cooperativity with the double-substitutions that is greater than either alone (ΔTm = 6.7 and 2.8 °C, respectively).
Table 4.
Melting temperature and cytotoxicity of double-substituted compounds against HCT-116 (colon) and Raji (lymphoma) cell lines.
| Compound | ΔTm in °C (1 equiv.)a | HCT-116 IC50 (μM) 96 h | Raji IC50 (μM) 96 h |
|---|---|---|---|
| 4 | 6.7 | 0.97 | 2.33 |
| 12 | 16.5 | 3.4 | 3.1 |
| 13 | 6.5 | 3.7 | 5.8 |
| 14 | 13.1 | 3.8 | 10.8 |
| 15 | 8.6 | 20.9 | 45 |
Increase in melting temperature of c-MYC G-quadruplex in the presence of one equivalent of ligand (DNA:Ligand = 1:1).
Cellular Cytotoxicity
Each of the four double-substituted compounds displayed a greater than three-fold decrease in potency in the HCT-116 colon cancer cell line, with compound 15 showing the least potency at an IC50 (96 h) of 20.9 μM (Table 4). Accordingly, this compound showed poor efficacy in the Raji lymphoma cell line, whereas the other three compounds showed moderate activity with 96 h IC50 values of 3.1–10.8 μM. Again, the best activity is seen with compound 12, with a 96 h IC50 value of 3.4 and 3.1 μM in HCT-116 and Raji cells, respectively. Thus, compound 12 was selected as the disubstituted quindoline analog for further examination.
Comparison of a Mono-substituted Compound (4) with the Disubstituted Compound (12)
Competition Dialysis
Competition dialysis shows that compound 4 binds with higher affinity to G-quadruplexes than compound 12. Competition dialysis results on compounds 4 and 12 are shown in Figure 1. Both compounds bind to folded G-quadruplex structures with greater affinity than double-stranded DNA (dGdC or dAdT). Overall, compound 4 showed higher binding to G-quadruplex structures than compound 12, although inclusion of a second substitution appeared to increase G-quadruplex selectivity, but in this case not for the c-MYC G-quadruplex structure. Overall, compound 12 shows the highest binding affinity for the HIF-1α G-quadruplex, which has a similar parallel structure to the c-MYC G-quadruplex, whereas compound 4 shows more promiscuous binding properties, with the highest affinity for the c-MYC G-quadruplex.
Figure 1.

Affinity of compounds 4 and 12 to various DNA topologies. DNA structures (G-quadruplex or dsDNA) were induced as described, placed in individual dialysis tubes, and incubated with 2 μM of compound for 48 h. [Compound bound] was determined for each binding site of the relevant structure. Compound 4 (white bars) demonstrates promiscuous binding to G-quadruplex structures with the strongest affinity for c-MYC, whereas compound 12 (black bars) shows notable binding to the parallel HIF-1α structure and minor affinity toward the more complicated hTERT and PDGFR-β formations.
Transcriptional Regulation
Compounds 4 and 12 were examined for their effect on transcriptional regulation of c-MYC in the HCT-116 colon cancer cell line (Figure 2). Using their respective 24 h IC50 doses (23 and 31 μM), mRNA was quantified at 6, 24, and 48 h post-treatment, and gene expression was normalized to the DMSO vehicle control. At all time points, compound 4 significantly lowered c-MYC expression, whereas compound 12 demonstrated an intriguing pattern of modulation of c-MYC expression with a significant downregulation early (i.e., 6 h), which was reversed to a significant increase by 48 h.
Figure 2.

Transcriptional regulation of c-MYC by compounds 4 and 12 in HCT-116 colon cancer cells. Cells were exposed for a day to their respective 24 h IC50 concentrations of 23 μM and 31 μM and examined for transcriptional regulation of c-MYC over time, normalized to GAPDH, and compared to the DMSO-vehicle control (6–48 h). Expression was decreased early by both compounds 4 and 12, but a sustained decrease was only noted with compound 4. *p<0.05 as compared to time-matched vehicle control.
