

Abstract
We recently described the discovery of oxygenated N-alkyl deoxynojirimycin (DNJ) derivative 7 (CM-10-18) with antiviral activity against dengue virus (DENV) infection both in vitro and in vivo. This imino sugar was promising, but had an EC50 against DENV in BHK cells of 6.5 μM, which limited its use in in vivo. Compound 7 presented structural opportunities for activity relationship analysis, which we exploited and report here. These structure-activity relationship studies led to analogs 2h, 2l, 3j, 3l, 3v and 4b–4c with nanomolar antiviral activity (EC50 = 0.3–0.5 μM) against DENV infection, while maintaining low cytotoxicity (CC50 > 500 μM, SI > 1000). In male Sprague-Dawley rats, compound 3l was well tolerated at a dose up to 200 mg/kg and displayed desirable PK profiles, with significantly improved bioavailability (F = 92 ± 4%).
INTRODUCTION
Dengue virus (DENV), a mosquito-borne flavivirus, is a major public health threat to 2.5 billion people worldwide and causes 50 to 100 million human infections annually.1 DENV infection could develop into dengue hemorrhagic fever or dengue shock syndrome, either of which is a life-threatening disease. Unfortunately, no effective vaccines or therapeutics are currently available for prevention or treatment of DENV infection. Due to the challenges associated with vaccine development,2 anti-DENV drug discovery is becoming increasingly important.1, 3
Imino sugars, with the endocyclic oxygen replaced by a basic nitrogen, are recognized as an attractive class of carbohydrate mimics.4 For example, deoxynojirimycin (DNJ, Figure 1) is a glucose mimetic that acts as a competitive inhibitor of endoplasmic reticulum (ER)-resident α-glucosidases I and II. ER α-glucosidases are glycoside processing enzymes responsible for removing glucose residues from N-linked glycans attached to nascent glycoproteins.5 This process is required for the proper folding and function of many glycoproteins, including envelope glycoproteins of many viruses, by allowing their interaction with ER chaperones. Therefore, it has been reasoned that inhibition of α-glucosidases might disturb the maturation, secretion, and function of viral envelope glycoproteins and, as a result, inhibit viral particle assembly and/or secretion of enveloped viruses, but not the non-enveloped viruses.6–11 Indeed, using RNAi knockdown technology, we have demonstrated the essential role of cellular ER α-glucosidases I and II in DENV replication (Figure 2). Therefore, ER α-glucosidases I and II have been validated as anti-DENV targets, and their inhibitor, imino sugar (e.g. 7), as a lead class for the development of anti-DENV therapy.
Figure 1.

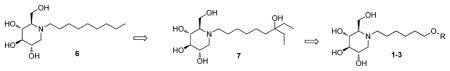
Structure of deoxynojirimycin (DNJ) and its derivatives 6–7.
Figure 2.

Validation of α-glucosidases I and II as anti-DENV targets. Establishment of stable cell lines expressing shRNA based on Huh7.5 cells was described previously.12 Indicated stable cell lines expressing small hairpin RNA (shRNA) targeting human α-glucosidase I (sh-glucosidase I), II (sh-glucosidase II) or non-targeting shRNA (control) were infected with DENV at MOI of 0.01 for 2 days. Virus titers in the supernatant were assayed by standard plaque assay. Experiment was performed in triplicate. Values represent average and standard deviation of DENV titer. p values were calculated using student t test (* indicates p < 0.05 compared to control).
Attempts to explore the antiviral application of imino sugars have been hampered by the lack of potency. For example, N-butyl deoxynojirimycin (NBDNJ) has been approved for the management of the lysosomal storage disease, Gauchers13. However, NBDNJ failed in an HIV clinical trial, because of low potency and difficulty in reaching therapeutic concentration.14 We and others have been actively engaged in the therapeutic application of imino sugars against enveloped virus (e.g. HBV, HCV, HIV, HSV, DENV, WNV, etc.) infection.7, 8, 10–12, 15–24 A general strategy for modifying imino sugar DNJ is to incorporate a terminal group tethered through a linker. The activities will, therefore, depend on the selection of both linkers and terminal groups. Recently, a heteroatom, nitrogen, has been used by Butters’ group to introduce phenyl based terminals and showed good enzymatic activities against α-glucosidases.25 In the other approach, we have employed a heteroatom, oxygen, containing group and successfully increased potencies in cell assay and reduced cell toxicities.8, 17 For example, an oxygenated N-alkyl DNJ derivative 7 (CM-10-18, Figure 1) has been found to have significantly improved antiviral potency compared to NBDNJ12 and demonstrated in vivo antiviral efficacy by reducing viremia in DENV infected AG129 mice. Moreover, although as much as 75 mg/kg dose was required for in vivo efficacy, presumably due to the limited potency and oral bioavailability, compound 7 represents the first compound in this class, with protection of mice from death in mouse models of lethal dengue virus infection.17, 18 Building on these encouraging results, an extensive structure-activity relationship (SAR) campaign was carried out based on the structure of 7, aiming at further increasing the antiviral potency, as well as improving the pharmacokinetic profile.
From a structural perspective, compound 7 offers two points for modification: the DNJ head group and an N-alkyl side chain. Although X-ray structure of DNJ derivatives with α-glucosidases has not been reported, it is widely accepted that the DNJ head group could be recognized by endoplasmic reticulum (ER) glucosidases and alterations of the head group may reduce the compound’s glucosidase inhibitory activity.26 The role of the tail is still unclear, but it may enhance the cellular uptake by increasing the lipophilicity.27 Extending the length of tail group could improve potency, however, this results in increased toxicity.6, 27, 28 According to our and others’ earlier work, the linker’s optimal length is consistently 8–9 carbons, as determined by the cell based assays.25, 28, 29 Therefore, in our newly designed analogs 1–5 (Scheme 1), the total length of the side chain (6-C linker + “O” + terminal ring) is remained at 8–9 carbons. Our empirically derived SAR, based on our past work8, 10, 28 indicates that: 1) adopting conformational restriction strategy through addition of a terminal ring structure reduces the toxicity while sustaining antiviral activity as compared to analogs with a straight alkyl chain, 2) introducing heteroatoms, e.g. oxygen atom, to the side chain results in an improved toxicity profile, and 3) there is a correlation between the hydrophobicity and potency for the imino sugars.
Scheme 1.

Structure Exploration Strategy of N-Alkylated DNJ Derivatives
In this current study, we focused on a structural exploration of the tail group in compound 7 to further improve its antiviral profile (Scheme 1). The tail structural feature relevant to our approach is an alkylated side chain with a tertiary alcohol functional group, while the new molecules bear an ether bond where we postulate that this change will modulate the physicochemical properties of the tail and increase their cell permeabilities.30 Compounds were first evaluated against bovine viral diarrhea virus (BVDV) using a yield reduction assay, which is considered as a model virus for DENV and other viruses of flaviviridae family. Selected analogs with good activity and low toxicity profile as determined by the BVDV assay were further evaluated against DENV. Overall, these efforts led to the discovery of analogs (2h, 2l, 3j, 3l, 3v and 4b–4c) with significantly improved antiviral activity and low cytotoxicity (CC50 > 500 μM, SI > 1000) profile, compared with compound 7. Selected compounds, with improved antiviral activity and representing unique structure features, were assessed for their in vitro absorption, distribution, metabolism, and excretion (ADME) properties, hence identifying a few promising leads (2l, 3j, 3l, 3v and 4b) for further investigation. In addition, a 7-day in vivo toxicity study indicated that compound 3l was well tolerated in male Sprague-Dawley rats at a dose up to 200 mg/kg. 3l also displayed desirable pharmacokinetics (PK) profiles, with much improved bioavailability (F = 92 ± 4%) in the same species. The improved in vitro antiviral profile, favorable ADME, in vivo toxicity, and PK profile provide a strong support for the development of these analogs as a potential therapy for the treatment of DENV infection.
CHEMISTRY
Scheme 1 illustrates our strategy for the lead optimization of molecule 7 in this study: 1) the tertiary hydroxyl group in 7 was incorporated into the side chain as an ether bond in the newly synthesized derivative 1; 2) the terminal secondary alkyl group [CH(R1)2] was fused to form a cycloalkyl or benzene ring system as shown in analogs 2 or 3, respectively, which could tolerate a larger scope of substituents (R3) for SAR investigation; 3) the high conformation freedom for the alkyl side chain of compound 7 was restrained by introducing a benzene ring structure (4) and/or unsaturated bonds (5).
Compound 7 was synthesized as outlined in Scheme 2. Cycloheptanone 8 was treated with mCPBA to provide lactone 9 through Baeyer-Villiger oxidation. This eight-member ring lactone was reacted with ethyl Grignard reagent to give diol 10. Selective oxidation of the primary alcohol by pyridinium chlorochromate (PCC) formed aldehyde 11. Finally, coupling of DNJ with 11 in the presence of Pd/C as catalyst under hydrogenation condition afforded the target molecule 7 in moderate yield. With this efficient and scalable synthetic route, compound 7 was prepared in gram scale for the animal study.17, 18 The synthesis of analogs 1–2 proceeded from mono-protection of 1,6-hexanediol 12 with benzyl bromide to give intermediate 13. Compound 13 was converted to mesylate (14) followed by reaction with corresponding alcohol 15 in presence of sodium hydride to form compound 16. Removal of the benzyl group in 16 under hydrogenation provided alcohol 17, which was oxidized to aldehyde 18 by PCC. Finally, reductive amination of DNJ and 18 under hydrogenation conditions afforded the target molecule 1 or 2. The synthesis of analog 3 commenced with direct alkylation of corresponding phenol 20 with 6-bromohexanol 19 using potassium carbonate as a base to yield alcohol 21. PCC oxidation of 21 followed by coupling with DNJ gave the target molecule. Analog 4 was synthesized in a similar fashion as compound 3. To avoid reduction of unsaturated bonds under the hydrogenation condition, two alternative methods were developed for the synthesis of DNJ derivatives 5. Analog 5a was prepared through reductive amination of DNJ and aldehyde 26 in the presence of sodium cyanoborohydride. 5b was synthesized by direct N-alkylation of DNJ with tosylate 28, which was easily prepared from commercially available alcohol 27. Similarly, analogs 5c–5e were synthesized from corresponding tosylates 32, which could be prepared from mesylate 29 via three step reactions (for detailed chemistry, see “Experimental Section”).
Scheme 2.

