

Summary
Cancer cells typically exhibit aberrant DNA methylation patterns that can drive malignant transformation. Whether cancer cells are dependent on these abnormal epigenetic modifications remains elusive. We used experimental and bioinformatics approaches to unveil genomic regions that require DNA methylation for survival of cancer cells. First, we surveyed the residual DNA methylation profiles in cancer cells with highly impaired DNA methyltransferases. Then, we clustered these profiles according to their DNA methylation status in primary normal and tumor tissues. Finally, we used gene expression meta-analysis to identify regions that are dependent on DNA methylation-mediated gene silencing. We further showed experimentally that these genes must be silenced by DNA methylation for cancer cell survival, suggesting these are key epigenetic events associated with tumorigenesis.
INTRODUCTION
During tumorigenesis cancer cells acquire, through a multistep process, a new set of properties that allow them to overcome physiological homeostasis. These properties include unlimited proliferation potential, self-sufficiency in growth signals, resistance to anti-proliferative and apoptotic signals and immune system evasion, among others (Hanahan and Weinberg, 2000; Hanahan and Weinberg, 2011). These alterations, on the other hand, contribute to a process known as the stress phenotype of cancer (Luo et al., 2009), which includes DNA damage/replication stress, proteotoxic stress, mitotic stress, metabolic stress and oxidative stress.
To survive the tumorigenic process, a cancer cell undergoes several modifications to its genomic circuitry, such as activating mutations in oncogenes and aberrant activation of non-oncogenic pathways. These adaptations lead to oncogene addiction (Weinstein, 2002) and non-oncogene addiction (Solimini et al., 2007), respectively. Because of this aberrant circuitry, cancer cells become hypersensitive to the effects of classic tumor suppressor genes (Luo et al., 2009; Weinstein, 2002) and, potentially, to genes that can inhibit the non-oncogenic signaling pathways that cancer cells rely on to survive.
Changes in the cancer cell transcriptome can be driven by genetic and epigenetic alterations (Baylin and Ohm, 2006; Jones and Baylin, 2007). DNA methylation is an epigenetic process that can heritably change gene expression without altering the DNA sequence. In normal somatic cells, most DNA methylation occurs at CpG dinucleotides within CpG poor sequences, while CpG rich sequences, also known as CpG islands, are usually unmethylated (Sharma et al., 2010). DNA methylation is a vital mechanism of epigenetic gene silencing, playing key roles in X-chromosome inactivation, genomic imprinting, embryonic development, silencing of repetitive elements and germ cell specific genes, differentiation and maintenance of pluripotency (De Carvalho et al., 2010; Meissner, 2010; Robertson, 2005). Besides these physiological roles, deregulated DNA methylation can also be a major driver of pathological conditions, including neurological and autoimmune diseases, as well as cancer (Kelly et al., 2010; Portela and Esteller, 2010; Taberlay and Jones, 2011). During tumorigenesis, global DNA methylation patterns change, resulting in hypomethylation of non-CpG islands and hypermethylation of CpG islands (Sharma et al., 2010). DNA hypermethylation has been shown to result in abnormal silencing of several tumor suppressor genes in most types of cancer (Jones and Baylin, 2002; Jones and Baylin, 2007).
Recently, several efforts to examine the cancer methylome, utilizing genome-wide techniques, have revealed that a large number of genes exhibit aberrant DNA methylation profiles in cancer (Figueroa et al., 2010; Irizarry et al., 2009). These changes can be used to stratify sub-types of cancers (Figueroa et al., 2010; Noushmehr et al., 2010) and to predict cancer outcomes (Portela and Esteller, 2010), among other uses. Distinguishing which genes play key “driver” roles via DNA methylation-mediated gene silencing in cancer initiation, progression and maintenance and those genes that are only “passengers” in the tumorigenic process would be extremely useful in developing more targeted epigenetic therapies (Kelly et al., 2010). However, making this distinction has proven extremely difficult due to the large number of differentially DNA methylated genes in human cancers (Kalari and Pfeifer, 2010).
We, and others, have suggested that cancer cells may become addicted to an aberrant epigenetic landscape, especially with respect to DNA methylation (Baylin and Ohm, 2006; Kelly et al., 2010). However, as of yet, there is no direct evidence for such an addiction. Furthermore, mining the thousands of genomic regions that are de-novo DNA methylated in cancer and identifying those required for cancer cell survival has proven extremely challenging (Kalari and Pfeifer, 2010). Here, we describe an approach to identify driver epigenetic events associated with cancer cell survival. Our Findings pave the way for new generations of epigenetic therapies, which target the genes cancer cells rely on being silenced by DNA methylation in order to survive.