Compounds 4 and 12 showed nonselective binding to the different G-quadruplex structures, as shown in the competition dialysis experiments (Figure 1). To correlate the cytotoxicity noted with compound 4 with alterations in c-MYC expression and to elucidate potential mechanisms of cell killing with compound 12, both compounds were also examined for their effect on transcription of various genes: HIF-1α, VEGFA, PDGFR-β, and hTERT in HCT-116 cell lines. Compounds were tested at their 24 h IC50 values, mRNA was quantified, and gene expression was normalized to DMSO vehicle control (Figure 3). Although compounds 4 and 12 both bind to the HIF-1α G-quadruplex in competition dialysis assay, these compounds do not show any notable effect on HIF-1α expression. Compound 4 demonstrates moderate binding to the hTERT G-quadruplex, with concurrent, significant, time-dependent lowering of mRNA expression, whereas the lower binding affinity compound 12 significantly lowers hTERT mRNA at 6 and 24 h, but expression normalizes by 48 h. This pattern is again seen with PDGFR-β by compound 12, which is in accordance with similar binding profiles as measured by competition dialysis. This compound does not demonstrate binding to the VEGFA G-quadruplex, and qPCR reveals only a late, but significant, increase in expression. Compound 4 shows strong binding to the G-quadruplexes in both the PDGFR-β and VEGFA promoters and significant effects on mRNA expression of each target. While there is no direct correlation between G-quadruplex affinity, as measured by the competition dialysis assay, and intracellular transcriptional regulation, it is clear that these compounds do not have specific effects on c-MYC transcription.
Figure 3.

Transcriptional regulation of different genes by compounds 4 (top) and 12 (bottom) in HCT-116 colon cancer cells. Cells were exposed for a day to their respective 24 h IC50 concentrations and examined at 6, 24, and 48 h for transcriptional regulation by qPCR. *p<0.05 as compared to time-matched vehicle control.
Structure–Activity Relationships
Melting temperature
Melting temperature data (ΔTm) of quindoline compounds 2–16 are correlated with the ability of these compounds to stabilize and interact with the c-MYC G-quadruplex and can be used to deduce some preliminary conclusions about structure–activity relationships as shown in Figure 4.
Figure 4.

Schematic depiction of structure–activity (ΔTm) relationships for the quindoline analogs. Text in brackets is a description of the structure–cytotoxicity (HCT-116) relationships for the quindoline analogs.
The quindoline N5 atom is highly basic due to the presence of a piperazine ring at position C11. This nitrogen atom is protonated at physiological pH and this ionization is important for G-quadruplex binding. Compound 16, a phenyl analog of compound 5, shows very low ΔTm value 1.9 °C.
The presence of a basic amino group in the side chain is important for binding. Compound 4 shows better stabilization than compound 3. Similarly, compound 8 (dimethylamino group) shows higher ΔTm than compounds 9 or 10 (methoxy or hydroxyl).
A compound with a longer linker length, compound 8 (three carbons), shows higher ΔTm than the two-carbon linker, compound 4.
Substitution of a second alkyl amino chain on indole nitrogen coupled with the presence of a basic amino group on piperazine increases G-quadruplex stabilization. Compound 12 shows higher ΔTm than compound 4. However, compound 11, an analog of compound 3, does not show any increase in ΔTm after the addition of a second group at R2.
Similar SAR conclusions can be drawn for cytotoxicity data in HCT-116 cell lines. A schematic depiction is shown in Figure 4. Compounds 4, 6, and 8, with amino side chains at R1, lead to better cytotoxicity in HCT-116 cell lines. Substitution at the R2 position (indole nitrogen) leads to lower cytotoxicity.
Exon-Specific Assay in CA46 Cells
Historically, compounds with purported mechanisms of action attributable to the c-MYC G-quadruplex were confirmed in vitro with a pair of Burkitt’s cell lines that were oncogenically transformed and driven by a translocation between an immunoglobin gene and the c-MYC gene. This translocation either maintains, in the case of RAJI cells, or loses, in the case of CA46 cells, the endogenous promoter region of c-MYC, including the G-quadruplex-forming NHE III1 region through exon 1. While these cell lines harbor similar basal levels of c-MYC expression, and are a good indication of G-quadruplex-mediated effects on c-MYC, they are not an isogenic pair and are fundamentally different in a number of aspects unrelated to either c-MYC or the G-quadruplex.