Synthetic Procedure for DNJ Derivative 7
RESULTS AND DISCUSSIONS
Antiviral Activity against BVDV Infection
Our ultimate goal is to develop a therapy for DENV infection. At first, the BVDV assay was used to screen analogs, as we have observed good correlation between BVDV and DENV assay.8 Compounds with improved activity against BVDV were further evaluated by DENV antiviral assays. Anti-BVDV activity was determined with a virus yield reduction assay using MDBK cell line, and expressed as concentrations inhibiting 50% of virus titer (EC50). Cytotoxicity was measured by MTT assay using the same cell line and expressed as concentrations inhibiting 50% of cell viability (CC50) (Table 1 and Table 2).
Table 1.
Antiviral Profiles of DNJ Derivatives 1–3 & 6–7 against BVDV Infection in MDBK Cellsa
| |||
|---|---|---|---|
| Compounds (R =) | EC50 (μM) | CC50 (μM) | SI |
| 6 | 1.1 | 350 | 318 |
| 7 | 6.2 | > 500 | > 81 |
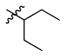 1a
1a
|
3.0 | 450 | 150 |
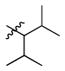 1b
1b
|
7.5 | > 500 | > 67 |
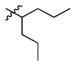 1c
1c
|
0.6 | > 500 | > 833 |
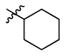 2a
2a
|
0.4 | 480 | 1200 |
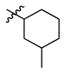 2b
2b
|
0.3 | > 500 | > 1667 |
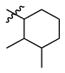
 2c
2c
|
1.5 | 450 | 300 |
|
|
0.4 | > 500 | > 1250 |
|
|
0.5 | 150 | 300 |
|
|
1.8 | 190 | 106 |
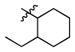
 2g
2g
|
1.2 | > 500 | > 417 |
|
|
3.5 | > 500 | > 143 |
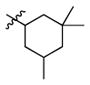 2i
2i
|
0.8 | 250 | 312 |
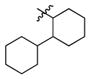 2j
2j
|
0.5 | 200 | 400 |
|
|
0.3 | > 500 | > 1667 |
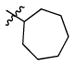 2l
2l
|
1.4 | > 500 | > 357 |
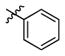 3a
3a
|
0.9 | > 500 | > 556 |
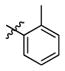 3b
3b
|
0.4 | > 500 | > 1250 |
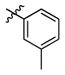 3c
3c
|
0.6 | > 500 | > 833 |
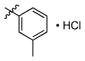 3d
3d
|
0.4 | > 500 | > 1250 |
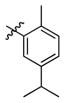 3e
3e
|
0.7 | 90 | 129 |
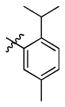 3f
3f
|
0.6 | 62 | 103 |
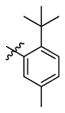 3g
3g
|
0.6 | 85 | 142 |
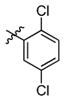 3h
3h
|
1.5 | 320 | 213 |
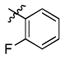 3i
3i
|
2.8 | > 500 | > 179 |
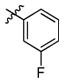 3j
3j
|
0.5 | > 500 | > 1000 |
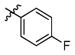 3k
3k
|
0.3 | > 500 | > 1667 |
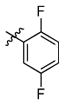 3l
3l
|
0.5 | > 500 | > 1000 |
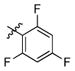 3m
3m
|
5.0 | > 500 | > 100 |
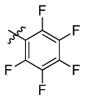 3n
3n
|
1.0 | > 500 | > 500 |
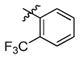 3o
3o
|
1.0 | > 500 | > 500 |
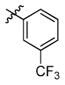 3p
3p
|
1.5 | > 500 | > 333 |
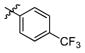 3q
3q
|
1.1 | > 500 | > 455 |
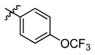 3r
3r
|
1.3 | > 500 | > 385 |
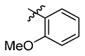 3s
3s
|
1.0 | > 500 | > 500 |
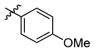 3t
3t
|
1.0 | > 500 | > 500 |
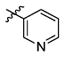 3u
3u
|
2.5 | > 500 | > 200 |
|
|
1.5 | > 500 | > 333 |
EC50: 50% effective concentration, measured by virus yield reduction assay; CC50: 50% cytotoxic concentration, measured by MTT assay; SI: selective index, SI = CC50/EC50.
Table 2.
Antiviral Profiles of DNJ Derivatives 4–5 & 7 against BVDV Infection in MDBK Cellsa
| Compounds | EC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|
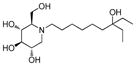 7
7
|
3.0 | > 500 | > 167 |
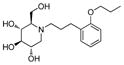 4a
4a
|
0.8 | > 500 | > 625 |
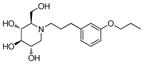 4b
4b
|
1.2 | > 500 | > 417 |
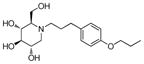 4c
4c
|
0.2 | > 500 | > 2500 |
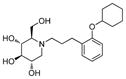 4d
4d
|
24 | 420 | 175 |
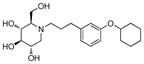 4e
4e
|
3.5 | 390 | 111 |
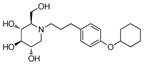 4f
4f
|
5.5 | 295 | 54 |
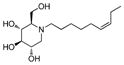 5a
5a
|
11 | > 500 | > 45 |
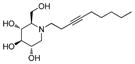 5b
5b
|
> 100 | > 500 | 5 |
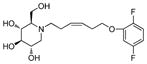 5c
5c
|
2.0 | > 500 | > 250 |
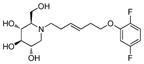 5d
5d
|
0.9 | > 500 | > 556 |
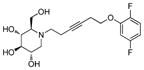 5e
5e
|
47 | > 500 | > 11 |
EC50: 50% effective concentration, measured by virus yield reduction assay; CC50: 50% cytotoxic concentration, measured by MTT assay; SI: selective index, SI = CC50/EC50.
In order to confirm that the mechanism-of-action of all the analogs was as proposed through inhibition of glucosidases, we performed a cell-based surrogate assay, in which the BVDV glycosylated envelop protein (BVDV E2 protein) was analyzed by western blot analysis. In the presence of imino sugar compounds, alteration of the glycan structure on the BVDV E2 protein, as a consequence of glucosidase inhibition, results in a slower mobility rate of BVDV E2 protein (Figure 3, pointed by arrows). Following this change, the glycoprotein undergoes misfolding and degradation leading to reduced protein density on the blot. We have demonstrated that the analogs with anti-BVDV activity were able to change the mobility of the E2 protein, and reduce the quantity of the protein correspondingly, supporting their inhibitory effect on the target enzymes. Figure 3 shows representative results obtained with analogs from each of the structure families.
Figure 3.

Effects of the analogs on the mobility rate of BVDV E2 protein in western blot assay. MDBK cells were infected with BVDV at an MOI of 1 for 1 hour followed by mock-treatment or treatment with indicated compounds at concentrations of 0.8, 4.0, 20 or 100 μM, respectively. Cells were harvested at 22 hour post infection and aliquots of cell lysates were analyzed by electrophoresis followed by western blotting to simultaneously detect BVDV E2 glycoprotein (green), and β-actin (red). Arrow heads indicated E2 protein with slower mobility rate.
Incorporation of the tertiary alcohol group in compound 7 into the side chain as an ether bond (1a–1c) led to analog 1c, with a 10-fold improved EC50 (0.6 μM) and no cytotoxicity observed at the highest tested concentration (up to 500 μM). Built on this observation, the 4-heptyl group in 1c was conformationally restrained to terminal cycloalkyl rings (2a–2l). The antiviral activity was found to correlate to the size of the terminal ring, with cyclopentyl (2k) or cyclohexyl ring (2a–2b and 2d) being optimal and cycloheptyl ring detrimental to the activity. Steric effects were also probed here. Substitution at meta- and para- position (2b and 2d) resulted in better antiviral profiles compared to the one at ortho- position (2c). Analogs with multiple or bulky group substitutions on the terminal cyclohexyl ring displayed decreased activity and/or increased toxicity (2e–2j). We further expanded the scope of the SAR by replacement of the cycloalkyl group with an aromatic ring system (3a–3v) and both electronic and steric factors were examined. Similarly, compounds with small mono-substituent on the benzene ring (3a–3c, 3i–3k and 3o–3t) showed moderate to good antiviral activity. Compounds with bulky substitutions (3e–3h) displayed severe cytotoxicity. Interestingly, for the fluoride substituted analogs, even the pentafluoro- one (3n) did not cause toxicity and exhibited moderately improved activity, which could be attributed to the relative small size of fluorine atom. Compounds with mono- or di-fluorination (3i–3l) of the terminal ring maintained the good antiviral activity. Analogs with fluorine atom at meta- and para- position (3j–3k) gave better EC50 than the one with fluorine atom at ortho- position (3i). For other substituents, such as methyl (3b–3c), trifluoromethyl (3o–3q) and methoxy (3s–3t), there was no position preference. The HCl salt (3d) and its neutral molecule (3c) possessed equally good antiviral activity. The activity of both pyridyl (3u) and benzyl (3v) analogs was slightly decreased, but without observable cytotoxicity. In a summary, 5-membered and 6-membered cycloalkyl, and phenyl derivatives with smaller substitution groups exhibited better antiviral activity relative to compound 7.
To restrain and optimize the conformational freedom of the linker, a benzene ring (4) or unsaturated bond (5) was incorporated into the side chain. Analogs 4a–4c exhibited good anti-BVDV activity with no observable cytotoxicity. However, the antiviral activity was decreased with slight cytotoxicity (4d–4f) when a terminal cyclohexyl ring structure was introduced. It was therefore hypothesized that introduction of bulky groups in the region are not favored. The clopP calculation demonstrated that compounds 4d–4f possessed distinctly higher clogP value (3.69) than that of analogs 4a–4c (2.77). Actually, all the toxic compounds (6, 2e–2f, 2i–2j, 3e–3h and 4d–4f in Table 1 and Table 2) had relatively higher clogP values (3.55–5.13), which could be due to the bulky hydrophobic substituents. Since the DNJ head group is hydrophilic, the hydrophobic tail group makes the molecule detergent-like, which might disrupt the cell or ER membrane, and cause the cytotoxicity; alternatively, higher clogP could also increase the potential off-target protein binding, thus leading to the side-effect. Further restriction of the chain conformation with double or triple bonds (5a–5e) did not improve EC50s compared with their parent compound 3l, which indicated that conformation of the linker with all the sp3 carbons might already be optimal. The X-ray crystallography of 3l (Figure 4) demonstrated the six-carbon linker adopted a linear conformation with no folding observed in the solid state, which is similar to the reported structure of compound 6 (NNDNJ), in which only the terminal C-C bond is in a folding conformation.31
Figure 4.

