

Abstract
The cytotherapeutic potential of mesenchymal stem cells (MSCs) has been evaluated in various disorders including those involving inflammation, autoimmunity, bone regeneration, and cancer. Multiple myeloma (MM) is a systemic malignancy associated with induction of osteolytic lesions that often are not repaired even after prolonged remission. The aims of the study were to evaluate the effects of intrabone and systemic injections of mesenchymal stem cells (MSCs) on MM bone disease, tumor growth, and tumor regrowth in the SCID-rab model and to shed light on the exact localization of systemically injected MSCs. Intrabone injection of MSCs, but not hematopoietic stem cells, into myelomatous bones prevented MM-induced bone disease, promoted bone formation, and inhibited MM growth. After remission was induced with melphalan treatment, intrabone-injected MSCs promoted bone formation and delayed myeloma cell regrowth in bone. Most intrabone or systemically injected MSCs were undetected 2–4 weeks after injection. The bone-building effects of MSCs were mediated through activation of endogenous osteoblasts and suppression of osteoclast activity. While a single intravenous injection of MSCs had no effect on MM, sequential weekly intravenous injections of MSCs prevented MM-induced bone disease but had no effect on tumor burden. MSCs expressed high levels of anti-inflammatory (e.g. HMOX1), and bone remodeling (e.g. Decorin, CYR61) mediators. In vitro, MSCs promoted osteoblast maturation and suppressed osteoclast formation, and these effects were partially prevented by blocking decorin. A subset of intravenously or intracardially injected MSCs trafficked to myelomatous bone in SCID-rab mice. While the majority of intravenously injected MSCs were trapped in lungs, intracardially injected MSCs were mainly localized in draining mesenteric lymph nodes. This study shows that exogenous MSCs act as bystander cells to inhibit MM-induced bone disease and tumor growth and that systemically injected MSCs are attracted to bone by myeloma cells or conditions induced by MM and inhibit bone disease.
Keywords: myeloma, mesenchymal stem cells, bone disease, osteoblasts, homing, lymph nodes
INTRODUCTION
Multiple myeloma (MM) is unique among most hematologic malignancies because it induces lytic bone disease, which is caused by stimulation of osteoclastogenesis and suppression of osteoblastogenesis in areas adjacent to tumor foci in bone marrow.(1) Bone disease in MM is life threatening due to the increasing severity of skeletal complications that occur because large osteolytic lesions often remain unrepaired even after long-term remission.(2) Numbers of osteolytic or focal lesions are significantly associated with poor outcomes for patients with MM,(3;4) supporting experimental findings that bone disease drives MM progression.(1;5–7)
Recent compelling evidence indicates that myeloma cells suppress osteoblastogenesis through contact-dependent cell–cell interaction(8–10) and production of soluble factors.(10–14) These studies revealed that osteolytic bone disease in MM is primarily a reflection of osteoblast deactivation, prompting major efforts for developing bone-anabolic interventions for this disease. While certain in vitro studies suggest that mesenchymal stem cells (MSCs) from patients with MM are functionally abnormal,(15;16) recent experimental and clinical studies clearly demonstrated that osteoblast precursors in myelomatous bone are functional because bortezomib, a clinically used proteasome inhibitor, was able to overcome MM-induced osteoblast deactivation and to increase bone formation and bone mass.(17;18) Although novel osteoblast-activating agents, such as DKK1-neutralizing antibody(19) and inhibitor of activin A signaling,(20) are being explored for clinical treatment of MM, there is an urgent need for additional approaches to achieve systemic bone anabolism and repair of large osteolytic lesions.
Clinical trials have investigated the use of mesenchymal cell cytotherapy for various regenerative and inflammatory diseases and for enhancing engraftment of hematopoietic stem cells.(21–23) These adherent mesenchymal cells, often referred to as MSCs or multipotent mesenchymal stromal cells, are traditionally isolated from bone marrow and expanded in culture before use in cytotherapy.(24) Despite concerns associated with their in vivo stemness characteristics (e.g., differentiation and self-renewal potential), low engraftment, and homing capability,(25;26) these cells have been used successfully to treat patients with osteogenesis imperfecta(27) and inflammatory disorders,(28) and they have been evaluated in experimental disorders such as myocardial infarction,(29) renal injury,(30) diabetes(31) and osteonecrosis.(32) The main mechanisms of action of MSC cytotherapy are attributed to the cells’ immunomodulatory properties(23;33) and production of anti-inflammatory mediators.(34;35)
In the present study, we exploited our animal model for MM to evaluate the potential use of MSC cytotherapy to induce recovery of osteolytic lesions and inhibit MM progression during active and remission stages. We also studied homing of systemically injected MSCs into pathologic (i.e., myelomatous bone) and non-pathologic tissues, as well as the consequences of systemic cytotherapy on MM bone disease and tumor growth.
MATERIALS AND METHODS
Myeloma cells, MSCs and Hematopoietic cell cells (HSC)
The IgG λ Hg myeloma cell line was obtained by passaging primary myeloma cells in SCID-hu or SCID-rab mice, as previously described.(36) The Hg cells are incapable of growing in vitro independently or in coculture with feeder cells (e.g., stromal cells, osteoclasts). In our animal models, Hg cells grow and propagate in the implanted bone but not in any murine organs, and they produce typical bone disease in SCID-hu and SCID-rab mice.(37) Hg cells are molecularly classified in the MMSET subtype of MM(38) and express significant levels of DKK1.(37) Therefore, the Hg myeloma cell line is clinically relevant and suitable for studying myeloma bone disease and myeloma-bone marrow microenvironment interaction.
MSCs from human fetal bone were prepared as previously described.(7;39;40) Briefly, fetal fibula (Advanced Bioscience Resources, Alameda, CA) were crushed into small pieces and were cultured in Dulbecco’s modified Eagle’s medium, low glucose, supplemented with 10% FBS and antibiotics (MSC medium). One-half of the medium was replaced every 4–6 days, and adherent cells were allowed to reach 80% confluency before they were subcultured with trypsin-EDTA (ethylenediaminetetraacetic acid). In vitro, MSCs were capable of differentiation into osteoblasts or adipocytes in specific induction conditions. MSCs were infected with lentivirus expressing luciferase and enhanced green fluorescent protein (EGFP) constructs, as previously described.(36)
HSC were obtained by purifying CD34+ cells from human cord blood (Lonza, Walkersville, MD) using CD34 immunomagnetic bead separation (Miltenyi Biotec, Cambridge, MA). MSCs and HSC, each from two different sources, and the Hg myeloma cells were subjected to global gene expression profile (GEP) as described.(38)
SCID-rab MM model
SCID-rab mice were prepared as previously described.(41) The Institutional Animal Care and Use Committee approved all experimental procedures and protocols. For engraftment of MM, 0.5 × 106 Hg myeloma cells were injected directly into the open (cut) side of the implanted rabbit bone in SCID-rab mice. Mice were periodically bled from the tail vein, and changes in levels of circulating human immunoglobulins (hIg, indicator of MM burden) were determined by enzyme-linked immunosorbent assay (ELISA), as previously described.(42) Cytotherapy in hosts with active MM was initiated when hIg levels were ≥1.5 μg/ml. Radiographs were taken with an AXR Minishot-100 beryllium source instrument (Associated X-Ray Imaging Corp., Haverhill, MA). Changes in bone mineral density (BMD) of the implanted bone were determined using a PIXImus DEXA (GE Medical Systems LUNAR, Madison, WI).(18)
Induction of remission
For evaluating the effects of MSC cytotherapy on relapse, SCID-rab mice engrafted with Hg myeloma cells each received four subcutaneous injections of melphalan (10 mg/kg every 4 days). Remission was determined by lack of human λ light chain in mouse serum, assessed by ELISA.
Cytotherapy
MSCs were maximally passaged eight times before being used in vivo. MSCs were trypsinized and diluted in phosphate buffered saline (PBS). For intrabone cytotherapy in hosts with active MM, MSCs (1 × 106 MSCs/mouse/100 μl PBS, n = 20) or PBS (100 μl/mouse, n = 10) were injected directly into the open end of the implanted rabbit bone in each SCID-rab mouse. In indicated experiment, intrabone cytotherapy was performed using HSC (1 × 106 MSCs/mouse/100 μl PBS, n=8). For intrabone cytotherapy in hosts in remission or for intravenous cytotherapy in hosts with active MM, in each experiment, 10 mice were injected with PBS (100 μl/mouse) and 10 mice were injected with MSCs (1 × 106 MSCs/mouse/100 μl).
Live-animal and ex vivo imaging
Hg myeloma cell-bearing SCID-rab mice each were given an intravenous (n = 6) or intracardiac (n = 4) injection of EGFP/luciferase-expressing MSCs (1 × 106 MSCs in 100 μl PBS per mouse). Similar number on MSCs were also intravenously injected into nonmyelomatous SCID-rab mice (n=3). In indicated experiments, intracardiac injections of MSCs were also administered to CB.17/Icr-SCID mice (n = 8) and immunocompetent C57BL/6 mice (n = 6, Harlan Sprague Dawley, Indianapolis, IN). Intracardiac injection was performed with the use of Dovetail Slide Micromanipulator (Stoelting Inc., Wood Dale, IL) to ensure accurate injection.