Recently, on the basis of the observation that one can separate the mRNA products from the translocated and non-translocated chromosomes in the CA46 cells, we described the “CA46 exon-specific” test.14 In this test, amplification of exon 1 is used to monitor the effects on the non-translocated, G-quadruplex-maintaining c-MYC gene, while amplification of exon 2 will predominantly represent c-MYC expression from the translocated, G-quadruplex-lost chromosome (Figure 5). To determine if the c-MYC-lowering effect of compound 4 is mediated through the c-MYC G-quadruplex, and to further examine the mechanism of compound 12, we tested the compounds in this exon-specific assay in CA46 cells. If the mechanism of c-MYC transcriptional modulation is mediated through stabilization of the G-quadruplex, a preferential decrease in exon 1, but not exon 2, would be noted, as previously reported for GQC-05.14 While this alone is not conclusive for a G-quadruplex-mediated compound effect, and further analysis with chromatin immunoprecipitation for displacement of transcription factors would be needed as confirmatory data, an absence of this exon-specific effect would enable a confident refusal of a c-MYC G-quadruplex-mediated action.
Figure 5.

The exon-specific expression in CA46 cells based on translocation events. (A) Due to the reciprocal translocation between chromosomes 8 and 14, there are varying resultant c-MYC mRNAs produced. The NT products are normal, with a functional c-MYC under the control of a G-quadruplex, whereas the functional c-MYC produced from the fragment (14;8) on the T allele lacks G4-mediated control. The G-quadruplex was removed, along with exon 1, and produces no known product from the fragment (8;14). Measurements of mRNAs containing exon 1 will mirror the NT allele; mRNAs containing exon 2 will show both the T and the NT products. This figure and legend were originally published in reference 14, © the American Society for Biochemistry and Molecular Biology.
Although the initial report of quindoline-i suggested targeting of this element,11 the CA46 test (measured at 24 h in a previous report) did not.14 Here we describe a more thorough time-course with quindoline-i as the parent compound for these new analogs, as well as for compounds 4 and 12 (Figure 6). Each compound was tested at its respective 24 h IC50 in CA46 cells (19.4, 2.8, and 10 μM for 4, 12, and quindoline-i, respectively). As anticipated by the previous results shown in Figure 2, there was no change in c-MYC expression, on either exon, induced by compound 12 at 24 h and 48 h, but contrary to the results in Figure 2, no lowering of c-MYC was seen at 6 h. For compound 4, which showed a significant change in c-MYC expression (Figure 2), there was no reduction in c-MYC expression in exon 1 corresponding to the non-translocated allele still maintaining the G-quadruplex, whereas c-MYC lowering was evident in the translocated allele. Thus, the c-MYC lowering seen by compound 4 in any cell line is due to a non-G-quadruplex-specific effect.
Figure 6.

The exon-specific assay data for compounds 4, 12, GQC-05, and quindoline-i. CA46 Burkitt’s lymphoma cells were exposed to 19.4, 2.8, 13, and 10 μM of each compound, respectively, for 6–48 h. If an exon-specific effect in c-MYC expression were noted, a preferential decrease in the non-translocated exon 1 would be expected, as the G-quadruplex is maintained. Compound 12 served as the negative control in this experiment, as predicted by the lack of c-MYC lowering ability in HCT-116 cells, and accordingly there was no noted change in expression. Quindoline-i significantly decreased expression from both exons, whereas compound 4 led to a significant decrease in exon 2 through 24 h, with significant increases in exon 1 from 24 to 48 h. Exon 1 is expressed 1/1000 as compared to exon 2, so while this increase is significant from the baseline, there will be no notable increase in intracellular c-MYC because of this. GQC-05 significantly decreased MYC mRNA expression in exon 1 but not in exon 2 at the 24-h time point. *p<0.05 as compared to time-matched vehicle control. **p<0.05 between exons.