X-ray Crystal Structure of 3l
Overall, evaluation of these analogs (1–7) in the BVDV system led to a clear SAR map and a number of analogs (1c, 2b, 2d, 2k, 3b–3d, 3j–3l and 4c) with 10~20-fold improvement in their antiviral profiles (EC50 = 0.3~0.6 μM, SI > 833~1667) compared with compound 7 (EC50 = 6.2 μM, SI > 81).
Antiviral Activity against DENV Infection
According to the SAR results above, as well as considering structural diversity, analogs 1c, 2a, 2d, 2h, 2k–2l, 3a–3c, 3j–3l, 3n, 3p–3t, 3v, 4a–4b and 5c–5e were selected and further tested against DENV infection in BHK cells. The antiviral activity was measured by yield reduction assay and expressed as EC50 values, and cytotoxicity was measured by MTT assay and expressed as CC50 values (Table 3).
Table 3.
Antiviral Profiles of Selected Compounds against DENV Infection in BHK Cellsa
| Compounds | EC50 (μM) | CC50 (μM) | SI |
|---|---|---|---|
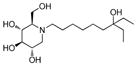 7
7
|
6.5 | > 500 | > 77 |
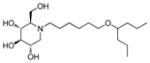 1c
1c
|
0.3 | 425 | 1417 |
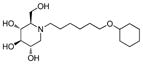 2a
2a
|
1.6 | > 500 | > 312 |
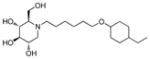 2d
2d
|
0.3 | 475 | 1583 |
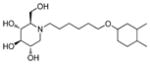 2h
2h
|
0.4 | > 500 | > 1250 |
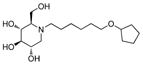 2k
2k
|
1.4 | > 500 | > 357 |
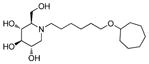 2l
2l
|
0.5 | > 500 | > 1000 |
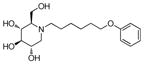 3a
3a
|
1.7 | > 500 | > 294 |
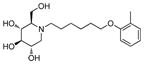 3b
3b
|
0.5 | 450 | 900 |
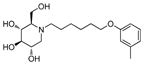 3c
3c
|
0.6 | > 500 | > 833 |
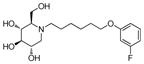 3j
3j
|
0.4 | > 500 | > 1250 |
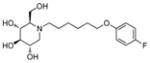 3k
3k
|
1.1 | > 500 | > 455 |
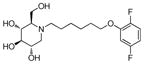 3l
3l
|
0.4 | > 500 | > 1250 |
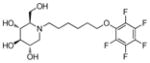 3n
3n
|
1.7 | > 500 | > 294 |
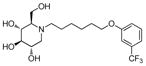 3p
3p
|
0.4 | 400 | 1000 |
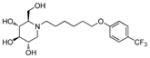 3q
3q
|
1.3 | 470 | 362 |
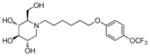 3r
3r
|
0.6 | > 500 | > 833 |
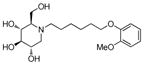 3s
3s
|
0.8 | > 500 | > 625 |
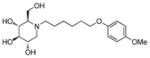 3t
3t
|
4.0 | > 500 | > 125 |
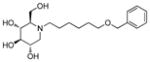 3v
3v
|
0.4 | > 500 | > 1250 |
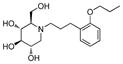 4a
4a
|
3.2 | > 500 | > 156 |
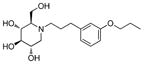 4b
4b
|
0.3 | > 500 | > 1667 |
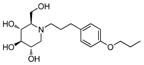 4c
4c
|
0.5 | > 500 | > 1000 |
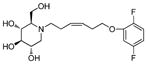 5c
5c
|
8.5 | > 500 | > 59 |
 5d
5d
|
2.2 | > 500 | > 227 |
 5e
5e
|
20 | > 500 | > 25 |
EC50: 50% effective concentration, measured by virus yield reduction assay; CC50: 50% cytotoxic concentration, measured by MTT assay; SI: selective index, SI = CC50/EC50.
Consistent with the results obtained from BVDV infection, none of these selected compounds showed significant toxicity on BHK cells. Analog 1c gave a more than 20-fold better EC50 (0.3 μM, SI = 1417) than compound 7. In compound 2 series, compounds with 4-ethylcylohexyl (2d) and 3,4-dimethylcylohexyl (2h) terminal structure showed nanomolar range anti-DENV activity with slight or no cytotoxicity. Interestingly, compound 2l with a bigger terminal ring possessed ~3-fold better activity than analogs 2a and 2k. Meta- methylation on the terminal benzene ring resulted in equally good activity, but lower toxicity than ortho- methylation did (3c vs 3b). Fluorine or trifluoromethyl groups at the meta- position led to better EC50 than that at the para- position (3j vs 3k and 3p vs 3q). Further fluorination of 3j formed analog 3l, which maintained good antiviral profile. However, penta-fluorination (3n) slightly decreased the activity. In the case of methoxy group, ortho- substitution (3s) afforded a 5-fold improved EC50 compared with para- substitution (3t). Replacement of the methoxy group in 3t with trifuoromethoxy group (3r) afforded a more than 6-fold improved EC50, which could be contributed to its strong electron withdrawing effect. Analog 3v with one extra carbon between the ether oxygen and the terminal ring also possessed excellent activity. Analog 4b, with a benzene ring in the middle of the side chain and the n-propoxy group at meta- position, gave the best anti-DENV profile (EC50 = 0.3 μM, CC50 > 500 μM), which indicated that the best length of the linker between DNJ head group and the ether bond was six-carbon. Similar to the results obtained from BVDV assay, linker restrained analogs 5c–5e showed poor antiviral activity against DENV infection, which further confirmed that the linker of six-sp3 carbon was optimal. In summary, analogs 2h, 2l, 3j, 3l, 3v and 4b–4c were identified with nanomolar range anti-DENV activity (EC50 = 0.3–0.5 μM) with no cytotoxicity observed at the highest tested concentration up to 500 μM (SI > 1000).
Assessment of In Vitro ADME Properties
In addition to the efficacy, optimization of the absorption, distribution, metabolism and excretion (ADME) properties of drug candidates also plays an important role in increasing the rate of drug discovery successes and advancing high quality candidates to further pre-clinical studies. Early assessment and optimization of these properties could reduce the risk of drug failure in advanced phases. In this regard, compounds, 1c, 2l, 3c, 3j, 3l, 3p, 3s, 3v and 4b, were selected as representatives of each structural classes (1, 2, 3 and 4) for in vitro ADME experiments: aqueous solubility, plasma protein binding, cytochrome P450 (CYP) inhibition, Caco-2 permeability, and liver microsome (LM) stability determination (Table 4). These experiments have been proven to be reliable indicators of plasma exposure level after oral administration.32 Compound 7 and all its new analogs possessed favorable aqueous solubility (271–315 μM in PBS) at both pH 4.0 and 7.4. New compounds showed increased plasma protein binding percentage (human, rat and mouse), compared with the parent compound 7, suggesting their potential longer serum half-life. In general, the new compounds exhibited lower CYP inhibition than analog 7. Compounds, 3j, 3s, 3v or 4b, had no inhibition on any of the five major drug metabolizing human CYP450s (1A2, 2C9, 2C19, 2D6, and 3A4) at the concentration of 10 μM, implying low drug-drug interaction potential. 2l and 3l exhibited weak inhibition on CYP3A4 (21%, midazolam as substrate) and CYP1A2 (24%), respectively. At the same concentration, 1c showed 44% inhibition on CYP2C97; 3c 21% on CYP1A2 and 29% on CYP3A4 (midazolam as substrate); 3p 38% on CYP2C9; 7 37% on CYP2C19 and 21% on CYP3A4 (midazolam or testosterone as substrate). Caco-2 permeability assay confirmed that all the compounds had reasonable efflux ratio of 2.3–4.7, hence predicting reasonable intestinal absorption. In the liver microsome of three different species (human, rat and mouse), all the new compounds exhibited good stability, although some of them were slightly less stable compared to compound 7. Overall, compounds (2l, 3j, 3l, 3v and 4b) possessed both reasonable ADME properties and good antiviral profiles against DENV infection (EC50 = 0.3–0.5 μM, CC50 > 500 μM), and could be considered as leads for advanced stage studies.
Table 4.
In Vitro ADME Properties of Compounds 7, 1c, 2l, 3c, 3j, 3l, 3p, 3s, 3v and 4b
| ADME Properties | 7 | 1c | 2l | 3c | 3j | 3l | 3p | 3s | 3v | 4b |
|---|---|---|---|---|---|---|---|---|---|---|
| Solubility in PBS at pH 4.0 (μM) | 315 | 290 | 286 | 279 | 271 | 296 | 281 | 286 | 282 | 281 |
| Solubility in PBS at pH 7.4 (μM) | 315 | 302 | 295 | 286 | 283 | 292 | 278 | 293 | 288 | 288 |
| Human Plasma Protein Binding (% bound) | 33 | 91 | 71 | 80 | 78 | 74 | 93 | 65 | 52 | 68 |
| Rat Plasma Protein Binding (% bound) | 30 | 77 | 65 | 79 | 71 | 68 | 90 | 70 | 49 | 57 |
| Mouse Plasma Protein Binding (% bound) | 40 | 83 | 85 | 83 | 79 | 77 | 92 | 69 | 53 | 60 |
| CYP1A2 Inhibition at 10 μM (%) | 7.9 | 2.0 | 9.0 | 21.4 | 0.2 | 24.4 | 0.2 | − 1.6 | 17.3 | 15.9 |
| CYP2C9 Inhibition at 10 μM (%) | 9.6 | 43.5 | 5.0 | 8.9 | 6.2 | 7.1 | 37.6 | 11.7 | 10.7 | 10.5 |
| CYP2C19 Inhibition at 10 μM (%) | 37.0 | − 1.1 | − 4.9 | 3.9 | 0.7 | 1.6 | − 2.5 | − 4.4 | 14.2 | 2.8 |
| CYP2D6 Inhibition at 10 μM (%) | 1.2 | 9.1 | 2.5 | 12.8 | 10.3 | 11.4 | 3.4 | 8.7 | 9.1 | 11.0 |
| CYP3A4 Inhibition 1a at 10 μM (%) | 21.0 | 2.5 | 21.2 | 28.7 | − 2.3 | 10.9 | − 18.5 | 12.1 | 6.6 | 8.6 |
| CYP3A4 Inhibition 2 at 10 μM (%) | 20.5 | 4.2 | 3.1 | 2.3 | − 2.3 | − 1.9 | 3.2 | − 2.4 | 3.3 | 0.7 |
| Caco-2 Permeability (Efflux Ratio) | 2.9 | 2.9 | 3.9 | 2.3 | 2.4 | 4.7 | 2.3 | 3.9 | 2.3 | 2.4 |
| Caco-2 Permeability (Recovery %, A-Bb) | 82 | 55 | 69 | 70 | 67 | 59 | 51 | 61 | 71 | 66 |
| Caco-2 Permeability (Recovery %, B-A) | 117 | 76 | 97 | 95 | 91 | 83 | 79 | 92 | 93 | 91 |
| Human LMc Stability (% @60min) | 99 | 73 | 89 | 87 | 87 | 91 | 94 | 99 | 104 | 99 |
| Rat LM Stability (% @60min) | 98 | 80 | 90 | 81 | 91 | 79 | 81 | 98 | 91 | 97 |
| Mouse LM Stability (% @60min) | 104 | 77 | 92 | 96 | 104 | 91 | 92 | 92 | 97 | 87 |
CYP3A4 Inhibition 1, using midazolam as substrate; CYP3A4 Inhibition 2, using testosterone as substrate.