Luciferase-labeled cells were traced within mice by using live-animal imaging and, at experiment’s end, within the implanted bones and mouse tissues by using ex vivo imaging. For live-animal imaging, mice were anesthetized with ketamine plus xylazine and were injected intraperitoneally with D-luciferin firefly (150 mg/kg; Xenogen Corp., Alameda, CA). Luciferase activity was localized and quantified using an IVIS 200 imaging system (Xenogen) as previously described.(36) For ex vivo imaging, the implanted rabbit bone was removed and cut in half, and indicated murine organs were removed and cut into small pieces and placed into a 6-well plate. D-luciferin (100 μl of 15 mg/ml diluted in PBS) at room temperature was added to each well; imaging analysis was performed with the IVIS 200 imaging system.
Microscopic examination of MSC localization
SCID mice each received an intracardiac injection of EGFP/luciferase-expressing MSCs (1 × 106 MSCs in 100 μl per mouse; n = 8). Indicated organs and lymph nodes were examined for localization of GFP-expressing MSCs using the ZEISS AXIO Observer.A1 microscope (Delta Optical Instruments, North Little Rock, AR). Intracardiac injections of similar numbers of MSCs were administered to C57BL/6 mice (n = 6), which are immunocompetent and have functional lymph nodes. Evans blue dye (Sigma-Aldrich Inc, St. Louis, MO) which is known to accumulate in and identify lymph nodes,(43) was injected (5% Evans Blue in Hank’s buffered salt solution, 25 μl) into the rear footpad or lateral tail base of the mice, three hours after MSC injection and 30 minutes before bioluminescence and florescence analyses.
Immunohistochemistry and histochemistry
Immunohistochemistry for λ light chain, osteocalcin, or GFP, tartrate-resistant acid phosphatase (TRAP) staining and quantification of numbers of osteoclasts and osteoblasts were performed as previously described.(7;18;19;37;40;42;44;45) BH2 microscope (Olympus, Melville, NY) was used to obtain images with a SPOT 2 digital camera (Diagnostic Instruments Inc., Sterling Heights, MI).
Differentiation of osteoclasts and osteoblasts
Human osteoclast precursors were prepared as previously described.(1;5–7) Briefly, human blood mononuclear cells were cultured in 24-well plates at 2.5 × 106 cells/ml in α-minimal essential medium supplemented with 10% FBS, RANKL (receptor activator for nuclear factor κ B ligand, 50 ng/ml, PeproTech Inc., Rocky Hill, NJ), macrophage colony-stimulating factor (25 ng/ml, PeproTech), and antibiotic cocktail (penicillin, streptomycin, and neomycin; Gibco, Grand Island, NY) (osteoclast medium) for 3–4 days, at which time nonadherent cells were removed; the remaining adherent cells were used as osteoclast precursors. Osteoclast medium was used for the entire study. For co-culture experiments, MSCs were cultured in the upper chamber of 1-micrometer Transwell inserts (non-contact conditions) and placed in 24-well plates containing osteoclast precursors in the bottoms of the wells. Osteoclast precursors were cultured in the absence or presence of MSCs conditioned media (50%) or co-cultured with MSCs in non-contact conditions for 6 days and then subjected to tartrate-resistant acid phosphatase (TRAP) staining with the use of an acid phosphatase kit (Sigma, St. Louis, MO).
The hFOB 1.19 human osteoblastic cell line was purchased from ATCC (Manassas, VA) and was maintained in culture according to the manufacture’s protocol. The hFOB 1.19 cells were cultured in osteogenic media containing 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.05 mM ascorbate, in the absence and presence of MSCs conditioned media (25%–50%) for 3 weeks and then stained for alizarin red with the use of Osteogenesis Quantitation Kit (Millipore, Billerica, MA). Decorin immunohistochemistry and the consequences of decorin neutralizing antibody on the effects of MSCs conditioned media on osteoblast or osteoclast differentiation were assessed as previously described.(46)
Statistical analysis
All values are expressed as mean ± standard error of the mean. Student’s unpaired t-test was used to analyze the effects of treatment on various in vivo and in vitro parameters.
RESULTS
Intrabone injection of MSCs nut not HSC promoted bone formation and inhibited myeloma-induced bone disease and tumor growth
For testing the therapeutic potential of intrabone injections of MSCs, 30 MM-bearing SCID-rab mice received injections of MSCs (n = 20, 1 × 106 cells/mouse) or PBS (n = 10) into implanted bones that were engrafted with Hg myeloma cells. Response to treatment was examined 4 weeks after initiation of treatment. BMD of the implanted bone in PBS-treated hosts (controls) was 14 ± 5% lower than pretreatment levels, but in MSC-treated hosts it was 43±11% higher than pretreatment levels (p < .001, Figure 1A, upper panel). X-ray radiographs also demonstrated that intrabone cytotherapy resulted in increased bone mass of myelomatous bones (Figure 1A, lower panel). At the end of the experiments, the implanted rabbit bones were removed from SCID-rab mice and histologically analyzed. As compared to PBS-injected bones, MSC-injected bones had lower numbers of TRAP-expressing osteoclasts (p < .03), and the higher numbers of osteocalcin-expressing osteoblasts (p < .001) (Figure 1B). Four weeks after initiation of treatment, the mouse serum levels of hIg (indicative of MM burden) were significantly lower in hosts treated with MSCs (p < .001, Figure 1C). In contrast to MSCs cytotherapy, intrabone injection of HSC had no effect MM bone disease and tumor growth in SCID-rab mice engrafted with Hg myeloma cells (Figure 1D, E).
Figure 1. MSCs injected into myelomatous bone did not survive for a long period of time but induced bone formation and inhibited growth of myeloma.
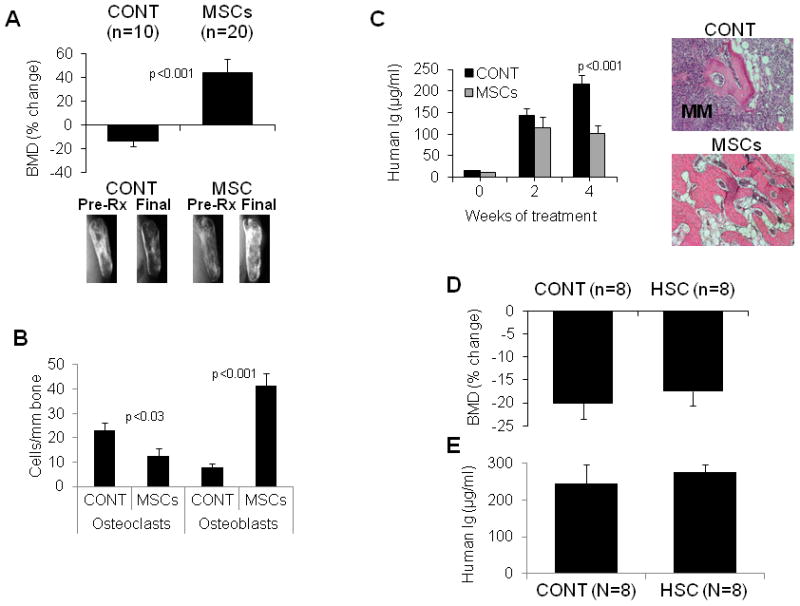
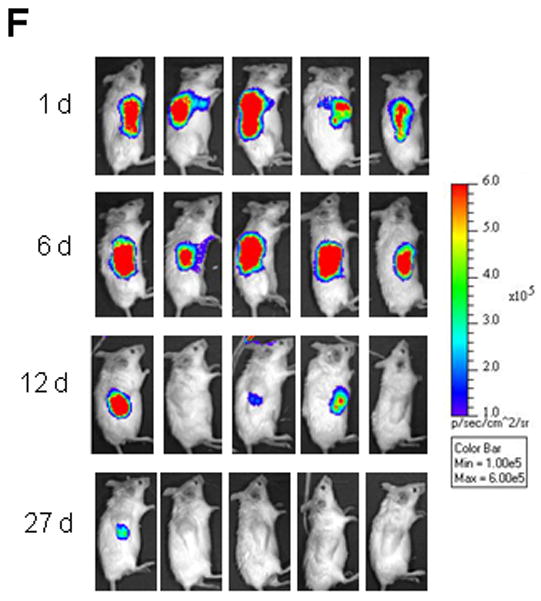
SCID-rab mice engrafted with the Hg myeloma cell line were injected with PBS (CONT), MSCs (1 × 106 MSCs/bone) or HSC (1 × 106 HSC/bone) directly into the implanted myelomatous bone. (A) Effect of MSCs on BMD and bone mass. Upper panel: Changes from pretreatment levels of BMD of the implanted myelomatous bone. Lower panel: Representative X-ray radiographs of the myelomatous bones before treatment (Pre-Rx) and at experiment’s end (Final). Note that BMD and bone mass were reduced in CONT bones but were markedly increased in bones treated with MSCs. (B) MSCs cytotherapy reduced numbers of TRAP-expressing osteoclasts and increased numbers of osteocalcin-expressing osteoblasts in the myelomatous bone sections at experiment’s end (4 weeks). (C) Effect of MSCs on MM growth. Left panel: Circulating hIg levels from myeloma-engrafted SCID-rab mice prior to treatment (week 0) and 2 and 4 weeks after treatment. Right panel: representative histological bone sections stained with hematoxylin and eosin (H&E), demonstrating infiltration of myeloma cells (purple) in control bone but not in MSC-treated bone. (D, E) intrabone injection of HSC had no effect on Hg myeloma cell growth and bone disease in SCID-rab mice. Changes from pretreatment levels of BMD of the implanted bone (D) and circulating hIg levels at experiment’s (E) of myelomatous SCID-rab mice injected with or PBS (CONT) or HSC (0.5 × 106 MSCs/bone) and analyzed 4 weeks after treatment. (F) Luciferase/EGFP-expressing MSCs were injected into the implanted bones and analyzed with live-animal imaging; five representative mice are shown. Note that MSCs gradually disappeared from the majority of the injected bones within 4 weeks.
