

Site-directed mutagenesis (1), slightly more than two decades old, changed the way protein biochemists do their business. Now comes the sequel—patterned library analysis—in another landmark paper, reported in a recent issue of PNAS by Lahr et al. (2), and closed the millennium on a high note. But what is patterned library analysis?
A protein's conformation is fixed by a large number of weak interactions. Like Gulliver immobilized by the Lilliputians, the fold is substantially overdetermined, and, typically, the breaking of a few individual bonds here and there fails to produce global change. There are 306 structures of phage lysozyme (3) in the protein databank (4), all within a few residues of the wild-type protein. Local details aside, all look just like wild-type lysozyme. For both lysozyme and Gulliver, conformation is inherently a multifactorial issue.
At the opposite extreme, numerous examples demonstrate that proteins with scant sequence identity can also have similar structure and function. Thus, the stereochemical code that determines protein conformation must be highly degenerate. How can such a multifactorial and degenerate code be probed effectively? Approaches using single-site mutagenesis, which seemed so promising initially, were quickly overwhelmed by the combinatorics of the problem. Later approaches using saturation mutagenesis of various sorts (e.g., ref. 5) were a large step in the right direction, but a gap remains between current methods and the full repertoire of site-diversity and combinatorial complexity that is ultimately desired. That gap has now been filled by Lahr et al. (2).
Patterned library analysis was devised to test hypotheses about protein folding and function. The method uses resin-splitting technology to create libraries of arbitrary complexity by generating mutants at any number of residue positions, with each position varied according to its own menu of residue choices. For example, in the sequence below, four residue positions (2, 5, 7, and i) are selected for mutation. Each position is varied within a different list of residue options (lists 1, 2, 3, and 4, with n1, n2, n3, and n4 members, respectively), resulting in a library with n1 × n2 × n3 × n4 = N possible transformants.
 |
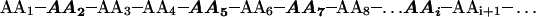 |
Given multiple libraries, a population of transformants can be selected at random from each library and then compared by using regression analysis. In particular, each transformant is evaluated for some activity of interest, using any convenient assay. For an activity that can be quantified, the transformants are, in effect, vectors, which are given by a linear combination of residue substitutions over available positions. This approach is a natural way to turn mutation data into a statistical model, and it is well suited to multifactorial phenomena like protein structure and function.
To validate their approach, Lahr et al. (2) assessed helix propensities in eglin c, a 70-residue proteinase inhibitor with a solitary helix. A close eglin homologue, CI2, has been utilized extensively in folding studies (6). Using three separate libraries, four surface positions in the helix were varied, with seven possible substitutions at each position. Six of the seven—K, Q, E, D, N, and H—were identical, but the seventh was chosen to be either P, A, or G, respectively. Binding activity was used as an assay. In eglin, the helix is situated at the opposite end of the molecule from the inhibitor binding site (Fig. 1), supporting the assumption that the only route from increased helix stability to tighter binding is via global molecular stabilization. Accordingly, representative populations were chosen from each library and were screened for binding activity.
Figure 1.
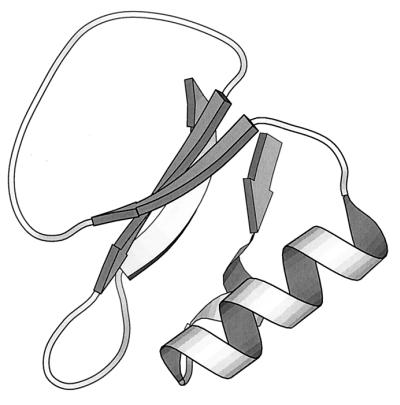
X-ray structure of eglin c (1CSE) (24), a 70-residue proteinase inhibitor. The solitary helix is at the opposite end of the molecule from the large binding loop.
Helix propensity is a well studied phenomenon (7–10). Do results from patterned library analysis correlate with those obtained in biophysical methods? In Fig. 3 of their paper, Lahr et al. (2) show that their rank-ordered helix propensities agree with those of Chakrabartty et al. (11) to the same degree that the biophysically determined scales agree with each other.
Correlation with reliable biophysical methods is reassuring. But the real power of the approach lies in the companion regression analysis, with its ability to provide a numerical estimate of the degree to which the experimental data account for the model under consideration. Here, the helix propensities account for only 31% of the variance. The analysis informs us both that the result is significant and that the hypothesis is incomplete. We are tipped-off, as it were, that answers to further, unasked questions can be extracted from this system.