A-B = apical to basolateral; B-A = basolateral to apical.
LM = Liver Microsome.
Ranking and Prioritizing Lead Analogs
In order to further rank and prioritize the lead compounds obtained above, we performed side-by-side comparison in a high throughput in-cell western assay in DENV infected human hepatoma cell line Huh7.5. As shown in Figure 5A, all the five leads, 2l, 3j, 3l, 3v and 4b, demonstrated much improved antiviral activity compared to compound 7, in absence of cellular toxicity. Among them, 3l was the most potent one, which is consistent with the observation shown in Figure 3, where 3l efficiently changed the BVDV E2 mobility at lower concentration. Also supporting this observation is the result from DENV yield reduction assay on BHK cell line, which indicated that all of these compounds dose dependently reduced DENV titers, with 3l being the most potent (Figure 5B), over a range of doses from 0.8 to 20 μM, and with the highest maximum inhibitory capacity at 20 μM concentration. Taken together, we considered analog 3l as the most potent compound with favorable in vitro ADME profile, and therefore it was selected into further animal experiments.
Figure 5.

Side-by-side comparison of anti-DENV activity in in-cell western assay and yield reduction assay. (A) Huh 7.5 cells were infected and treated with indicated compounds, as described in “Experimental Section”. Green color represents the detection of DENV E protein, red for cell stain. (B) Standard yield reduction assay was performed using indicated concentration of compound. DENV titers were determined and expressed as % of control. Values plotted represent the average of three experimental repeats.
Assessment of Acute Toxicity In Vivo
Male Sprague-Dawley rats were orally administered with single dose of formulated compound 3l at 25, 50, 100, 200 mg/kg or vehicle alone, followed by 7-day observation. During the study period, no mortality or weight loss was observed. All the animals gained weight but with a decrease trend as the dose increased, which was consistent with the observed dose-related sign of gastrol-intestinal stress (soft feces with 50–100 mg/kg doses, and diarrhea with 200 mg/kg dose). These side-effects only occurred at 1–2 days post-treatment, and all the animals recovered after day 2 post-treatment. Generally, this 7-day in vivo toxicity study indicated that compound 3l was well tolerated in male Sprague-Dawley rats at a dose of up to 200 mg/kg.
Assessment of Pharmacokinetics (PK) In Vivo
Male Sprague-Dawley rats were given a 5 mg/kg intravenous (i.v.) or a 25 mg/kg oral dose of 3l. Following intravenous dosing at 5 mg/kg, the compound had an average plasma half-life (t1/2) of 1.44 ± 0.43 hours, and its average volume of distribution (Vd) was 3.1 L/kg. After oral dosing at 25 mg/kg, 3l reached a favorable maximum plasma concentration (Cmax) of 1340 ± 814 ng/mL (3.6 ± 2.2 μM, Vd = 17.2 L/kg) at an average time (Tmax) of 4.2 hours. The average oral bioavailability (F) was excellent, with an average value of 92 ± 4%, which is much improved compared with compound 7 (F = 56%17, Table 5).
Table 5.
Summary of Antiviral and other Profiles of Compounds 31 vs 7
| Compound | 3l | 7 |
|---|---|---|
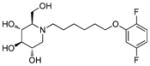
| Structure |
|
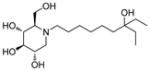
|
| Anti-BVDVa | EC50 = 0.5 ± 0.4 μM SI > 1000 |
EC50 = 6.2 ± 4.5 μM SI > 81 |
| Anti-DENVb | EC50 = 0.4 ± 0.2 μM SI > 1250 |
EC50 = 6.5 ± 0.6 μM SI > 77 |
| Solubility in PBS at pH 7.4 | 292 μM | 315 μM |
| CYP3A4 inhibitionc (at 10 μM) | 10.9%; - 1.9% | 21.0%; 20.5% |
| Bioavailability (F) | 92% | 56% |
tested in MDBK cells.
tested in BHK cells.
EC50: 50% effective concentration, measured by virus yield reduction assay; values represent average and standard deviation of results obtained from 3 independent experiments; SI: selective index, SI = CC50/EC50.
using midazolam and testosterone as substrates, respectively.
CONCLUSION
Building upon the encouraging in vivo efficacy observed for compound 7, we have designed and synthesized a wide range of ether tethered N-alkylated DNJ derivatives 1–5. BVDV virus yield reduction assay was used as a surrogate assay for SAR mapping. Among the newly synthesized imino sugars, compounds 2h, 2l, 3j, 3l, 3v and 4b–4c with cycloalkyl, phenyl derivatives, or middle phenyl alkyl ether displayed nanomolar range anti-DENV activity (EC50 = 0.3–0.5 μM), with no observable cytotoxicity at the highest tested concentration up to 500 μM (SI > 1000). The assessment of in vitro ADME properties identified a few optimized leads (2l, 3j, 3l, 3v and 4b) for advanced stage investigation. For a pilot animal toxicity and PK profiling studies, we selected compound 3l. This compound efficiently changed BVDV E2 glycoprotein mobility rate at lower concentration (Figure 3) indicating it is a more potent target enzyme inhibitor. Consistent with this observation, 3l is also demonstrated to be one of the most potent antiviral compounds against both BVDV and DENV in multiple antiviral assays. Interestingly, 3l was well tolerated in male Sprague Dawley rats at a dose up to 200 mg/kg and displayed desirable pharmacokinetics profiles, with much improved bioavailability compared to the hit 7 (92% vs 56%). Considering that 3l possesses significantly improved in vitro antiviral potency compared to compound 7, which has already achieved in vivo efficacy in animal models, these favorable ADME properties, toxicity and PK data of compound 3l justify its further development and possibly other analogs developed in this study, as potential anti-DENV therapy.
Experimental Section
Chemistry
1H NMR spectra were recorded on a 300 MHz INOVA VARIAN spectrometer. Chemical shifts values are given in ppm and referred as the internal standard to TMS (tetramethylsilane). The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quadruplet; m, multiplet and dd, doublet of doublets. The coupling constants (J) are reported in Hertz (Hz). Optical rotations were recorded on a JASCO P-2000 Digital Polarimeter. The data was collected at 24 °C in a 10 cm cell in methanol. [α]D values are given in 10−1 deg · cm2 g−1 (concentration c was given as g/100 mL). Mass Spectra were obtained on a 1200 Aligent LC-MS spectrometer (ES-API, Positive). Silica gel column chromatography was performed over silica gel 100–200 mesh, and the eluent was a mixture of ethyl acetate and hexanes, or mixture of methanol and ethyl acetate. All the tested compounds possess a purity of at least 95%. Analytical HPLC was run on the Agilent 1100 HPLC instrument, equipped with Agilent, ZORBAX SB-C18 column and UV detection at 210 nm. Eluent system was: A (Water, 0.1% Formic acid) and B (Methanol, 0.1% Formic acid); flow rate = 1 mL/min; Method A: 45%A, 55%B (3a–3d, 3i–3m, 3s–3v, 4a–4c and 5c–5e); Method B: 35%A, 65%B (3h, 3n–3r and 4d–4f); Method C: 25%A, 75%B (3e–3g). For compounds 1a–1c, 2a–2l, 5a–5b, 6 and 7, the purity was determined based on 1H NMR.
a. Synthesis of (2R,3R,4R,5S)-1-(7-Ethyl-7-hydroxynonyl)-2-(hydroxymethyl)piperidine-3,4,5-triol (7)
a.a Preparation of Oxocan-2-one (9)
Cycloheptanone (8, 5.60g, 50 mmol) in dichoromethane (75 mL) was added dropwise to a solution of meta-chloroperoxybenzoic acid (mCPBA, 16.80 g, 75 mmol) in dichloromethane (50 mL) at 0 °C. After addition, the reaction mixture was stirred at room temperature in the dark for 5 days. The solid was removed by filtration, and the filtrate was washed with sat. sodium bicarbonate solution (50 mL), followed by brine (50 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated to give a residue, which was slurried in a mixture of ethyl acetate and hexanes (10: 90). The solid was filtered off and the solvent was evaporated to afford the crude product, which was further purified through silica gel column chromatography (ethyl acetate: hexanes = 10: 90) to give 2.60 g of pure compound 9 as colorless oil. Yield, 41%.
a.b. Preparation of 7-Ethylnonane-1,7-diol (10)
Oxocan-2-one (9, 1.98 g, 15.5 mmol) in tetrahydrofuran (THF, 30 mL) was added dropwise to a solution of ethylmagnesium bromide in THF (1 M, 75.5 mL, 75.5 mmol) at 0 °C under Argon atmosphere. After addition, the reaction mixture was stirred and allowed to warm up to room temperature over 2 hours. The reaction was quenched with cold sat. ammonium chloride solution (50 mL), and then extracted with ethyl acetate (30 mL × 3). The combined organic layer was dried over anhydrous sodium sulfate and concentrated to give the crude product, which was further purified through silica gel column chromatography (ethyl acetate: hexanes = 30: 70) to afford 2.53 g of pure compound 10 as colorless oil. Yield, 87%.