To evaluate the survival of intrabone injected MSCs and to examine whether these cells are directly responsible for the bone-building effects, EGFP/luciferase-expressing MSCs were injected into the myelomatous bones in SCID-rab mice and detected by bioluminescence analysis of the live animals. Luciferase bioluminescence was detected at high levels in the implanted bones during the first 6 days and was markedly reduced 12 days after injection of MSCs. Within 4 weeks after intrabone cytotherapy, MSCs were not detected in the majority of mice (Figure 1F). These findings suggest that MSCs exert their bone-anabolic effects as bystander cells and that these bone-anabolic effects resulted from MSC interactions with bone marrow elements (e.g., endogenous osteoblasts) during the short period of their engraftment.
Intrabone injection of MSCs during remission promoted bone formation and delayed myeloma relapse
To investigate whether MSC cytotherapy affected the likelihood of experiencing relapse, remission was induced in SCID-rab mice engrafted with Hg myeloma cells by treating them with a total of four subcutaneous injections of melphalan (10 mg/kg/every 4 days), an agent clinically used to treat MM, which was followed by an intrabone injection of PBS or MSCs (10 mice/group). MM regrowth was monitored for 11 weeks, which was the time at which most control mice had detectable myeloma growth, based on circulating hIg levels in mice sera.
Three weeks after the cytotherapy, BMD was 23±5% higher in bones injected with MSCs and 23±3% lower in bones injected with PBS, as compared to levels before MSCs or PBS injections (p < .001, Figure 2A). Eleven weeks after cytotherapy, BMD was 9±7% lower in bones injected with MSCs and 33±6% lower in bones injected with PBS (p < .03, Figure 2A). X-rays radiographs of the implanted bones during remission and relapse correlated with BMD changes, demonstrating that during relapse bone mass was lower in control bones and was preserved in bones injected with MSCs (Figure 2B). After melphalan treatment, circulating hIg levels were undetectable, but 2 weeks after cytotherapy hIg was detected in 30% of hosts injected with MSCs and in 80% of hosts injected with PBS. At the end of the experiment, hIg levels were 6-fold lower in hosts treated with MSCs than in those treated with PBS (p < .01) (Figure 2C). Histological analysis and immunohistochemical staining for human λ revealed regrowth and massive infiltration of myeloma cells in bones of the control group but not of the MSC-treated group (Figure 2D). These data indicate that intrabone injection of MSCs at remission markedly increased bone mass, and the effect on bone mass was associated with delayed relapse.
Figure 2. Intrabone injected MSCs promoted bone formation during MM remission and delayed postrelapse MM growth.
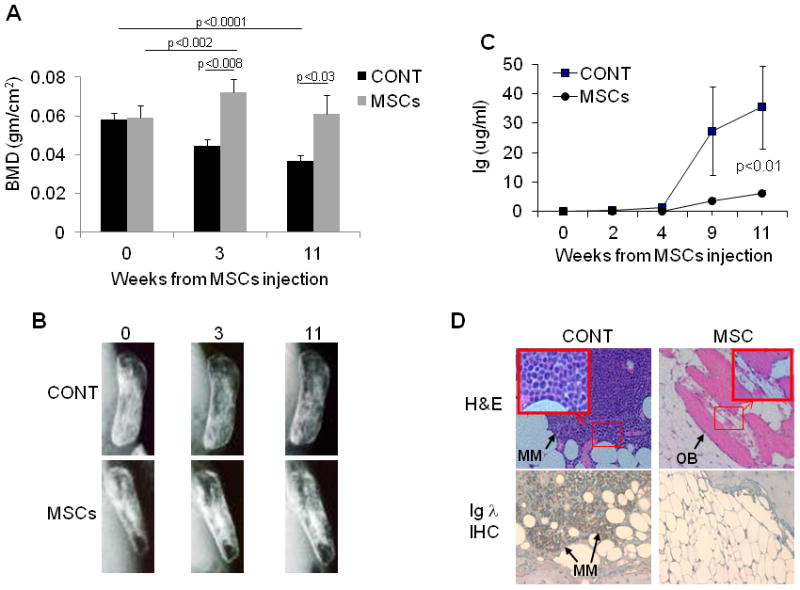
SCID-rab mice engrafted with Hg myeloma cell line each received four injections of melphalan (10 mg/kg/every 4 days, s.c.), which resulted in detection of no circulating hIg. After melphalan treatment, PBS or MSCs (1 × 106 cells/bone) were injected directly into the implanted bones (10 mice/group). (A) BMD levels of the implanted bones before (week 0) and 3 and 11 weeks after injection of PBS (CONT) or MSCs. Note marked increase in BMD after MSC treatment, an effect that remained evident 11 weeks after the injection of MSCs. (B) Representative X-ray radiographs of myelomatous bones. Note marked decalcification of bone in PBS-injected hosts and preservation of bone in MSC-treated hosts. (C) Circulating hIg levels after injection of PBS (squares) or MSCs (circles) revealed delayed postrelapse growth of MM. (D) Representative histological bones sections stained with H&E (upper panel; myeloma cells stained purple, bone stained pink and osteoblasts [OB] are indicated by arrow along the bone surface) or immunohistochemically (IHC) stained for hIg λ for detection of Hg myeloma cells (lower panel; myeloma cells stained brown). Note regrowth of Hg myeloma cells in CONT bone but not MSCs-treated bone.
Systemically injected MSCs trafficked to myelomatous bone
Our data demonstrate the ability of intrabone injections of MSCs to increase bone mass in osteolytic bones, but the clinically relevant approach of such intervention is to induce a similar effect by systemically administering the MSCs. Therefore, we initially evaluated the ability of intravenously injected EGFP/luciferase-expressing MSCs (1 × 106 cells/mouse) to traffic into myelomatous bones in SCID-rab mice. As expected, the majority of the injected MSCs were identified (“trapped”) in lungs 2–3 days after they were injected into hosts with active MM (Figure 3A, upper panel) or after treatment with melphalan (data not shown).
Figure 3. Intravenously injected MSCs trafficked to myelomatous bone during active stage of MM and after induction of remission with melphalan.
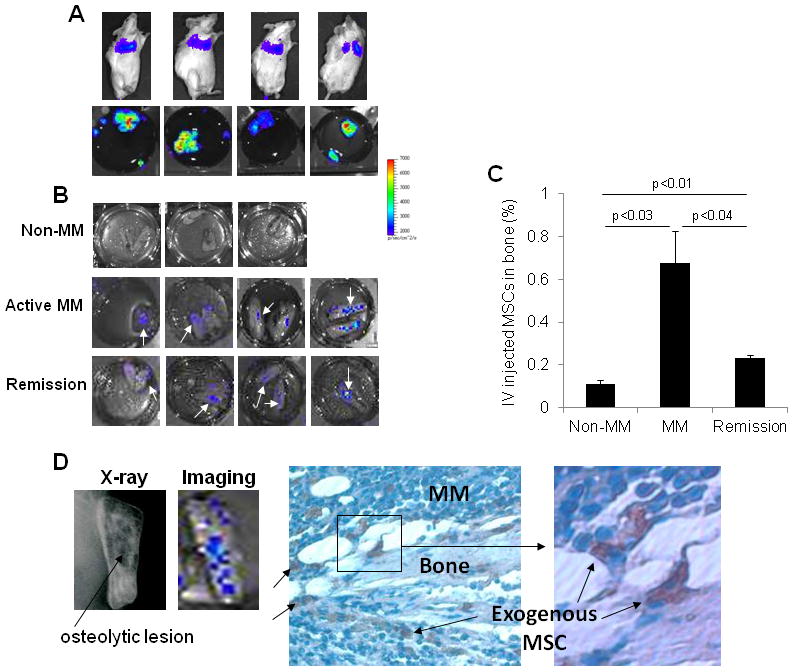
Luciferase/EGFP-expressing MSCs were injected into tail veins (1 × 106 cells/mouse) of nonmyelomatous SCID-rab mice (n = 3), myeloma-bearing SCID-rab mice (n = 6) or after induction of remission by melphalan (n = 4); mice were analyzed with live-animal and ex vivo imaging. (A) Representative live-animal (upper panel) and ex vivo (lower panel) imaging detected MSCs in lungs of myeloma-bearing SCID-rab mice. (B) Ex vivo imaging of the implanted bones from nonmyelomatous SCID-rab mice (non-MM), myelomatous SCID-rab mice (active MM) or after induction of remission by melphalan treatment; white arrows indicate detection of MSCs. (C) Analysis of percent exogenous MSCs detected in the implanted bones without MM, during active MM and remission, based on ex vivo imaging. Note higher percent of MSCs during the active stage of the disease. (D) Left panel: representative X-ray and bioluminescence analysis of the implanted bone, demonstrating localization of intravenously injected MSCs in area with osteolytic lesion (black arrow). Right panel: immunohistochemical detection of GFP in the same bone detected MSCs (pink) in focal area of MM 2 days after cytotherapy.