Experimental design is constrained not only by available methodology but also by our preconceptions. What we think affects what we think to measure. Mutations in the eglin c helix, phage lysozyme, and numerous other proteins alter the equilibrium position of the folding reaction, but rarely, if ever, do they change the overall fold in any appreciable way. Indeed, most mutagenesis studies seek to measure changes in the equilibrium position of the folding reaction
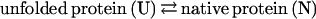 |
as a function of residue type and/or position. These studies can provide useful information about protein stability, the free energy difference between the folded and unfolded populations. But the measurements are indifferent to protein specificity, the extent to which the unfolded population is preorganized. Yet, both thought experiments (12) and actual experiments (13) concur that the reason lysozyme mutants all look like lysozyme originates primarily in their preorganization, and not in the subsequent folding reaction. Like the assembly of a prefabricated house, the folding reaction organizes subassemblies progressively, leading ultimately to a unique final structure—but much of the organization was already there at the onset (14, 15).
To put it blandly, not everyone agrees that unfolded proteins are already preorganized. Why then do experimentalists not simply design telling experiments that can distinguish among the possibilities? Some do, of course (13, 16–18). But the unfolded population is too disperse for crisp characterization using available tools (like optical spectroscopy and NMR). Until now.
Now the challenge is to design informative experiments that can exploit patterned library analysis. The process will provide us with an invaluable opportunity to test our premises in fundamental ways. Here's an example, just to start the ball rolling.
In the progenitor of all protein fragment complementation studies, Richards (19) showed that limited proteolysis of ribonuclease A cleaved the molecule into two fragments: S-peptide (residues 1–20) and S-protein (21–124) (Fig. 2A). Neither fragment exhibited structure in isolation, but, when mixed in stoichiometric proportions, the two fragments reassociated with exquisite fidelity, with recovery of native-like structure and biological activity. Later studies (20, 21) did, in fact, find evidence of helical structure in isolated S-peptide, which contains residues 1–12, a helix in the intact molecule.
Figure 2.

(A) X-ray structure of ribonuclease S (2RNS) (25). S-peptide (residues 1–20, dark shading), a helix, has been cleaved from S-protein (residues 21–124, light shading), the remainder of the molecule. (Residues 16–23, which are disordered in the crystal structure, are not shown.) (B) X-ray structure of two fragments from intact BPTI (1BPT) (26). The β-hairpin fragment, Pβ, comprises residues 20–33, and the helical fragment, Pα, comprises residues 43–58. Apart from these two segments, the rest of the molecule is not displayed.
In a similar vein, Oas and Kim (22) selected two fragments from bovine pancreatic trypsin inhibitor (Fig. 2B)—residues 43–58, dubbed Pα, and residues 20–33, dubbed Pβ. The isolated fragments appeared to lack structure, but the reassociated dimer was native-like.
Experiments such as these have been widely interpreted to mean that the long range interactions needed to stabilize the intact structure are contributed by sites from the separated fragments. Absent fragment association, these crucial interactions are necessarily thwarted.
However, there is another possible interpretation: Recovery of structure in the association complex is largely a nonspecific solvation effect. In this view, the separate fragments are preorganized (23), but their fluctuations are large in water, and their structures evade detection by optical spectroscopy or NMR. Upon complementation, nonspecific surfaces of compatible size and polarity associate, providing the fragments with a mutually stabilizing opportunity to be sequestered from solvent water. This energetically favored association pushes the equilibrium toward the folded form—but not as a consequence of specific long-range interactions.
In principle, these different interpretations could be tested quite easily by simple mix and match experiments. Does S-peptide complement Pβ, and, conversely, does Pα complement S-protein? Of course, there are technical reasons why such an experiment might fail even if the basic idea is correct, but the use of patterned libraries could overcome such obstacles. Specifically, libraries could be designed to test whether substitutions that favor association can be found without corresponding regression toward each fragment's native counterpart. If these complexes can associate successfully but nonspecifically, then our ideas about protein folding may require further evaluation.
Lahr et al. (2) introduce the beginning of an open-ended methodology, one that lends itself to chip technology and Baysean analysis. As Dr. Gulliver was soon to learn, many an adventure awaits beyond the shores of Lilliput.
Acknowledgments
This work was supported by grants from the National Institutes of Health and the Mathers Foundation.
Footnotes
See companion article on page 14860 in issue 26 of volume 96.