a.c. Preparation of 7-Ethyl-7-hydroxynonanal (11)
7-Ethylnonane-1,7-diol (10, 3.35 g, 17.8 mmol) in dry dichloromethane (40 mL) was added to a suspension of pyridinium chlorochromate (PCC, 4.60 g, 21.3 mmol) in dry dichloromethane (60 mL). The reaction mixture was stirred at room temperature for 6 hours, and then filtered through a silica gel pad and washed with ethyl acetate (50 mL). The filtrate was concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 20: 80) to afford 2.30 g of pure compound 11 as colorless oil. Yield, 69%.
a.d. Synthesis of (2R,3R,4R,5S)-1-(7-Ethyl-7-hydroxynonyl)-2-(hydroxymethyl)piperidine-3,4,5-triol (7)
A solution of 1-deoxynojirimycin (DNJ, 653 mg, 4.0 mmol) in acetic acid (8 mL) was stirred at room temperature overnight, and then the solvent was removed under reduced pressure. The resulting residue was treated with 200 proof ethanol (20 mL) and 7-ethyl-7-hydroxynonanal (11, 969 mg, 5.2 mmol), followed by acetic acid (0.5 mL). The reaction mixture was stirred at room temperature under Argon atmosphere for another 2 hours. Then, it was transferred to the hydrogenation bottle, followed by addition of 5% Pd/C (160 mg) and 200 proof ethanol (10 mL). The mixture was hydrogenated under 45 psi of H2 for 24 hours. After the reaction was complete, it was filtered through a silica gel pad. The filtrate was concentrated and purified through silica gel column chromatography (methanol: ethyl acetate = 30: 70) to afford 0.81 g of pure compound 7 as white solid. Yield, 61%. 1H NMR (300 MHz, CD3OD): δ 3.85 (d, J = 7.2 Hz, 2H, OCH2), 3.52-3.44 (m, 1H, OCH), 3.36 (t, J = 9.3 Hz, 1H, OCH), 3.13 (t, J = 9.0 Hz, 1H, OCH), 3.02 (dd, J = 11.4, 5.1 Hz, 1H, NCH), 2.89-2.79 (m, 1H, NCH), 2.67-2.57 (m, 1H, NCH), 2.27-2.15 (m, 2H, 2 x NCH), 1.52-1.33 (m, 14H), 0.84 (t, J = 7.5 Hz, 6H, 2 x CH3). MS: MH+ = 334.
b. Synthesis of DNJ Derivatives 1-2
b.a. Preparation of 6-(Benzyloxy)hexan-1-ol (13)
To a stirring solution of 1,6-hexanediol (12, 23.15 g, 196 mmol) and 18-crown-6 (2.15 g, 8 mmol) in THF (200 mL) at 0 °C, was added finely ground potassium hydroxide (10.06 g, 179 mmol), followed by the addition of benzyl bromide (19.4 mL, 163 mmol). The reaction mixture was stirred for 48 hours at room temperature, and then washed with brine (200 mL). The aqueous layer was extracted with ethyl acetate (100 mL × 2). All the organic layer was combined, dried over anhydrous sodium sulfate and concentrated to give the crude product, which was further purified through silica gel column chromatography (ethyl acetate: hexanes = 20: 80) to afford 22.42 g of pure compound 13 as colorless oil. Yield was 55%.
b.b. Preparation of 6-(Benzyloxy)hexyl Methanesulfonate (14)
To a stirring solution of 6-(benzyloxy)hexan-1-ol (13, 21.42 g, 103 mmol) in pyridine (100 mL) at 0 °C, methanesulfonyl chloride (9.6 mL, 123 mmol) was added dropwise. After addition, the reaction mixture was stirred at 0 °C for 3 hours. Then, the reaction was quenched into ice water (100 mL), and the pH was adjusted to 1~2 with 2 N hydrochloric acid (HCl). The mixture was extracted with dichloromethane (100 mL × 3). The combined organic layer was dried over anhydrous sodium sulfate, concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 10: 90 to 20: 80) to afford 28.50 g of pure compound 14 as colorless oil. Yield, 97%.
b.c. General Procedure for the Preparation of Intermediates 16
To a solution of the alcohol 15 (6 mmol) in anhydrous dimethylformamide (DMF, 10 mL) at room temperature, NaH (60%, 480 mg, 12 mmol) was added. The mixture was stirred at room temperature for 90 min and treated with a solution of 6-(benzyloxy)hexyl methanesulfonate (14, 1.43 g, 5mmol) in anhydrous DMF (5 mL). The reaction mixture was then heated to 75 °C for 1 hour. After cooling to room temperature, the reaction was quenched with ice/water (50 mL) and extracted with ethyl acetate (30 mL × 3). The combined organic layer was dried over anhydrous sodium sulfate, concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 2: 98) to afford the pure compound 16.
b.d. General Procedure for the Preparation of Alcohols 17
A reaction mixture of the compound 16 (606 mg) and 10% Pd/C (242 mg) in 200 proof ethanol (20 mL) was hydrogenated under 60 psi of H2 for 24 hours. After the reaction was complete, it was treated with celite (800 mg), and then filtered through a silica gel pad. The filtrate was concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 5: 95 to 30: 70) to afford the pure compound 17.
b.e. General Procedure for the Preparation of Aldehydes 18
To a stirred suspension of PCC (647 mg, 3 mmol) and silica gel (647 mg) in dry dichloromethane (10 mL), was added a solution of the alcohol 17 (2 mmol) in dichloromethane (10 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 4 hours. Then, it was filtered through a silica gel pad, and washed with ethyl acetate (20 mL). The filtrate was concentrated and then purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 15: 85) to afford the pure compound 18.
b.f. General Procedure for the Synthesis of DNJ Derivatives 1–2
A solution of DNJ (33 mg, 0.2 mmol) in acetic acid (1 mL) was stirred at room temperature overnight. The solvent was then removed under reduced pressure. The resulting residue was treated with 200 proof ethanol (5 mL) and the aldehyde 18 (0.26 mmol), followed by acetic acid (2 drops as catalyst). The reaction mixture was stirred at room temperature under argon atmosphere for another hour. Then, it was transferred to the hydrogenation bottle, followed by addition of 10% Pd/C (32 mg) and 200 proof ethanol (5 mL). The mixture was hydrogenated under 45 psi of H2 for 24 hours. After the reaction was complete, it was treated with celite (100 mg), and then filtered through a silica gel pad. The filtrate was concentrated and purified through silica gel column chromatography (methanol: ethyl acetate = 5: 95 to 20: 80) to afford the pure compound 1 or 2.
(2R,3R,4R,5S)-1-(6-(Cycloheptyloxy)hexyl)-2-(hydroxymethyl)piperidine-3,4,5-triol (2l)
White semi-solid. Yield, 93%. [α]D = − 4 (c = 2.0, methanol). 1H NMR (300 MHz, CD3OD): δ 3.86 (d, J = 2.7 Hz, 2H, OCH2), 3.53-3.34 (m, 5H, 5 x OCH), 3.14 (t, J = 9.3 Hz, 1H, OCH), 3.04 (dd, J = 11.4, 4.8 Hz, 1H, NCH), 2.89-2.81 (m, 1H, NCH), 2.69-2.64 (m, 1H, NCH), 2.30-2.19 (m, 2H, 2 x NCH), 1.94-1.29 (m, 20H). MS: calculated for MH+ = 360, found 360.
c. Synthesis of DNJ Derivatives 3
c.a. General Procedure for the Preparation of Alcohols 21
To a stirring solution of the substituted phenol 20 (1.5 mmol) and 6-bromo-1-hexanol (19, 0.14 mL, 1 mmol) in DMF (3 mL), was added potassium carbonate (207 mg, 1.5 mmol). The reaction mixture was stirred at 80 °C for 4 hours. After cooling to room temperature, the reaction was quenched with water (30 mL), and extracted with ethyl acetate (20 mL × 3). The combined organic layer was washed with brine (30 mL), dried over sodium sulfate, and then concentrated under reduced pressure. The resulting residue was purified through silica gel column chromatography (ethyl acetate: hexanes = 5: 95 to 20: 80) to afford the pure compound 21.
c.b. General Procedure for the Preparation of Aldehydes 22
To a stirring suspension of PCC (363 mg, 1.68 mmol) and silica gel (363 mg) in dry dichloromethane (10 mL), was added a solution of the alcohol 21 (1.40 mmol) in dichloromethane (5 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 4 hours. Then, it was filtered through a silica gel pad, and washed with ethyl acetate (20 mL). The filtrate was concentrated and then purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 10: 90) to afford the pure compound 22.
c.c. General Procedure for the Synthesis of DNJ Derivatives 3
A solution of DNJ (49 mg, 0.3 mmol) in acetic acid (1 mL) was stirred at room temperature overnight, and then the solvent was removed under reduced pressure. The resulting residue was treated with 200 proof ethanol (5 mL) and the aldehyde 22 (0.39 mmol), followed by acetic acid (2 drops as catalyst). The reaction mixture was stirred at room temperature under argon atmosphere for another hour. Then, it was transferred to the hydrogenation bottle, followed by addition of 10% Pd/C (30 mg) and 200 proof ethanol (5 mL). The mixture was hydrogenated under 45 psi of H2 for 24 hours. After the reaction was complete, it was treated with celite (100 mg), and then filtered through a silica gel pad. The filtrate was concentrated and purified through silica gel column chromatography (methanol: ethyl acetate = 5: 95 to 20: 80) to afford the pure compound 3.
(2R,3R,4R,5S)-1-(6-(3-Fluorophenoxy)hexyl)-2-(hydroxymethyl)piperidine-3,4,5-triol (3j)
White semi-solid. Yield, 96%. [α]D = − 6 (c = 1.5, methanol). 1H NMR (300 MHz, CD3OD): δ 7.23-7.18 (m, 1H, CHar), 6.71-6.60 (m, 3H, CHar), 3.94 (t, J = 6.3 Hz, 2H, ArOCH2), 3.85 (s, 2H, OCH2), 3.52-3.44 (m, 1H, OCH), 3.36 (t, J = 9.3 Hz, 1H, OCH), 3.13 (t, J = 9.3 Hz, 1H, OCH), 3.00 (dd, J = 11.1, 5.1 Hz, 1H, NCH), 2.84-2.77 (m, 1H, NCH), 2.65-2.60 (m, 1H, NCH), 2.23-2.12 (m, 2H, 2 x NCH), 1.79-1.35 (m, 8H, 4 x CH2). MS: calculated for MH+ = 358, found 358.