Due to the high bioluminescence intensity of MSCs in lungs, live-animal imaging could not be used to identify the relatively few MSCs in other organs; therefore, we attempted to identify the injected MSCs in other murine organs and in implanted bones ex vivo. For ex vivo detection, bioluminescence was analyzed in individual organs placed in separate wells. We found that, in addition to localization in lungs (Figure 3A, lower panel), MSCs were detected in implanted bones (Figure 3B) but not in any other murine tissues, including uninvolved mouse bone (data not shown). Based on ex vivo bioluminescence intensity, approximately 0.10 ± 0.02% of MSCs trafficked into nonmyelomatous implanted bones, 0.67 ± 0.15% of MSCs trafficked into implanted bones of hosts with active MM (p < .03 active MM vs. non-MM) and 0.23 ± 0.01% of MSCs trafficked into implanted bones of hosts in remission (p < .04 active MM vs. remission, (p < .01 remission vs. non-MM, Figure 3C). Furthermore, bioluminescence analysis, X-ray radiographs (Figure 3D, left panel) and immunohistochemistry for GFP (Figure 3D, right panel) indicated that MSCs seemed to traffic to areas of bone with existing lytic lesions. These data suggest that a subset of MSCs is attracted to myelomatous bones by myeloma cells or by conditions induced by MM or melphalan treatment.
Because most intravenously injected MSCs were “trapped” in the lungs, we sought to evaluate whether intracardiac injection of MSCs would bypass the lungs and result in increased homing of MSCs to bone. Surprisingly, live-animal imaging after intracardiac injection of MSCs indicated that most MSCs were in the mesenteric, abdominal area of myelomatous SCID-rab mice (Figure 4A). By using ex vivo imaging, we also detected small numbers of MSCs in implanted bones after intracardiac injection (Figure 4B). Quantitatively, intravenous and intracardiac injections yielded similar numbers of MSCs in myelomatous bones of hosts with active disease (Figure 4C). In contrast, after melphalan-induced remission, the number of intravenously injected MSCs in implanted bones was more than 60% lower than in those of hosts with active MM (p < .03) (Figure 4C, see also Figure 3C). The rest of the intracardially injected MSCs were localized in various abdominal organs including reproductive organs, intestine, pancreas, peritoneal membrane, liver, kidney, and spleen (Figure 4D). Of all organs, accumulation of MSCs in intestinal and reproductive organs was prominent (Figure 4E). These findings indicate that small subset of systemically injected MSCs are capable of transmigration and trafficking to myelomatous bone.
Figure 4. Intracardially injected MSCs were primarily localized in the abdomen, and few migrated to myelomatous bone.
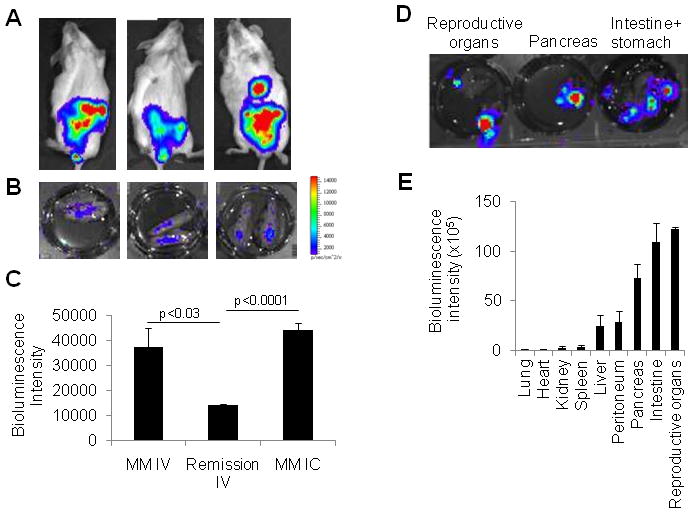
MSCs expressing luciferase/EGFP were intracardially injected into myelomatous SCID-rab mice (1 × 106 cells/mouse, n = 4) and were analyzed 3 days after injection. (A) Live-animal imaging demonstrated localization of MSCs in abdominal regions but not in implanted bones. (B) Ex vivo imaging detected MSCs in implanted bones. (C) Ex vivo bioluminescence intensities of implanted bones were compared among different experimental settings: intravenous injection in mice with active disease (MM IV; see Figure 3 for details), intravenous injection in mice with melphalan-induced remission (Remission IV; see Figure 3 for details), and intracardiac injection in mice with active disease (MM IC). Note similar numbers of MSCs in bones with active MM when exogenous MSCs were administered by intravenous and intracardiac injections, and lower number of MSCs in bones in remission. (D) Ex vivo imaging of indicated abdominal organs from hosts intracardially injected with MSCs. (E) Quantification of ex vivo bioluminescence intensity demonstrated distribution of MSCs in indicated organs. Note high bioluminescence intensity in reproductive organs, intestine, and pancreas.
Intracardially injected MSCs were mainly localized in lymph nodes
The live-animal imaging data on trafficking of intracardially injected MSCs prompted us to perform detailed analyses on specific tissue localization of these MSCs. Unconditioned SCID mice (n = 4) were intracardially injected with EGFP/luciferase-expressing MSCs. Similar to what was observed in MM-bearing SCID-rab mice (see Figure 4A), live-animal bioluminescence activity was mainly detected in the abdominal area but not in bones of SCID mice 3 days after MSC injection. Even after all internal abdominal organs were removed, bioluminescence was still detected along the skin and the peritoneal muscle membrane, and this bioluminescence localization changed each time the mouse position was slightly changed. These observations led us to speculate that most intracardially injected MSCs were localized in the vascular system. Postmortem lymphatic drainage was previously documented.(43) Indeed, careful microscopic examination revealed bioluminescent MSCs in draining lymph nodes attached by connective tissue to the removed organs (Figure 5A, B). The lymph nodes were cultured for 3–5 days, and the exogenous MSCs were released to the bottom surface of the culture plates, where they regained the fibroblast-like morphology typical of MSCs in culture (Figure 5C).
Figure 5. Intracardially injected MSCs primarily migrated to abdominal lymph nodes.
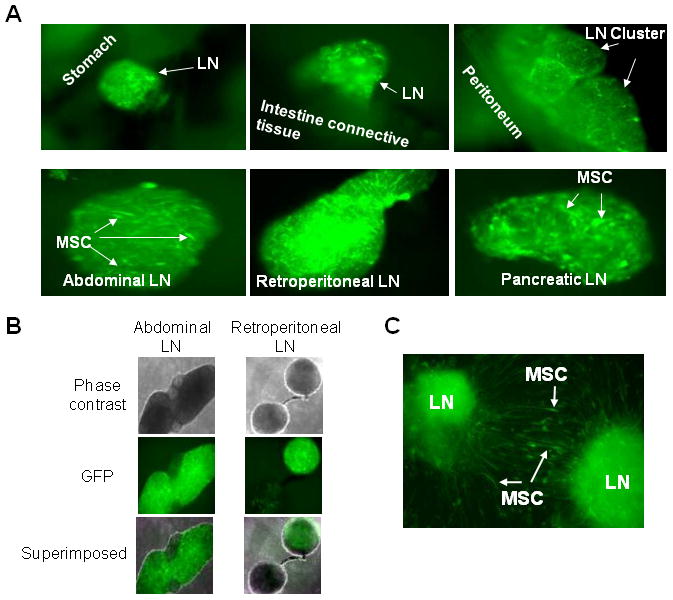
Luciferase/EGFP-expressing MSCs were intracardially injected into myelomatous SCID-rab mice (1 × 106 cells/mouse, n = 4). Three days after injection, the MSCs were identified in dissected abdominal organs using fluorescence microscopy. (A) MSCs primarily migrated to lymph nodes (LN; white arrows) attached to indicated abdominal organs but not within the organs. Note identification of MSCs with fibroblast-like shapes within lymph nodes. (B) Phase-contrast image and fluorescence image (GFP positivity) of abdominal and retroperitoneal lymph nodes are shown individually and superimposed. (C) Culture of GFP-positive lymph nodes for 3–5 days resulted in release of EGFP-expressing MSCs, which regained their typical culture morphology (white arrows). Photos taken with 200X original magnification.
Because lymph nodes of SCID mice are underdeveloped, further investigation was pursued in C57BL/6 mice, which have functional lymph nodes. Evans blue dye, known to accumulate in and identify lymph nodes,(43) was injected into the mouse rear footpad or lateral tail base, 3 hours after intracardiac injection of MSCs and 30 minutes before bioluminescence and florescence analyses. Microscopic examination revealed colocalization of Evans blue dye and GFP positivity, indicating specific trafficking of MSCs to lymph nodes (Figure 6A). Immunohistochemistry showed GFP-expressing MSCs within the lymph nodes (Figure 6B). These findings indicate that the majority of injected MSCs are “trapped” in lungs following intravenous injection or are localized in draining lymph nodes following intracardiac injection.
Figure 6. Intracardially injected MSCs migrated to abdominal lymph nodes of immunocompetent mice.