For quindoline-i the interpretation of the results is more complex. There is clearly no exon-specific effect, as we have observed previously for GQC-05, because lowering of c-MYC expression occurred for both exon 1 and exon 2 at all three times evaluated. In each case the extent of suppression of gene expression was about the same. Thus, the explanation for the decrease in c-MYC mRNA expression in the non-translocated allele where the G-quadruplex is retained can most likely be explained by the same, but as yet unknown, non-G-quadruplex-mediated effect seen in the translocated allele rather than a direct effect through the G-quadruplex in the c-MYC promoter.
11-Piperazinyl-substituted quindoline analogs were primarily designed as c-MYC G-quadruplex–interactive compounds. However, as evident from competition dialysis experiments, transcriptional data, and the exon-specific assay in CA46 cells, these compounds are predominantly non-selective. As observed with other G-quadruplex-interactive compounds, their anticancer activity in cells is due to the interaction with G-quadruplex in multiple oncogenes and cannot be attributed only to c-MYC or any other single gene.
CONCLUSIONS
We have synthesized a series of 11-piperazinyl-substituted quindoline analogs and tested two of these compounds further for cytotoxicity in HCT-116 and Raji cell lines. The most active compound (4) shows IC50s of 0.97 μM in HCT-116 cell lines and 2.33 μM in Raji cell lines. This compound also exhibits reduction in c-MYC mRNA expression in HCT-116 cells at 6 h, and this continues through 48 h. Despite the significant effect on c-MYC expression, further analysis using the exon-specific effect in CA46 did not support the mechanism of action of this compound to be through the G-quadruplex within the NHE III1. Compounds 4 and 12 are both non-selective and also affect the expression of other target genes. This work suggests that these G-quadruplex-interactive compounds are not selective and their anticancer activity could be due to a combination of effects on different genes in colon and lymphoma cell lines. While initial results led to excitement about new and potent compounds, complete cell-free and in vitro analyses enabled us to conclude that they are not working in the cell through the expected target mechanism of action. Thus it is imperative to fully characterize compounds using appropriate cellular systems before claims are made for putative G-quadruplex-interactive compounds that target specific G-quadruplexes in promoter elements. Proof of principle in cellular systems similar to the CA46 exon-specific assay described here are critical if such claims are to be made.
Supplementary Material
Acknowledgments
This research was supported by grants from Science Foundation Arizona (SBC 0016-07), the National Institutes of Health (CA95060), and the National Foundation for Cancer Research (VONHOFF0601). We thank David Bishop for preparing, proofreading, and editing the final version of the text and figures.
Footnotes
Spectral data (1H/13C–NMR, HRMS, and HPLC-MS) for compounds 2–16. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Eisenman RN. Deconstructing myc. Genes Dev. 2001;15:2023–2030. doi: 10.1101/gad928101. [DOI] [PubMed] [Google Scholar]
- 2.Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2:764–776. doi: 10.1038/nrc904. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004;23:3217–3221. doi: 10.1038/sj.onc.1207548. [DOI] [PubMed] [Google Scholar]
- 4.Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999;18:3004–3016. doi: 10.1038/sj.onc.1202746. [DOI] [PubMed] [Google Scholar]
- 5.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 6.Slamon DJ, deKernion JB, Verma IM, Cline MJ. Expression of cellular oncogenes in human malignancies. Science. 1984;224:256–262. doi: 10.1126/science.6538699. [DOI] [PubMed] [Google Scholar]