(2R,3R,4R,5S)-1-(6-(2,5-Difluorophenoxy)hexyl)-2-(hydroxymethyl)piperidine-3,4,5-triol (3l)
White semi-solid. Yield, 89%. [α]D = − 5 (c = 2.1, methanol). 1H NMR (300 MHz, CD3OD): δ 7.09-7.01 (m, 1H, CHar), 6.90-6.84 (m, 1H, CHar), 6.64-6.56 (m, 1H, CHar), 4.03 (t, J = 6.3 Hz, 2H, ArOCH2), 3.87 (d, J = 2.7 Hz, 2H, OCH2), 3.53-3.45 (m, 1H, OCH), 3.37 (t, J = 9.3 Hz, 1H, OCH), 3.15 (t, J = 9.3 Hz, 1H, OCH), 3.05 (dd, J = 11.4, 5.1 Hz, 1H, NCH), 2.93-2.83 (m, 1H, NCH), 2.71-2.61 (m, 1H, NCH), 2.31-2.20 (m, 2H, 2 x NCH), 1.86-1.77 (m, 2H, CH2), 1.62-1.49 (m, 4H, 2 x CH2), 1.43-1.33 (m, 2H, CH2). MS: calculated for MH+ = 376, found 376.
(2R,3R,4R,5S)-1-(6-(Benzyloxy)hexyl)-2-(hydroxymethyl)piperidine-3,4,5-triol (3v)
White semi-solid. Yield, 69%. [α]D = − 4 (c = 1.5, methanol). 1H NMR (300 MHz, CD3OD): δ 7.34-7.25 (m, 5H, CHar), 4.49 (s, 2H, ArCH2O), 3.86 (d, J = 2.7 Hz, 2H, OCH2), 3.53-3.45 (m, 3H, 3 x OCH), 3.37 (t, J = 9.6 Hz, 1H, OCH), 3.14 (t, J = 9.0 Hz, 1H, OCH), 3.04 (dd, J = 11.4, 5.1 Hz, 1H, NCH), 2.88-2.80 (m, 1H, NCH), 2.69-2.64 (m, 1H, NCH), 2.30-2.21 (m, 2H, 2 x NCH), 1.66-1.27 (m, 8H, 4 x CH2). MS: calculated for MH+ = 354, found 354.
d. Synthesis of DNJ Derivatives 4
d.a. General Procedure for the Preparation of Aldehydes 24
To a stirring suspension of PCC (647 mg, 3 mmol) and silica gel (647 mg) in dry dichloromethane (10 mL), was added a solution of the alcohol 23 (2 mmol) in dichloromethane (10 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 4 hours. Then, it was filtered through a silica gel pad, and washed with ethyl acetate (20 mL). The filtrate was concentrated and then purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 10: 90) to afford the pure compound 24.
d.b. General Procedure for the Synthesis of DNJ Derivatives 4
A solution of DNJ (33 mg, 0.2 mmol) in acetic acid (1 mL) was stirred at room temperature overnight. The solvent was then removed under reduced pressure. The resulting residue was treated with 200 proof ethanol (5 mL) and the aldehyde 24 (0.26 mmol), followed by acetic acid (2 drops as catalyst). The reaction mixture was stirred at room temperature under argon atmosphere for another hour. Then, it was transferred to the hydrogenation bottle, followed by addition of 10% Pd/C (33 mg) and 200 proof ethanol (5 mL). The mixture was hydrogenated under 45 psi of H2 for 24 hours. After the reaction was complete, it was treated with celite (100 mg), and then filtered through a silica gel pad. The filtrate was concentrated and purified through silica gel column chromatography (methanol: ethyl acetate = 5: 95 to 20: 80) to afford the pure compound 4.
(2R,3R,4R,5S)-2-(Hydroxymethyl)-1-(3-(3-propoxyphenyl)propyl)piperidine-3,4,5-triol (4b)
White semi-solid. Yield, 97%. [α]D = − 6 (c = 1.7, methanol). 1H NMR (300 MHz, CD3OD): δ 7.18-7.12 (m, 1H, CHar), 6.77-6.69 (m, 3H, CHar), 3.90 (t, J = 6.6 Hz, 2H, ArOCH2), 3.81 (d, J = 2.7 Hz, 2H, OCH2), 3.49-3.43 (m, 1H, OCH), 3.35 (t, J = 9.3 Hz, 1H, OCH), 3.13 (t, J = 9.3 Hz, 1H, OCH), 2.99 (dd, J = 11.4, 5.1 Hz, 1H, NCH), 2.87-2.84 (m, 1H, NCH), 2.67-2.54 (m, 3H, NCH and ArCH2, overlapped), 2.27-2.16 (m, 2H, 2 x NCH), 1.84-1.74 (m, 4H, 2 x CH2), 1.03 (t, J = 7.5 Hz, 3H, CH3). MS: calculated for MH+ = 340, found 340.
e. Synthesis of DNJ Derivative 5a
e.a. Preparation of (Z)-Non-6-enal (26)
To a stirring suspension of PCC (841 mg, 3.9 mmol) and silica gel (841 mg) in dry dichloromethane (20 mL), was added a solution of the alcohol 25 (427 mg, 3 mmol) in dichloromethane (10 mL). The reaction mixture was stirred at room temperature under argon atmosphere for 4 hours. Then, it was filtered through a silica gel pad, and washed with ethyl acetate (20 mL). The filtrate was concentrated and then purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 10: 90) to afford 375 mg of pure compound 26 as colorless oil. Yiled, 89%.
e.b. Synthesis of (2R,3R,4R,5S)-2-(Hydroxymethyl)-1-((Z)-non-6-en-1-yl)piperidine-3,4,5-triol (5a)
To a solution of DNJ (33 mg, 0.2 mmol) and (Z)-non-6-enal (26, 36 mg, 0.26 mmol) in methanol (5 mL), was added acetic acid (24 μL, 0.4 mmol), followed by the addition of sodium cyanoborohydride (1 M in THF, 0.3 mL, 0.3 mmol). The reaction mixture was stirred at room temperature under argon atmosphere for 24 hours. Then, it was quenched with water (5 mL), concentrated and purified through silica gel column chromatography (methanol: ethyl acetate = 5: 95 to 20: 80) to afford 43 mg of pure compound 5a as colorless semi-solid. Yield, 75%. 1H NMR (300 MHz, CD3OD): δ 5.38-5.29 (m, 2H, 2 x CH=), 3.86 (d, J = 1.8 Hz, 2H, OCH2), 3.54-3.46 (m, 1H, OCH), 3.38 (t, J = 9.6 Hz, 1H, OCH), 3.16 (t, J = 9.0 Hz, 1H, OCH), 3.04 (dd, J = 11.1, 4.8 Hz, 1H, NCH), 2.88-2.80 (m, 1H, NCH), 2.71-2.64 (m, 1H, NCH), 2.32-2.22 (m, 2H, 2 x NCH), 2.08-1.99 (m, 4H, 2 x CH2), 1.58-1.26 (m, 6H, 3 x CH2), 0.95 (t, J = 7.8 Hz, 3H, CH3). MS: calculated for MH+ = 288, found 288.
f. Synthesis of DNJ Derivative 5b
f.a. Preparation of Non-3-yn-1-yl 4-Methylbenzenesulfonate (28)
To a stirring solution of non-3-yn-1-ol (27, 421 mg, 3 mmol) in pyridine (6 mL) at 0 °C, was added 4-toluenesulfonyl chloride (686 mg, 3.6 mmol). The reaction mixture was stirred at 0 °C for 3 hours. Then, it was quenched with 2 N HCl, acidified to pH 1~2 at 0 °C, and extracted with ethyl acetate (20 mL × 3). The combined organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 10: 90) to afford 585 mg of pure compound 28 as colorless oil. Yield, 66%.
f.b. Synthesis of (2R,3R,4R,5S)-2-(Hydroxymethyl)-1-(non-3-yn-1-yl)piperidine-3,4,5-triol (5b)
A reaction mixture of non-3-yn-1-yl 4-methylbenzenesulfonate (28, 294 mg, 1 mmol), DNJ (49 mg, 0.3 mmol) and sodium carbonate (159 mg, 1.5 mmol) in DMF (3 mL) was stirred at 75 °C in a sealed tube for 24 hours. After cooling to room temperature, the reaction was quenched with water (10 mL), concentrated and slurried in a mixture of methanol and dichloromethane (40: 60, 10 mL). The solid was filtered off and the filtrate was concentrated to give the crude product, which was purified by preparative TLC plate (methanol: ethyl acetate = 10: 90) to afford 45 mg of pure compound 5b as white semi-solid. Yield, 49%. 1H NMR (300 MHz, CD3OD): δ 3.92-3.81 (m, 2H, OCH2), 3.49-3.41 (m, 1H, OCH), 3.33 (t, J = 9.0 Hz, 1H, OCH), 3.12 (t, J = 9.0 Hz, 1H, OCH), 3.01-2.81 (m, 3H), 2.35-2.09 (m, 6H), 1.48-1.28 (m, 6H, 3 x CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3). MS: calculated for MH+ = 286, found 286.
g. Synthesis of DNJ Derivatives 5c–5e
g.a. General Procedure for the Preparation of Intermediates 30
To a solution of the mesylate 29 (3 mmol) and 2,5-difluorophenol (585 mg, 4.5 mmol) in DMF (10 mL), was added potassium carbonate (622 mg, 4.5 mmol). The reaction mixture was stirred at 80 °C for 1 hour. After cooling to room temperature, it was quenched with water (20 mL) and extracted with ethyl acetate (20 mL × 3). The combined organic layer was washed with brine (20 mL), dried over anhydrous sodium sulfate, concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 5: 95) to afford the pure compound 30.