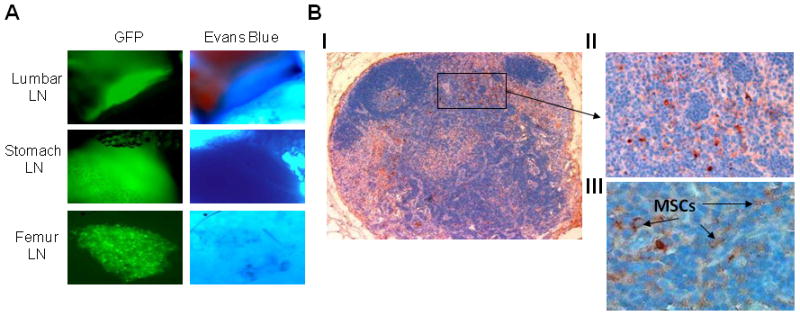
C57BL/6 mice (n = 6) were intracardially injected with luciferase/EGFP-expressing MSCs (1 × 106 MSCs/mouse) and 3 hours later were injected with Evans blue dye into the rear footpad or lateral tail base of the mice. (A) Bright-light and fluorescence microscopy revealed colocalization of Evans blue dye and EGFP-expressing MSCs in indicated lymph nodes (LN; 200X original magnification). (B) GFP immunohistochemical staining (brown indicates GFP positive) of mesenteric lymph node histological section: (I) whole lymph node (100X original magnification), (II) enlargement of indicated area (200X original magnification), (III) higher magnification (400X original magnification) demonstrating individual EGFP-expressing MSCs (black arrows). Note typical structure of a functional lymph node and detection of GFP-expressing MSCs.
Weekly intravenous injections of MSCs inhibit MM-induced bone disease
Because intracardiac injections did not significantly increase trafficking of MSCs to bone and the intravenous route is the more practical and clinically relevant approach, we compared the effects of single or four weekly (“sequential”) intravenous injections of MSCs (10 mice/group in each study) on MM-induced bone disease and tumor growth in SCID-rab mice engrafted with Hg myeloma cells. In contrast to a single intravenous injection, four weekly injections of MSCs significantly prevented BMD reduction of the myelomatous bone, while BMD was reduced by 14 ± 3% (p < .006 vs. pretreatment) in control hosts (Figure 7A, C). Prevention of bone loss by sequential intravenous MSC cytotherapy was also visualized by X-ray radiographs (Figure 7E). In myelomatous bone of hosts treated with sequential intravenous injections of MSCs, histological analysis revealed a reduction in TRAP-expressing osteoclasts (p < .001; Figure 7F) and a significant increase in numbers of osteocalcin-expressing osteoblasts (p < .03; Figure 7F), although the numbers of osteoblasts were significantly lower than in bones treated with intrabone injections of MSC as shown in Figure 1C. Myeloma growth was not affected by either single or sequential intravenous injections of MSCs (Figure 7B, D). These data demonstrate the ability of systemic MSC cytotherapy to inhibit MM-induced bone disease.
Figure 7. Weekly intravenous injections of MSCs inhibited myeloma-induced bone disease.
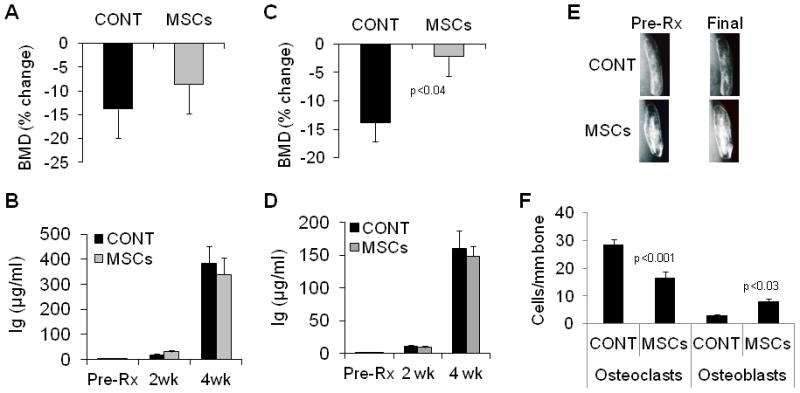
In two separate sets of experiments, myeloma-bearing SCID-rab mice received MSCs in either a single intravenous injection or four weekly intravenous injections. Changes from pretreatment levels of BMD of the implanted myelomatous bone (A) and circulating hIg levels at indicated times (B) were determined for hosts treated with a single injection of either PBS (CONT) or MSCs (10 mice/group); note no effects on BMD or myeloma growth. Changes from pretreatment levels of BMD of the implanted myelomatous bone (C) and circulating hIg levels at indicated times (D) were determined for hosts treated with four weekly injections of either PBS or MSCs (10 mice/group). (E) Representative x-ray radiographs of the implanted myelomatous bones before treatment (Pre-Rx) and at experiment’s end (Final) from mice treated with four weekly injections of either PBS or MSCs. Note prevention of reduced BMD and preservation of bone loss in hosts that received MSC cytotherapy. (F) Numbers of TRAP-expressing osteoclasts and osteocalcin-expressing osteoblasts in sections of the myelomatous implanted bones from mice treated with four weekly injections of either PBS or MSCs. Note that osteoclast numbers were reduced and osteoblast numbers were increased in myelomatous bones from hosts that received MSC cytotherapy.
MSCs express high level of potential anti-inflammatory and bone remodeling mediators and secrete decorin that inhibits osteoclast differentiation and promotes osteoblast maturation
To study the effect of MSCs on differentiation of osteoclast precursors, we used a non-contact coculture system, where human osteoclast precursors were generated in a 24-well plate and MSCs were cultured in 1-micrometer Transwell inserts (105 cells/Transwell) for 6 days. Coculturing of osteoclast precursors with MSCs in osteoclast medium resulted in fewer mature number of multinucleated osteoclasts than when they were cultured alone (Figure 8A). Similar results were obtained when conditioned medium from MSC was added to osteoclast precursors cultured alone (Figure 8A).
Figure 8. MSCs express high level of potential anti-inflammatory and bone remodeling mediators, and secrete factors such as decorin that inhibit osteoclast differentiation and promote osteoblast maturation.
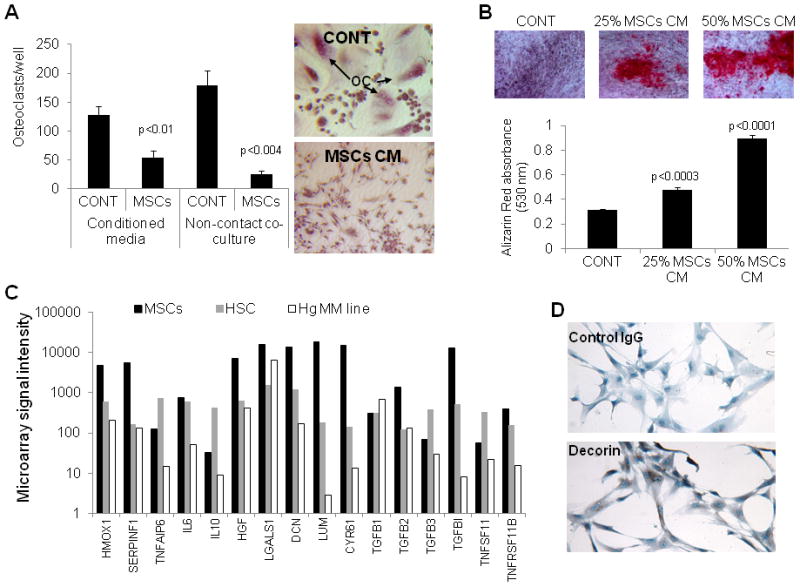

(A) Osteoclast precursors were cultured alone (CONT), treated with 50% MSC conditioned media (CM) or co-cultured with MSCs in non-contact conditions in osteoclast medium for 6 days and then subjected to TRAP staining and quantification of number of multinucleated osteoclasts (OC). (B) The hFOB 1.19 osteoblastic precursor cells were cultured in osteogenic medium in the absence and presence of 25% or 50% MSC condition media for 3 weeks and then stained for alizarin red. (C) Expression of selected genes in MSCs, HSC and Hg myeloma cells analyzed by GEP. Note higher expression of HMOX1, SERPINF1, DCN (decorin), LUM (lumican), CYR61 and TNFRSF11B (OPG) by MSCs. (D) Immunohistochemistry staining confirmed expression of decorin (stained brown) in MSCs. (E) Decorin neutralizing antibody (5 μg/ml) attenuated the inhibitory effect of 25% but not 50% MSC conditioned media on osteoclast formation. (F) Decorin neutralizing antibody (5 μg/ml) attenuated the stimulatory effects of 25% and 50% MSC conditioned media on osteoblastic differentiation of hFOB 1.19 cells.
To study the effect of MSCs on osteoblast differentiation, the human hFOB 1.19 osteoblastic cells were cultured in osteogenic media in the absence and presence of MSCs conditioned media for 3 weeks. In these conditions, mineralized bone nodules were apparent in cultures treated with MSCs conditioned media (25%–50%) but not with control media (Figure 8B).
To shed light on factors produced by MSCs and potentially impact bone remodeling and anti-MM response we performed GEP analysis on MSCs, HSC and Hg MM cells and compared expression of factors previously proposed to mediate MSCs cytotherapeutic effects or exert anti-tumor effects (Figure 8C).(1;35) Anti-inflammatory and wound healing associated factor such as HMOX1and SERPINF1 were highly expressed in MSCs compared to HSC or Hg MM cells. MSCs also expressed higher levels of secreted bone matrix proteins such as decorin (DCN), lumican (LUM) and CYR61, and the anti-osteoclastic factor, TNFSF11B (OPG).