- 7.Seenisamy J, Rezler EM, Powell TJ, Tye D, Gokhale V, Joshi CS, Siddiqui-Jain A, Hurley LH. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J Am Chem Soc. 2004;126:8702–8709. doi: 10.1021/ja040022b. [DOI] [PubMed] [Google Scholar]
- 8.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc Natl Acad Sci USA. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan DS, Yang H, Kwan MH, Cheng Z, Lee P, Bai LP, Jiang ZH, Wong CY, Fong WF, Leung CH, Ma DL. Structure-based optimization of FDA-approved drug methylene blue as a c-myc G-quadruplex DNA stabilizer. Biochimie. 2011;93:1055–1064. doi: 10.1016/j.biochi.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Ma Y, Ou TM, Tan JH, Hou JQ, Huang SL, Gu LQ, Huang ZS. Quinolino-benzo-[5, 6]-dihydroisoquindolium compounds derived from berberine: a new class of highly selective ligands for G-quadruplex DNA in c-myc oncogene. Eur J Med Chem. 2011;46:1906–1913. doi: 10.1016/j.ejmech.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 11.Ou TM, Lu YJ, Zhang C, Huang ZS, Wang XD, Tan JH, Chen Y, Ma DL, Wong KY, Tang JC, Chan AS, Gu LQ. Stabilization of G-quadruplex DNA and down-regulation of oncogene c-myc by quindoline derivatives. J Med Chem. 2007;50:1465–1474. doi: 10.1021/jm0610088. [DOI] [PubMed] [Google Scholar]
- 12.Peng D, Tan JH, Chen SB, Ou TM, Gu LQ, Huang ZS. Bisaryldiketene derivatives: A new class of selective ligands for c-myc G-quadruplex DNA. Bioorg Med Chem. 2010;18:8235–8242. doi: 10.1016/j.bmc.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 13.Guyen B, Schultes CM, Hazel P, Mann J, Neidle S. Synthesis and evaluation of analogues of 10H-indolo[3,2-b]quinoline as G-quadruplex stabilising ligands and potential inhibitors of the enzyme telomerase. Org Biomol Chem. 2004;2:981–988. doi: 10.1039/b316055f. [DOI] [PubMed] [Google Scholar]
- 14.Brown RV, Danford FL, Gokhale V, Hurley LH, Brooks TA. Demonstration that drug-targeted down-regulation of MYC in non-Hodgkins lymphoma is directly mediated through the promoter G-quadruplex. J Biol Chem. 2011;286:41018–41027. doi: 10.1074/jbc.M111.274720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaires JB. Current Protocols in Nucleic Acid Chemistry. Vol. 1. John Wiley and Sons, Inc; New York: 2003. A competition dialysis assay for the study of structure-selective ligand binding to nucleic acids; pp. 8.3.1–8.3.8. [DOI] [PubMed] [Google Scholar]
- 16.Mossman BT. In vitro approaches for determining mechanisms of toxicity and carcinogenicity by asbestos in the gastrointestinal and respiratory tracts. Environ Health Perspect. 1983;53:155–161. doi: 10.1289/ehp.8353155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bierer DE, Dubenko LG, Zhang P, Lu Q, Imbach PA, Garofalo AW, Phuan PW, Fort DM, Litvak J, Gerber RE, Sloan B, Luo J, Cooper R, Reaven GM. Antihyperglycemic activities of cryptolepine analogues: an ethnobotanical lead structure isolated from Cryptolepis sanguinolenta. J Med Chem. 1998;41:2754–2764. doi: 10.1021/jm970735n. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi Y, Kitaomo M, Chang M-r, Shirasaka S, Shimamura C, Okuno Y, Yamato M, Harayama T. Synthesis and antitumor activity of fused quinoline derivatives. V. Methylindolo[3,2-b]quinolines. Chem Pharm Bull (Tokyo) 1997;45:2096–2099. doi: 10.1248/cpb.45.2096. [DOI] [PubMed] [Google Scholar]
- 19.Dash J, Shirude PS, Balasubramanian S. G-quadruplex recognition by bis-indole carboxamides. Chem Commun (Camb) 2008:3055–3057. doi: 10.1039/b806042h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.El Bakali J, Klupsch F, Guédin A, Brassart B, Fontaine G, Farce A, Roussel P, Houssin R, Bernier JL, Chavatte P, Mergny JL, Riou JF, Hénichart JP. 2,6-Diphenylthiazolo[3,2-b][1,2,4]triazoles as telomeric G-quadruplex stabilizers. Bioorg Med Chem Lett. 2009;19:3434–3438. doi: 10.1016/j.bmcl.2009.05.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.