g.b. General Procedure for the Preparation of Alcohols 31
To a stirred solution of the intermediate 30 (2.44 mmol) in THF (20 mL), was added tetra-n-butylammonium fluoride (1 M in THF, 12.2 mL, 12.2 mmol). The reaction mixture was stirred at room temperature for 2 hours. Then, it was quenched with water (30 mL) and extracted with ethyl acetate (30 mL × 3). The combined organic layer was washed with brine (30 mL), dried over anhydrous sodium sulfate, concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 10: 90 to 25: 75) to afford the pure compound 31.
g.c. General Procedure for the Preparation of Tosylates 32
To a stirring solution of the alcohol 31 (2.15 mmol) in pyridine (5 mL) at 0 °C, 4-toluenesulfonyl chloride (492 mg, 2.58 mmol was added. The reaction mixture was stirred at 0 °C for 3 hours. Then it was quenched with 2 N HCl, acidified to pH 1~2 at 0 °C, and extracted with ethyl acetate (20 mL × 3). The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, concentrated and purified through silica gel column chromatography (ethyl acetate: hexanes = 0: 100 to 10: 90) to afford the pure compound 32.
g.d. General Procedure for the Synthesis of DNJ Derivatives 5c–5e
A reaction mixture of the tosylate 32 (126 mg, 0.33 mmol), DNJ (16 mg, 0.1 mmol) and sodium carbonate (53 mg, 0.5 mmol) in DMF (3 mL) was stirred at 75 °C in a sealed tube for 24 hours. After cooling to room temperature, the reaction was quenched with water (10 mL), concentrated and slurried in a mixture of methanol and dichloromethane (40: 60, 10 mL). The solid was filtered off and the filtrate was concentrated to give the crude product, which was purified by preparative thin layer chromatography (TLC) plate (methanol: ethyl acetate = 10: 90) to afford the pure compound 5c–5e.
(2R,3R,4R,5S)-1-((Z)-6-(2,5-Difluorophenoxy)hex-3-en-1-yl)-2-(hydroxymethyl)piperidine-3,4,5-triol (5c)
Colorless semi-solid. Yield, 30%. 1H NMR (300 MHz, CD3OD): δ 7.10-7.01 (m, 1H, CHar), 6.91-6.85 (m, 1H, CHar), 6.64-6.58 (m, 1H, CHar), 5.56-5.48 (m, 2H, 2 x CH=), 4.06-4.01 (m, 2H, ArOCH2), 3.87-3.84 (m, 2H, OCH2), 3.49-3.42 (m, 1H, OCH), 3.34 (t, J = 7.2 Hz, 1H, OCH), 3.13 (t, J = 9.0 Hz, 1H, OCH), 3.00 (dd, J = 11.4, 5.1 Hz, 1H, NCH), 2.82-2.77 (m, 1H, NCH), 2.69-2.67 (m, 1H, NCH), 2.60-2.54 (m, 2H), 2.34-2.14 (m, 4H). MS: calculated for MH+ = 374, found 374.
Western blotting
MDBK cells were infected with BVDV at MOI of 1 and incubated with compounds (at concentration of 0.8 μM, 4.0 μM, 20 μM and 100 μM) for 24 hours. Cells were lysed in Laemmili buffer prior to analysis on 12% polyacrylamide gel (NuPage, Invitrogen). After electrophoresis and electrotransfer to nitrocellulose membranes, proteins were detected with monoclonal antibody WB166 against BVDV E2 protein (Central Veterinary Laboratories, Surrey, UK) followed by goat anti-mouse infra-red dye-labeled secondary antibodies (LI-COR) incubation and LI-COR Odyssey imaging.
BVDV Virus Yield Reduction Assay
Antiviral effect on BVDV was performed with standard yield reduction assay. Briefly, For BVDV infection (NADL strain), 2 × 105 of Madin-Darby bovine kidney (MDBK) cells were seeded in 24-well plates. 24 hours later, the cells were infected with BVDV at a multiplicity of infection (MOI) of 1 in 100 μL complete medium. After adsorption for 1 hour at 37 °C, unbound virus was removed by washing with serum-free medium followed by addition of fresh medium and compounds at concentration ranging from 0.1 to 100 μM. To determine virus titers, culture media were harvested 22 hours after infection for a standard plaque forming assay: MDBK cells were infected with serial 10-fold dilutions of culture media harvested from infected cells with or without treatment. After 1 hour absorption, the cells were washed and overlaid with medium containing 1% methylcellulose and incubated at 37 °C for 3 days. Plaques were counted directly under microscope or after staining with neutral red. The dose-dependent plaque yield reduction curves were then plotted to determine the inhibitor concentration required to inhibit BDVD virus titer by 50% (EC50).
DENV Virus Yield Reduction Assay
Baby hamster kidney (BHK) cells were plated in 24-well plates and infected with dengue virus (serotype 2, New Guniea C strain) at an MOI of 0.01 for 1 hour. After the removal of the inoculums, the cells were treated with test compounds at concentrations ranging from 0.1 to 100 μM for 48 hours at 37 °C. The supernatants were then collected for detection of virus titer. Monolayer of Vero cells were infected for 1 hour with 10-fold dilutions of the culture media harvested from infected cells with or without treatment, followed by overlay with media containing 0.5% methylcellulose and incubated at 37 °C for 3 to 5 days. Viral foci were detected by immunostaining with 4G2 antibody (Millipore) or counting of the plaques after crystal violet staining. The dose-dependent plaque yield reduction curves were then plotted to determine the inhibitory concentration required to inhibit dengue virus titer by 50% (EC50).
Cytotoxicity Assay
To determine the cell viability, a MTT based assay (Sigma) was performed. Cells were set up and incubated with various concentrations of compounds under condition that was identical to that used for yield reduction assay, except that cells were not infected. The dose-dependent curves were generated to determine the inhibitory concentration required to inhibit cell viability by 50% (CC50).
DENV In-cell Western Assay
The assay was performed essentially as described for detection of hepatitis C virus protein in human hepatoma cell line Huh7.5,12 except that a DENV envelop protein (E protein) monoclonal antibody (4G2) was used. Cells were infected at MOI of 0.01 for 1 hour, followed by incubation in absence or presence of doses of test compounds (ranging from 0.1 to 100 μM) for 2 days. DENV E protein was detected following incubation with 4G2 antibody and IRD-conjugated anti-mouse immunoglobulin G, shown in green color (LI-COR). The cell viability was simultaneously determined by DRAQ5 (Biostatus Limited, UK) and Sapphire 700 (LI-COR, Lincoln, Nebraska) staining, shown in red color. The fluorescence signal intensity was determined with LI-COR Odyssey.
ADME Profiling
Selected compounds were analyzed for ADME characteristics in standard in vitro assays (Pharmaron). The following assays were performed: 1) solubility determination, to analyze aqueous solubility at physiological and acidic pH (7.4 and 4.0, mimicking serum and gut conditions, respectively); 2) plasma protein binding measurement, to determine binding of compound to human and rodent serum protein, which relates to serum stability and availability to target tissues, 3) metabolic stability, to determine the stability of compound in human and rodent liver cell extracts, which relates to pharmacokinetics and excretion; 4) inhibition of different isozymes of human cytochrome P450 (CYP), which relates to compound stability and the potential for multi-drug interactions; and 5) cell membrane permeability and intestinal uptake through polarized (human intestinal) Caco-2 cell monolayers. Compound analysis in all assays was carried out by LC-MS/MS (ESI).
Formulation
In order to dose the animal at high dose in the in vivo toxicity assay and PK study, we intended to dissolve compound 3l at concentration of 20 mM in PBS. This was achieved by using 10% Solutol HS15 (BASF) solution in PBS and shaking at 37 °C for 6 hours to give a clear solution (20 mM). The stability and biological activity were confirmed with LC-MS and standard antiviral assays.
Single Dose Oral Toxicity Study in Rats
Single dose oral toxicity study of compound 3l was performed in 10-week old Sprague-Dawley rats, at 25, 50, 100 or 200 mg/kg doses (BASi). Each treatment group included 2 rats. The rats were observed for 7 days (general clinical observation, body weight, food consumption) and sacrificed at day 8.
Pharmacokinetics Study
In male Sprague-Dawley rats, the oral bioavailability and pharmacokinetics of compound 3l were evaluated (BASi). Three rats were used in each of the study. Rats were given a 5 mg/kg intravenous (i.v.) dose of compound 3l via an implanted venous catheter. After a washout period of at least 48 hours following the i.v. dose, animals were given a 25 mg/kg oral dose of 3l via gavage. Plasma concentrations of the test compound were determined by LC-MS/MS. Non-compartmental pharmacokinetic analysis was performed for plasma concentrations of each animal in Watson (v7.3.0.01, Thermo Inc.)
Supplementary Material
Scheme 3.

General Synthetic Procedure for DNJ Derivatives 1–2
Scheme 4.

Synthetic Procedure for DNJ Derivatives 3–4
Scheme 5.

Synthetic Procedure for DNJ Derivatives 5
Acknowledgments
This project was supported by Transformational Medical Technologies program contract [HDTRA1-09-CHEM-BIO-BAA] from the Department of Defense Chemical and Biological Defense program through the Defense Threat Reduction Agency (DTRA), NIH grants (AI061441 and AI084267-0109) and by the Hepatitis B Foundation through an appropriation from the Commonwealth of Pennsylvania. We thank Dr. Robert Moriarty (University of Illinois at Chicago) for chemistry consultation.