We previously demonstrated that decorin produced by osteoblasts inhibits osteoclast differentiation.(46) Since MSCs also produce high level of decorin (Figure 8D), we tested whether decorin mediates the effect of MSCs on osteoclast and osteoblast differentiation, using neutralizing antibody against decorin.(46) Our data revealed that decorin neutralizing antibody, partially but significantly prevented the inhibitory effect of MSCs conditioned media on osteoclast formation (Figure 8E) and their stimulatory effect on osteoblast differentiation (Figure 8F). These results suggest that MSCs express and produce soluble factors such as decorin that directly and indirectly inhibit osteoclastogenesis and promote osteoblastogenesis.
DISCUSSION
We demonstrated, for the first time, the ability of weekly systemic injections of MSCs to traffic to myelomatous bone, survive for short period of time and inhibit MM-induced bone disease. Intrabone injections of MSCs not only promoted bone formation and inhibited tumor growth in bone with active MM but also effectively promoted bone formation during remission and delayed MM relapse. Both systemic and intrabone cytotherapeutic strategies are clinically applicable because typical MM is a systemic disease and large osteolytic lesions often are not repaired even in patients who are disease-free for lengthy periods of time.(2) Our study also provides insight into the exact tissue/organ localization of intravenously and intracardially injected MSCs, suggesting that, although few MSCs transmigrate to the pathological site, most MSCs that pass the lung barrier do not traffic to internal nonpathological organs but, rather, are localized within lymph nodes attached to these organs.(47;48) These findings are relevant not only to MM but also to other pathological disorders to which MSC cytotherapy is potentially beneficial.
The mechanisms of action by which MSC cytotherapy stimulates bone formation and inhibits MM-induced bone tumor growth are partially understood. The notion that exogenous, expanded MSCs lack typical in vivo stemness properties is considerately recognized.(21) Endogenous bone marrow MSCs are relatively few, particularly in bone of elderly persons (the median age of patients with MM is 68). Therefore, it is likely that the apparent differentiated mesenchymal-lineage cells (e.g., osteoblasts, adipocytes and their committed precursors) are the main mesenchymal cellular elements that directly interact with bone marrow elements and impact hematopoiesis in bone, so the cytotherapy markedly increased the proportion of MSCs in bone, at least for a short period of time. As deduced from our in vitro studies, during this short time, the injected MSCs are likely to interact with endogenous osteoblast precursors and secrete factors that induce their differentiation into bone-building osteoblasts, while simultaneously directly interacting with osteoclast precursors and secrete factors that attenuate formation of bone-resorbing osteoclasts.(40) Our study also point out into several factors that are highly produced by MSCs and reportedly shown to impact osteoclast and/or osteoblast formation including HMOX1,(49;50) SERPINF1(51;52) and the bone matrix proteins decorin(46) and CYR61.(53;54) Our functional study further demonstrated that, similar to osteoblasts,(46) MSCs also produce high level of decorin protein which inhibits osteoclast formation and promotes differentiation osteoblast differentiation. Studies are underway to validate the role of other factors in the MSCs cytotherapeutic effects on bone.
The mechanisms by which MSC cytotherapy inhibits myeloma cell growth is puzzling because in vitro MSCs (often referred as “stromal cells”) are known to support survival and growth of myeloma cell lines with their production of growth factors and cell–cell-contact interactions,(55;56) but the ability of cultured MSCs to support long-term growth of primary myeloma cells is often limited and nonreproducible. We previously demonstrated the ability of mesenchymal cell cytotherapy to impact bone remodeling and increase bone formation in nonmyelomatous SCID-rab mice.(40) These findings suggest that MSCs cytotherapy primarily change bone remodeling which creates an inhospitable environment for myeloma cells, resulting in reduced MM growth.
In addition to increasing osteoblast activity and suppressing osteoclast activity, MSCs may recruit hematopoietic elements that inhibit inflammatory conditions typically associated with myeloma growth in bone. We speculate that MM progression is restrained, directly and indirectly, by anti-inflammatory factors produced by the injected MSCs or by endogenous cells recruited to myelomatous bone after cytotherapy. Our findings that MSCs express high levels of anti-inflammatory and anti-neoplastic factors such as SERPINF1 and decorin support this concept. We have previously demonstrated that decorin also attenuates myeloma cell growth.(46) Although certain soluble factors produced by MSCs may mediate part of their therapeutic activities, we previously showed that cytotherapy at a remote site (subcutaneous) had no effect on MM bone disease or growth,(40) suggesting that MSCs must be present in bone marrow to elicit their antimyeloma effects. The number of exogenous MSCs present in the bone marrow seems to be critical for effectively stimulating bone formation and inhibiting MM; while MSCs injected directly into bone efficiently induced an antimyeloma environment, relatively few systemically injected MSCs trafficked to bone and were unable to significantly promote bone formation or restrain MM growth, although MM-induced bone disease was prevented.
Our findings that MSCs are not detectable in vivo for long periods of time support the notion that most of their activities are mediated through “touch-and-go” mechanisms as bystander cells.(23) These findings are highly clinically relevant and might be advantageous for various reasons. First, this phenomenon limits the duration of treatment effects, allowing control of this intervention. Secondly, this observation suggests the feasibility of using allogeneic MSCs, particularly those from healthy donors or sources from which abundant MSCs can be expanded (e.g., human placenta).(40) Third, our findings that most exogenous MSCs do not participate in building bone also suggest that using expanded autologous MSCs from patients with MM may not necessarily be more efficacious. This is an important finding because there has been a concern that in vivo use of patient MSCs, due to their abnormal phonotypic and genetic characteristics, (15;16;57) may support growth and proliferation of myeloma cells. In support of use of allogeneic MSCs for MM, we recently demonstrated that intralesionally injected human placenta mesenchymal cells exert similar therapeutic effects in SCID-rab mice.(40)
Increasing trafficking of systemically injected MSCs into the pathologic site is a major challenge in this field. Although previous reports suggested that a small subset of MSCs express the chemokine receptor CXCR4,(58) enforced expression of CXCR4 in MSCs(59;60) or glycan enzymatic engineering of CD44(61) improved MSC homing to bone, but the absolute number of MSCs that traffic to pathologic sites remained relatively small. Thus, in contrast to hematopoietic stem cells, MSCs are poorly motile or transmigratory, probably due to their size, adherent growth pattern, and lack of molecular machinery to allow efficient chemoattraction. Nevertheless, our findings clearly demonstrate the ability of a subset of systemically injected MSCs to transmigrate and traffic to a bone with active MM and, to a lesser extent, after melphalan-induced remission. These results suggest that MSCs are attracted to bone by myeloma cells or by conditions induced by MM or melphalan treatment. Alternatively MSCs may be cleared in various tissues but exhibit higher survival rates in the implanted bone or lymph nodes and therefore could be detected in these tissues 2–3 days after intravenous or intracardiac injections, respectively.
Live-animal imaging and microscopic examination of EGFP-expressing MSCs allowed us to evaluate localization of MSCs more precisely. After intracardiac injection, MSCs were localized in lymph nodes adjacent to internal organs. Although previous reports using RT-PCR,(31;62) GFP,(63) or luciferase analysis(64;65) showed systemically injected MSCs localized in whole organs, their exact locations within these organs were not thoroughly characterized. In pathological conditions such as MM, renal injury,(30) myocardial infarction,(29) or diabetes(31), a small subset of MSCs seems to be attracted and to traffic to the actual cellular compartment of the pathologic organs, but our study suggests that the majority of MSCs are trapped in draining lymph nodes. Accumulation of MSCs in lymph nodes may partially explain their immunomodulatory properties; indeed, recent studies demonstrated localization of intravenously injected MSCs in lymph nodes of experimental mouse models of autoimmunity.(47;48) These observations may also explain the increased interest in using mesenchymal cell cytotherapy in clinical treatment of Crohn’s disease.
An important yet unresolved question raised by our study is whether the subset of MSCs that transmigrated to the myelomatous bone after intravenous is the same as migrated there after intracardiac injection and, if so, whether this MSC subset has unique morphologic, molecular, and phenotypic properties. Based on trafficking behavior of intracardially injected MSCs, we expected that, of those that passed the pulmonary vascular system, the majority of intravenously injected MSCs would traffic to lymph nodes. Instead, our findings indicated that a similar number of MSCs trafficked to the myelomatous bone by both routes, suggesting that the same subpopulation of MSCs actively trafficked to myelomatous bone whether injected intravenously or intracardially.
In summary, we have shown that intrabone injected MSCs acted as bystander cells to promote bone formation, inhibit osteolysis, and delay MM growth and regrowth. Sequential intravenous injections of MSCs effectively prevented MM-induced bone disease but had no effect on MM growth. While a small subset of MSCs are capable of transmigration and homing to myelomatous bone, the majority of systemically injected MSCs were localized in lungs or in draining lymph nodes. Our study provides a proof-of-concept for the use of MSC cytotherapy to treat large, unhealed, osteolytic lesions and for systemic inhibition of MM bone disease.
Acknowledgments
This work was supported by grants CA-093897 (SY) and CA55819 (SY) from the National Cancer Institute, by Senior and Translational Research Awards from the Multiple Myeloma Research Foundation (SY), and by a Junior Award from the International Myeloma Foundation (XL). The manuscript was edited by Peggy Brenner, Office of Grants and Scientific Publications, University of Arkansas for Medical Sciences.