References
- 1.Hutchison C A, III, Phillips S, Edgell M H, Gillam S, Janke P, Smith M. J Biol Chem. 1978;253:6551–6560. [PubMed] [Google Scholar]
- 2.Lahr S J, Broadwater A, Carter C W, Jr, Collier M L, Hensley L, Waldner J C, Pielak G J, Edgell M H. Proc Natl Acad Sci USA. 1999;96:14860–14865. doi: 10.1073/pnas.96.26.14860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matthews B W. In: Studies on Protein Stability with T4 Lysozyme. Eisenberg D S, Richards F M, editors. Vol. 46. San Diego: Academic; 1995. pp. 249–278. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein F C, Koetzle T G, Williams G J B, Meyer E F, Jr, Brice M D, Rogers J R, Kennard O, Shimanouchi T, Tasumi M. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 5.Lim W A, Sauer R T. J Mol Biol. 1991;219:359–376. doi: 10.1016/0022-2836(91)90570-v. [DOI] [PubMed] [Google Scholar]
- 6.Itahaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 7.O'Neil K T, DeGrado W F. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 8.Lyu P C, Liff M I, Marky L A, Kallenbach N R. Science. 1990;250:669–673. doi: 10.1126/science.2237416. [DOI] [PubMed] [Google Scholar]
- 9.Padmanabhan S, Marqusee S, Ridgeway T, Laue T M, Baldwin R L. Nature (London) 1990;344:268–270. doi: 10.1038/344268a0. [DOI] [PubMed] [Google Scholar]
- 10.Merutka G, Lipton W, Shalongo W, Park S H, Stellwagen E. Biochemistry. 1990;29:7511–7515. doi: 10.1021/bi00484a021. [DOI] [PubMed] [Google Scholar]
- 11.Chakrabartty A, Kortemme T, Baldwin R L. Protein Sci. 1994;3:843–852. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lattman E E, Rose G D. Proc Natl Acad Sci USA. 1993;90:439–441. doi: 10.1073/pnas.90.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinclair J F, Shortle D. Protein Sci. 1999;8:991–1000. doi: 10.1110/ps.8.5.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldwin R L, Rose G D. Trends Biochem Sci. 1999;24:26–33. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 15.Baldwin R L, Rose G D. Trends Biochem Sci. 1999;24:77–83. doi: 10.1016/s0968-0004(98)01345-0. [DOI] [PubMed] [Google Scholar]
- 16.Cocco M J, Lecomte J T. Biochemistry. 1990;29:11067–11072. doi: 10.1021/bi00502a008. [DOI] [PubMed] [Google Scholar]
- 17.Feng Y, Sligar S G, Wand A J. Nat Struct Biol. 1994;1:30–35. doi: 10.1038/nsb0194-30. [DOI] [PubMed] [Google Scholar]
- 18.Eliezer D, Yao J, Dyson H J, Wright P E. Nat Struct Biol. 1998;5:148–155. doi: 10.1038/nsb0298-148. [DOI] [PubMed] [Google Scholar]
- 19.Richards F M. Proc Natl Acad Sci USA. 1958;44:162–166. doi: 10.1073/pnas.44.2.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown J E, Klee W A. Biochemistry. 1971;10:470–476. doi: 10.1021/bi00779a019. [DOI] [PubMed] [Google Scholar]
- 21.Bierzynski A, Kim P S, Baldwin R L. Proc Natl Acad Sci USA. 1982;79:2470–2474. doi: 10.1073/pnas.79.8.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oas T G, Kim P S. Nature (London) 1988;336:42–48. doi: 10.1038/336042a0. [DOI] [PubMed] [Google Scholar]
- 23.Srinivasan R, Rose G D. Proc Natl Acad Sci USA. 1999;96:14258–14263. doi: 10.1073/pnas.96.25.14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bode W, Papamokos E, Musil D. Eur J Biochem. 1987;166:673–692. doi: 10.1111/j.1432-1033.1987.tb13566.x. [DOI] [PubMed] [Google Scholar]
- 25.Kim E E, Varadarajan R, Wyckoff H W, Richards F M. Biochemistry. 1992;31:12304–12314. doi: 10.1021/bi00164a004. [DOI] [PubMed] [Google Scholar]
- 26.Danishefsky A T, Housset D, Kim K-S, Tao F, Fuchs J, Woodward C, Wlodawer A. Protein Sci. 1993;4:577–587. doi: 10.1002/pro.5560020409. [DOI] [PMC free article] [PubMed] [Google Scholar]