ABBREVIATIONS
- DNJ
deoxynojirimycin
- DENV
dengue virus
- BVDV
bovine viral diarrhea virus
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HIV
human immunodeficiency virus
- HSV
herpes simplex virus
- WNV
West Nile virus
- SAR
structure-activity relationship
- ER
endoplasmic reticulum
- EC50
50% effective concentration
- CC50
50% cytotoxic concentration
- SI
selectivity index
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- MDBK
Madin-Darby bovine kidney
- BHK
baby hamster kidney
- MOI
multiplicity of infection
- ADME
absorption, distribution, metabolism, and excretion
- PBS
phosphate buffered saline
- CYP
cytochrome P450
- LM
liver microsome
- PK
pharmacokinetics
- t1/2
plasma half-life
- Vd
volume of distribution
- Cmax
maximum plasma concentration
- Tmax
time to reach Cmax
- F
bioavailability
- mCPBA
meta-chloroperoxybenzoic acid
- PCC
pyridinium chlorochromate
- HCl
hydrochloric acid
- THF
tetrahydrofuran
- DMF
dimethylformamide
- TLC
thin layer chromatography
Footnotes
Supporting Information. Characterization data and NMR spectra of compounds 1–5 & 7; X-ray data of 3l (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Noble CG, Chen YL, Dong H, Gu F, Lim SP, Schul W, Wang QY, Shi PY. Strategies for development of Dengue virus inhibitors. Antiviral Res. 2010;85:450–462. doi: 10.1016/j.antiviral.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Whitehead SS, Blaney JE, Durbin AP, Murphy BR. Prospects for a dengue virus vaccine. Nat Rev Microbiol. 2007;5:518–528. doi: 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 3.Yin Z, Chen YL, Schul W, Wang QY, Gu F, Duraiswamy J, Kondreddi RR, Niyomrattanakit P, Lakshminarayana SBGA, Xu HY, Liu W, Liu B, Lim JY, Ng CY, Qing M, Lim CC, Yip A, Wang G, Chan WL, Tan HP, Lin K, Zhang B, Zou G, Bernard KAGC, Beltz K, Dong M, Weaver M, He H, Pichota A, Dartois V, Keller TH, Shi PY. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci U S A. 2009;106:20435–20439. doi: 10.1073/pnas.0907010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Compain P, Martin OR. Iminosugars: From synthesis to therapeutic applications. Wiley; 2007. [Google Scholar]
- 5.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 6.Block TM, Lu X, Mehta AS, Blumberg BS, Tennant B, Ebling M, Korba B, Lansky DM, Jacob GS, Dwek RA. Treatment of chronic hepadnavirus infection in a woodchuck animal model with an inhibitor of protein folding and trafficking. Nat Med. 1998;4:610–614. doi: 10.1038/nm0598-610. [DOI] [PubMed] [Google Scholar]
- 7.Block TM, Lu X, Platt FM, Foster GR, Gerlich WH, Blumberg BS, Dwek RA. Secretion of human hepatitis B virus is inhibited by the imino sugar N-butyldeoxynojirimycin. Proc Natl Acad Sci U S A. 1994;91:2235–2239. doi: 10.1073/pnas.91.6.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J, Wang L, Ma D, Qu X, Guo H, Xu X, Mason PM, Bourne N, Moriarty R, Gu B, Guo JT, Block TM. Novel imino sugar derivatives demonstrate potent antiviral activity against flaviviruses. Antimicrob Agents Chemother. 2009;53:1501–1508. doi: 10.1128/AAC.01457-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dwek RA, Butters TD, Platt FM, Zitzmann N. Targeting glycosylation as a therapeutic approach. Nat Rev Drug Discov. 2002;1:65–75. doi: 10.1038/nrd708. [DOI] [PubMed] [Google Scholar]
- 10.Gu B, Mason P, Wang L, Norton P, Bourne N, Moriarty R, Mehta A, Despande M, Shah R, Block T. Antiviral profiles of novel iminocyclitol compounds against bovine viral diarrhea virus, West Nile virus, dengue virus and hepatitis B virus. Antivir Chem Chemother. 2007;18:49–59. doi: 10.1177/095632020701800105. [DOI] [PubMed] [Google Scholar]
- 11.Jordan R, Nikolaeva OV, Wang L, Conyers B, Mehta A, Dwek RA, Block TM. Inhibition of host ER glucosidase activity prevents Golgi processing of virion-associated bovine viral diarrhea virus E2 glycoproteins and reduces infectivity of secreted virions. Virology. 2002;295:10–19. doi: 10.1006/viro.2002.1370. [DOI] [PubMed] [Google Scholar]
- 12.Qu X, Pan X, Weidner J, Yu W, Alonzi D, Xu X, Butters T, Block T, Guo JT, Chang J. Inhibitors of endoplasmic reticulum alpha-glucosidases potently suppress hepatitis C virus virion assembly and release. Antimicrob Agents Chemother. 2011;55:1036–1044. doi: 10.1128/AAC.01319-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butters TD, Dwek RA, Platt FM. Therapeutic applications of imino sugars in lysosomal storage disorders. Curr Top Med Chem. 2003;3:561–574. doi: 10.2174/1568026033452483. [DOI] [PubMed] [Google Scholar]
- 14.Fischl MA, Resnick L, Coombs R, Kremer AB, Pottage JC, Jr, Fass RJ, Fife KH, Powderly WG, Collier AC, Aspinall RL, et al. The safety and efficacy of combination N-butyl-deoxynojirimycin (SC-48334) and zidovudine in patients with HIV-1 infection and 200–500 CD4 cells/mm3. J Acquir Immune Defic Syndr. 1994;7:139–147. [PubMed] [Google Scholar]
- 15.Block TM, Jordan R. Iminosugars as possible broad spectrum anti hepatitis virus agents: the glucovirs and alkovirs. Antivir Chem Chemother. 2001;12:317–325. doi: 10.1177/095632020101200601. [DOI] [PubMed] [Google Scholar]
- 16.Bridges CG, Ahmed SP, Kang MS, Nash RJ, Porter EA, Tyms AS. The effect of oral treatment with 6-O-butanoyl castanospermine (MDL 28,574) in the murine zosteriform model of HSV-1 infection. Glycobiology. 1995;5:249–253. doi: 10.1093/glycob/5.2.249. [DOI] [PubMed] [Google Scholar]
- 17.Chang J, Schul W, Butters TD, Yip A, Liu B, Goh A, Lakshminarayana SB, Alonzi D, Reinkensmeier G, Pan X, Qu X, Weidner JM, Wang L, Yu W, Borune N, Kinch MA, Rayahin JE, Moriarty R, Xu X, Shi PY, Guo JT, Block TM. Combination of α-glucosidase inhibitor and ribavirin for the treatment of dengue virus infection in vitro and in vivo. Antiviral Res. 2011;89:26–34. doi: 10.1016/j.antiviral.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang J, Schul W, Yip A, Xu X, Guo JT, Block TM. Competitive inhibitor of cellular α-glucosidases protects mice from lethal dengue virus infection. Antiviral Res. 2011;92:369–371. doi: 10.1016/j.antiviral.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Datema R, Romero PA, Rott R, Schwarz RT. On the role of oligosaccharide trimming in the maturation of Sindbis and influenza virus. Arch Virol. 1984;81:25–29. doi: 10.1007/BF01309294. [DOI] [PubMed] [Google Scholar]
- 20.Malvoisin E, Wild F. The role of N-glycosylation in cell fusion induced by a vaccinia recombinant virus expressing both measles virus glycoproteins. Virology. 1994;200:11–20. doi: 10.1006/viro.1994.1157. [DOI] [PubMed] [Google Scholar]
- 21.Steinmann E, Whitfield T, Kallis S, Dwek RA, Zitzmann N, Pietschmann T, Bartenschlager R. Antiviral effects of amantadine and iminosugar derivatives against hepatitis C virus. Hepatology. 2007;46:330–338. doi: 10.1002/hep.21686. [DOI] [PubMed] [Google Scholar]
- 22.Taylor DL, Sunkara PS, Liu PS, Kang MS, Bowlin TL, Tyms AS. 6–0-butanoylcastanospermine (MDL 28,574) inhibits glycoprotein processing and the growth of HIVs. AIDS. 1991;5:693–698. doi: 10.1097/00002030-199106000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Wu SF, Lee CJ, Liao CL, Dwek RA, Zitzmann N, Lin YL. Antiviral effects of an iminosugar derivative on flavivirus infections. J Virol. 2002;76:3596–3604. doi: 10.1128/JVI.76.8.3596-3604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zitzmann N, Mehta AS, Carrouée S, Butters TD, Platt FM, McCauley J, Blumberg BS, Dwek RA, Block TM. Imino sugars inhibit the formation and secretion of bovine viral diarrhea virus, a pestivirus model of hepatitis C virus: implications for the development of broad spectrum anti-hepatitis virus agents. Proc Natl Acad Sci U S A. 1999;96:11878–11882. doi: 10.1073/pnas.96.21.11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rawlings AJ, Lomas H, Pilling AW, Lee MJ, Alonzi DS, Rountree JS, Jenkinson SF, Fleet GW, Dwek RA, Jones JH, Butters TD. Synthesis and biological characterisation of novel N-alkyl-deoxynojirimycin alpha-glucosidase inhibitors. Chembiochem. 2009;17:1101–1105. doi: 10.1002/cbic.200900025. [DOI] [PubMed] [Google Scholar]
- 26.Mehta A, Carrouée S, Conyers B, Jordan R, Butters T, Dwek RA, Block TM. Inhibition of hepatitis B virus DNA replication by imino sugars without the inhibition of the DNA polymerase: therapeutic implications. Hepatology. 2001;33:1488–1495. doi: 10.1053/jhep.2001.25103. [DOI] [PubMed] [Google Scholar]
- 27.Tan A, van den Broek L, van Boeckel S, Ploegh H, Bolscher J. Chemical modification of the glucosidase inhibitor 1-deoxynojirimycin. Structure-activity relationships. J Biol Chem. 1991;266:14504–14510. [PubMed] [Google Scholar]
- 28.Mehta A, Ouzounov S, Jordan R, Simsek E, Lu X, Moriarty RM, Jacob G, Dwek RA, Block TM. Imino sugars that are less toxic but more potent as antivirals, in vitro, compared with N-n-nonyl DNJ. Antivir Chem Chemother. 2002;13:299–304. doi: 10.1177/095632020201300505. [DOI] [PubMed] [Google Scholar]
- 29.Durantel D, Branza-Nichita N, Carrouée-Durantel S, Butters TD, Dwek RA, Zitzmann N. Study of the mechanism of antiviral action of iminosugar derivatives against bovine viral diarrhea virus. J Virol. 2001;75:8987–8998. doi: 10.1128/JVI.75.19.8987-8998.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naccache P, Sha’afi RI. Patterns of nonelectrolyte permeability in human red blood cell membrane. J Gen Physiol. 1973;62:714–736. doi: 10.1085/jgp.62.6.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brumshtein B, Greenblatt HM, Butters TD, Shaaltiel Y, Aviezer D, Silman I, Futerman AH, Sussman JL. Crystal structures of complexes of N-butyl- and N-nonyl-deoxynojirimycin bound to acid beta-glucosidase: insights into the mechanism of chemical chaperone action in Gaucher disease. J Biol Chem. 2007;282:29052–29058. doi: 10.1074/jbc.M705005200. [DOI] [PubMed] [Google Scholar]
- 32.Di LK, EH Application of pharmaceutical profiling assays for optimization of drug-like properties. Curr Opin Drug Discov Devel. 2005;8:495–504. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.