Footnotes
Supplemental Data: none included
Conflicts of Interest: All authors state that they have no conflicts of interest
Author contributions: XL prepared luciferase/EGFP-expressing MSCs, performed ex vivo and in vivo studies, analyzed and interpreted the data and wrote the paper; WL and SK performed cell culture, ex vivo, and in vivo studies; SY designed and performed research, conceptualized the work, analyzed and interpreted the data, and wrote the paper. SY takes responsibility for the integrity of the data analysis.
References
- 1.Yaccoby S. Advances in the understanding of myeloma bone disease and tumour growth. Br J Haematol. 2010;149:311–321. doi: 10.1111/j.1365-2141.2010.08141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roodman GD. Skeletal imaging and management of bone disease. Hematology Am Soc Hematol Educ Program. 2008:313–319. doi: 10.1182/asheducation-2008.1.313. [DOI] [PubMed] [Google Scholar]
- 3.Walker R, Barlogie B, Haessler J, Tricot G, Anaissie E, Shaughnessy JD, Jr, Epstein J, van HR, Erdem E, Hoering A, Crowley J, Ferris E, Hollmig K, van RF, Zangari M, Pineda-Roman M, Mohiuddin A, Yaccoby S, Sawyer J, Angtuaco EJ. 2007 Magnetic Resonance Imaging in Multiple Myeloma: Diagnostic and Clinical Implications. J Clin Oncol. 2007 Dec 2;25:1121–8. doi: 10.1200/JCO.2006.08.5803. [DOI] [PubMed] [Google Scholar]
- 4.Bartel TB, Haessler J, Brown TL, Shaughnessy JD, Jr, van RF, Anaissie E, Alpe T, Angtuaco E, Walker R, Epstein J, Crowley J, Barlogie B. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood. 2009;114:2068–2076. doi: 10.1182/blood-2009-03-213280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epstein J, Yaccoby Consequences of interactions between the bone marrow stroma and myeloma. Hematol J. 2003;4:310–314. doi: 10.1038/sj.thj.6200313. [DOI] [PubMed] [Google Scholar]
- 6.Yaccoby S, Wezeman MJ, Henderson A, Cottler-Fox M, Yi Q, Barlogie B, Epstein J. Cancer and the microenvironment: myeloma-osteoclast interactions as a model. Cancer Res. 2004;64:2016–2023. doi: 10.1158/0008-5472.can-03-1131. [DOI] [PubMed] [Google Scholar]
- 7.Yaccoby S, Wezeman MJ, Zangari M, Walker R, Cottler-Fox M, Gaddy D, Ling W, Saha R, Barlogie B, Tricot G, Epstein J. Inhibitory effects of osteoblasts and increased bone formation on myeloma in novel culture systems and a myelomatous mouse model. Haematologica. 2006;91:192–199. [PMC free article] [PubMed] [Google Scholar]
- 8.Pennisi A, Ling W, Li X, Khan S, Shaughnessy JD, Jr, Barlogie B, Yaccoby S. The ephrinB2/EphB4 axis is dysregulated in osteoprogenitors from myeloma patients and its activation affects myeloma bone disease and tumor growth. Blood. 2009;114:1803–1812. doi: 10.1182/blood-2009-01-201954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barille S, Collette M, Bataille R, Amiot M. Myeloma cells upregulate interleukin-6 secretion in osteoblastic cells through cell-to-cell contact but downregulate osteocalcin. Blood. 1995;86:3151–3159. [PubMed] [Google Scholar]
- 10.Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, Grano M, Colucci S, Svaldi M, Rizzoli V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. 2005;106:2472–2483. doi: 10.1182/blood-2004-12-4986. [DOI] [PubMed] [Google Scholar]
- 11.Ehrlich LA, Chung HY, Ghobrial I, Choi SJ, Morandi F, Colla S, Rizzoli V, Roodman GD, Giuliani N. IL-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood. 2005;106:1407–1414. doi: 10.1182/blood-2005-03-1080. [DOI] [PubMed] [Google Scholar]
- 12.Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD., Jr The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 13.Vallet S, Pozzi S, Patel K, Vaghela N, Fulciniti MT, Veiby P, Hideshima T, Santo L, Cirstea D, Scadden DT, Anderson KC, Raje N. A novel role for CCL3 (MIP-1alpha) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leukemia. 2011;25:1174–81. doi: 10.1038/leu.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Standal T, Abildgaard N, Fagerli UM, Stordal B, Hjertner O, Borset M, Sundan A. HGF inhibits BMP-induced osteoblastogenesis: possible implications for the bone disease of multiple myeloma. Blood. 2007;109:3024–3030. doi: 10.1182/blood-2006-07-034884. [DOI] [PubMed] [Google Scholar]
- 15.Corre J, Mahtouk K, Attal M, Gadelorge M, Huynh A, Fleury-Cappellesso S, Danho C, Laharrague P, Klein B, Reme T, Bourin P. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia. 2007;21:1079–1088. doi: 10.1038/sj.leu.2404621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garayoa M, Garcia JL, Santamaria C, Garcia-Gomez A, Blanco JF, Pandiella A, Hernandez JM, Sanchez-Guijo FM, Del Canizo MC, Gutierrez NC, San Miguel JF. 2009 Mesenchymal stem cells from multiple myeloma patients display distinct genomic profile as compared with those from normal donors. Leukemia. 2009 Sep 4;23:1515–27. doi: 10.1038/leu.2009.65. [DOI] [PubMed] [Google Scholar]
- 17.Zangari M, Yaccoby S, Pappas L, Cavallo F, Kumar NS, Ranganathan S, Suva LJ, Gruenwald JM, Kern S, Zhan F, Esseltine D, Tricot G. A prospective evaluation of the biochemical, metabolic, hormonal and structural bone changes associated with bortezomib response in multiple myeloma patients. Haematologica. 2011;96:333–336. doi: 10.3324/haematol.2010.031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pennisi A, Li X, Ling W, Khan S, Zangari M, Yaccoby S. The proteasome inhibitor, bortezomib suppresses primary myeloma and stimulates bone formation in myelomatous and nonmyelomatous bones in vivo. Am J Hematol. 2009;84:6–14. doi: 10.1002/ajh.21310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD., Jr Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109:2106–2111. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallet S, Mukherjee S, Vaghela N, Hideshima T, Fulciniti M, Pozzi S, Santo L, Cirstea D, Patel K, Sohani AR, Guimaraes A, Xie W, Chauhan D, Schoonmaker JA, Attar E, Churchill M, Weller E, Munshi N, Seehra JS, Weissleder R, Anderson KC, Scadden DT, Raje N. Activin A promotes multiple myeloma-induced osteolysis and is a promising target for myeloma bone disease. Proc Natl Acad Sci U S A. 2010;107:5124–5129. doi: 10.1073/pnas.0911929107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prockop DJ. Repair of tissues by adult stem/progenitor cells (MSCs): controversies, myths, and changing paradigms. Mol Ther. 2009;17:939–946. doi: 10.1038/mt.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prockop DJ, Olson SD. Clinical trials with adult stem/progenitor cells for tissue repair: let’s not overlook some essential precautions. Blood. 2007;109:3147–3151. doi: 10.1182/blood-2006-03-013433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 24.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 25.Prockop DJ. “Stemness” does not explain the repair of many tissues by mesenchymal stem/multipotent stromal cells (MSCs) Clin Pharmacol Ther. 2007;82:241–243. doi: 10.1038/sj.clpt.6100313. [DOI] [PubMed] [Google Scholar]
- 26.Karp JM, Leng Teo GS. Mesenchymal stem cell homing: the devil is in the details. Cell Stem Cell. 2009;4:206–216. doi: 10.1016/j.stem.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 27.Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, Sussman M, Orchard P, Marx JC, Pyeritz RE, Brenner MK. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat Med. 1999;5:309–313. doi: 10.1038/6529. [DOI] [PubMed] [Google Scholar]
- 28.Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng. 2010;12:87–117. doi: 10.1146/annurev-bioeng-070909-105309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, Delafontaine P, Prockop DJ. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Togel F, Cohen A, Zhang P, Yang Y, Hu Z, Westenfelder C. Autologous and allogeneic marrow stromal cells are safe and effective for the treatment of acute kidney injury. Stem Cells Dev. 2009;18:475–485. doi: 10.1089/scd.2008.0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee RH, Seo MJ, Reger RL, Spees JL, Pulin AA, Olson SD, Prockop DJ. Multipotent stromal cells from human marrow home to and promote repair of pancreatic islets and renal glomeruli in diabetic NOD/scid mice. Proc Natl Acad Sci U S A. 2006;103:17438–17443. doi: 10.1073/pnas.0608249103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kikuiri T, Kim I, Yamaza T, Akiyama K, Zhang Q, Li Y, Chen C, Chen W, Wang S, Le AD, Shi S. Cell-based immunotherapy with mesenchymal stem cells cures bisphosphonate-related osteonecrosis of the jaw-like disease in mice. J Bone Miner Res. 2010;25:1668–1679. doi: 10.1002/jbmr.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones BJ, McTaggart SJ. Immunosuppression by mesenchymal stromal cells: from culture to clinic. Exp Hematol. 2008;36:733–741. doi: 10.1016/j.exphem.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 34.Ohtaki H, Ylostalo JH, Foraker JE, Robinson AP, Reger RL, Shioda S, Prockop DJ. Stem/progenitor cells from bone marrow decrease neuronal death in global ischemia by modulation of inflammatory/immune responses. Proc Natl Acad Sci U S A. 2008;105:14638–14643. doi: 10.1073/pnas.0803670105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pittenger M. Sleuthing the source of regeneration by MSCs. Cell Stem Cell. 2009;5:8–10. doi: 10.1016/j.stem.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 36.Li X, Pennisi A, Zhan F, Sawyer JR, Shaughnessy JD, Yaccoby S. Establishment and exploitation of hyperdiploid and non-hyperdiploid human myeloma cell lines. Br J Haematol. 2007;138:802–811. doi: 10.1111/j.1365-2141.2007.06742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pennisi A, Ling W, Li X, Khan S, Wang Y, Barlogie B, Shaughnessy JD, Jr, Yaccoby S. Consequences of daily administered parathyroid hormone on myeloma growth, bone disease, and molecular profiling of whole myelomatous bone. PLoS ONE. 2010;5:e15233. doi: 10.1371/journal.pone.0015233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, Tricot G, van RF, Walker R, Zangari M, Crowley J, Barlogie B, Shaughnessy JD., Jr The molecular classification of multiple myeloma. Blood. 2006;108:2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ge Y, Zhan F, Barlogie B, Epstein J, Shaughnessy J, Yaccoby Y. Fibroblast activation protein (FAP) is upregulated in myelomatous bone and supports myeloma cell survival. Br J Haematol. 2006;133:83–92. doi: 10.1111/j.1365-2141.2006.05976.x. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Ling W, Pennisi A, Wang Y, Khan S, Heidaran M, Pal A, Zhang X, He S, Zeitlin A, Abbot S, Faleck H, Hariri R, Shaughnessy JD, Jr, van RF, Nair B, Barlogie B, Epstein J, Yaccoby S. Human Placenta-Derived Adherent Cells Prevent Bone loss, Stimulate Bone formation, and Suppress Growth of Multiple Myeloma in Bone. Stem Cells. 2011;29:263–273. doi: 10.1002/stem.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yata K, Yaccoby S. The SCID-rab model: a novel in vivo system for primary human myeloma demonstrating growth of CD138-expressing malignant cells. Leukemia. 2004;18:1891–1897. doi: 10.1038/sj.leu.2403513. [DOI] [PubMed] [Google Scholar]
- 42.Yaccoby S, Barlogie B, Epstein J. Primary myeloma cells growing in SCID-hu mice: a model for studying the biology and treatment of myeloma and its manifestations. Blood. 1998;92:2908–2913. [PubMed] [Google Scholar]
- 43.Harrell MI, Iritani BM, Ruddell A. Lymph node mapping in the mouse. J Immunol Methods. 2008;332:170–174. doi: 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yaccoby S, Pearse RN, Johnson CL, Barlogie B, Choi Y, Epstein J. Myeloma interacts with the bone marrow microenvironment to induce osteoclastogenesis and is dependent on osteoclast activity. Br J Haematol. 2002;116:278–290. doi: 10.1046/j.1365-2141.2002.03257.x. [DOI] [PubMed] [Google Scholar]
- 45.Yaccoby S, Epstein J. The proliferative potential of myeloma plasma cells manifest in the SCID-hu host. Blood. 1999;94:3576–3582. [PubMed] [Google Scholar]
- 46.Li X, Pennisi A, Yaccoby S. Role of decorin in the antimyeloma effects of osteoblasts. Blood. 2008;112:159–168. doi: 10.1182/blood-2007-11-124164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, Giunti D, Ceravolo A, Cazzanti F, Frassoni F, Mancardi G, Uccelli A. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106:1755–1761. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- 48.Parekkadan B, Tilles AW, Yarmush ML. Bone marrow-derived mesenchymal stem cells ameliorate autoimmune enteropathy independently of regulatory T cells. Stem Cells. 2008;26:1913–1919. doi: 10.1634/stemcells.2007-0790. [DOI] [PubMed] [Google Scholar]
- 49.Barbagallo I, Vanella A, Peterson SJ, Kim DH, Tibullo D, Giallongo C, Vanella L, Parrinello N, Palumbo GA, Di RF, Abraham NG, Asprinio D. Overexpression of heme oxygenase-1 increases human osteoblast stem cell differentiation. J Bone Miner Metab. 2010;28:276–288. doi: 10.1007/s00774-009-0134-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sakai E, Shimada-Sugawara M, Nishishita K, Fukuma Y, Naito M, Okamoto K, Nakayama K, Tsukuba T. Suppression of RANKL-dependent heme oxygenase-1 is required for high mobility group box 1 release and osteoclastogenesis. J Cell Biochem. 2012;113:486–498. doi: 10.1002/jcb.23372. [DOI] [PubMed] [Google Scholar]
- 51.Akiyama T, Dass CR, Shinoda Y, Kawano H, Tanaka S, Choong PF. PEDF regulates osteoclasts via osteoprotegerin and RANKL. Biochem Biophys Res Commun. 2010;391:789–794. doi: 10.1016/j.bbrc.2009.11.139. [DOI] [PubMed] [Google Scholar]
- 52.Broadhead ML, Akiyama T, Choong PF, Dass CR. The pathophysiological role of PEDF in bone diseases. Curr Mol Med. 2010;10:296–301. doi: 10.2174/156652410791065345. [DOI] [PubMed] [Google Scholar]
- 53.Crockett JC, Schutze N, Tosh D, Jatzke S, Duthie A, Jakob F, Rogers MJ. The matricellular protein CYR61 inhibits osteoclastogenesis by a mechanism independent of alphavbeta3 and alphavbeta5. Endocrinology. 2007;148:5761–5768. doi: 10.1210/en.2007-0473. [DOI] [PubMed] [Google Scholar]
- 54.Si W, Kang Q, Luu HH, Park JK, Luo Q, Song WX, Jiang W, Luo X, Li X, Yin H, Montag AG, Haydon RC, He TC. CCN1/Cyr61 is regulated by the canonical Wnt signal and plays an important role in Wnt3A-induced osteoblast differentiation of mesenchymal stem cells. Mol Cell Biol. 2006;26:2955–2964. doi: 10.1128/MCB.26.8.2955-2964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mitsiades CS, McMillin DW, Klippel S, Hideshima T, Chauhan D, Richardson PG, Munshi NC, Anderson KC. The role of the bone marrow microenvironment in the pathophysiology of myeloma and its significance in the development of more effective therapies. Hematol Oncol Clin North Am. 2007;21:1007–viii. doi: 10.1016/j.hoc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 56.Caligaris-Cappio F, Bergui L, Gregoretti MG, Gaidano G, Gaboli M, Schena M, Zallone AZ, Marchisio PC. Role of bone marrow stromal cells in the growth of human multiple myeloma. Blood. 1991;77:2688–2693. [PubMed] [Google Scholar]
- 57.Wallace SR, Oken MM, Lunetta KL, Panoskaltsis-Mortari A, Masellis AM. Abnormalities of bone marrow mesenchymal cells in multiple myeloma patients. Cancer. 2001;91:1219–1230. doi: 10.1002/1097-0142(20010401)91:7<1219::aid-cncr1122>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 58.Wynn RF, Hart CA, Corradi-Perini C, O’Neill L, Evans CA, Wraith JE, Fairbairn LJ, Bellantuono I. A small proportion of mesenchymal stem cells strongly expresses functionally active CXCR4 receptor capable of promoting migration to bone marrow. Blood. 2004;104:2643–2645. doi: 10.1182/blood-2004-02-0526. [DOI] [PubMed] [Google Scholar]
- 59.Cheng Z, Ou L, Zhou X, Li F, Jia X, Zhang Y, Liu X, Li Y, Ward CA, Melo LG, Kong D. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol Ther. 2008;16:571–579. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- 60.Lien CY, Chih-Yuan HK, Lee OK, Blunn GW, Su Y. Restoration of bone mass and strength in glucocorticoid-treated mice by systemic transplantation of CXCR4 and cbfa-1 co-expressing mesenchymal stem cells. J Bone Miner Res. 2009;24:837–848. doi: 10.1359/jbmr.081257. [DOI] [PubMed] [Google Scholar]
- 61.Sackstein R, Merzaban JS, Cain DW, Dagia NM, Spencer JA, Lin CP, Wohlgemuth R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–187. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 62.Shi M, Li J, Liao L, Chen B, Li B, Chen L, Jia H, Zhao RC. Regulation of CXCR4 expression in human mesenchymal stem cells by cytokine treatment: role in homing efficiency in NOD/SCID mice. Haematologica. 2007 Jul;92:897–904. doi: 10.3324/haematol.10669. [DOI] [PubMed] [Google Scholar]
- 63.Devine SM, Cobbs C, Jennings M, Bartholomew A, Hoffman R. Mesenchymal stem cells distribute to a wide range of tissues following systemic infusion into nonhuman primates. Blood. 2003 Apr 15;101:2999–3001. doi: 10.1182/blood-2002-06-1830. [DOI] [PubMed] [Google Scholar]
- 64.Togel F, Yang Y, Zhang P, Hu Z, Westenfelder C. Bioluminescence imaging to monitor the in vivo distribution of administered mesenchymal stem cells in acute kidney injury. Am J Physiol Renal Physiol. 2008 Jul;295:F315–F321. doi: 10.1152/ajprenal.00098.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, Battula VL, Weil M, Andreeff M, Marini FC. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells. 2009 Oct;27:2614–2623. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]