RESULTS
Identification of the minimum DNA methylation profile required for cancer cell survival
We hypothesized that cancer cells depend on DNA methylation of a few key regions for survival and that these regions would preferentially maintain methylation when artificially reducing global DNA methylation. To test this hypothesis, we profiled HCT116 colon cancer cells and HCT116 cells with a genetic disruption of DNMT3B and DNMT1 (DKO) (Rhee et al., 2002). This genetic disruption led to a complete knockout of DNMT3B and a truncated DNMT1 transcript, expressed at very low levels (Egger et al., 2006; Rhee et al., 2002; Spada et al., 2007). For this study, we used two DKO subclones, DKO8 and DKO1, which retain approximately 45% and 5% of the HCT116 wild type global DNA methylation levels, respectively (Rhee et al., 2002; Sharma et al., 2011). It is important to note that a further reduction of DNMT1 levels, by RNAi, in cells with a genetic disruption of DNMT1 results in demethylation and a massive reduction of cell viability and immediate induction of cell death (Spada et al., 2007), suggesting that DNA methylation is required for cancer cell survival.
We profiled promoter DNA methylation of HCT116, DKO8 and DKO1 cell lines, using the Illumina Infinium platform (HumanMethylation27) and observed a reduction in global DNA methylation levels in DKO8 cells compared to HCT116 wild type cells and an even greater reduction in DKO1 cells (Figure 1A), consistent with previous data (Rhee et al., 2002; Sharma et al., 2011). Surprisingly, we found a collection of 566 CpG sites, spanning 490 genes that despite the strong impairment in DNA methyltransferase activity, still retained a high level of DNA methylation in DKO1 cells, with a beta value higher than 0.6 (see Table S1 for gene/probe list). These regions were also highly methylated in HCT116 and DKO8 cells, and none showed a difference in their beta values greater than 0.2 among the three cell lines.
Figure 1. Clustering of DNMT deficient cells identifies three classes of putative driver genes marked by DNA methylation. See also Figure S1 and Table S1.
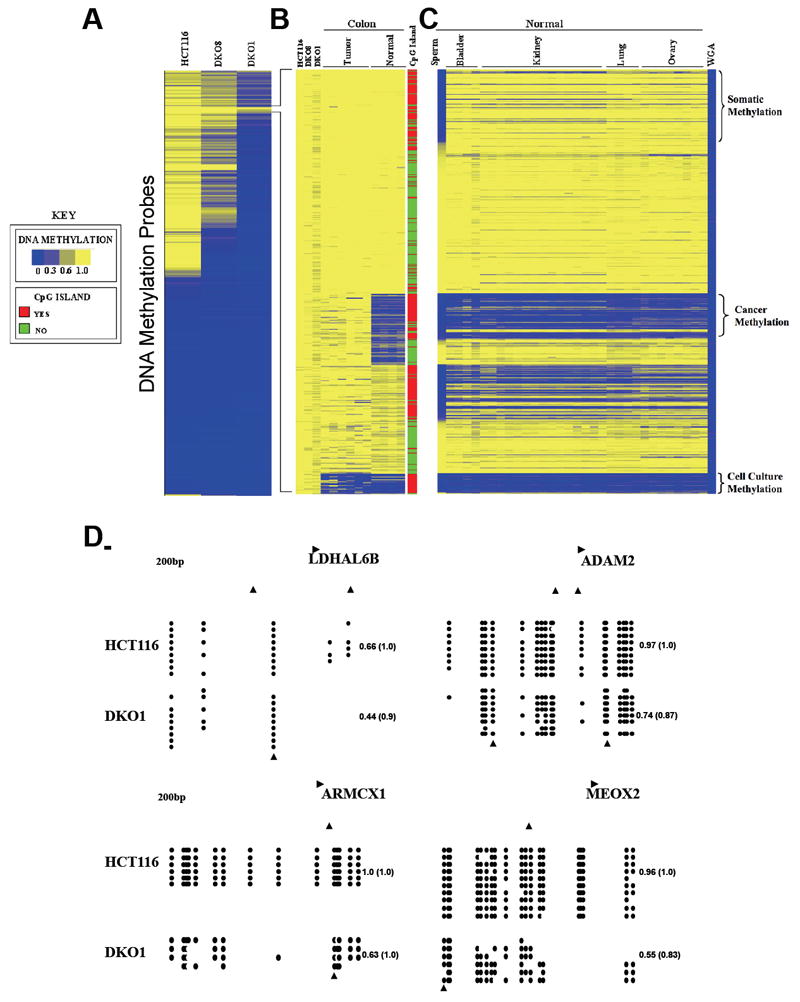
(A) One-dimensional hierarchical clustering using Euclidean distance and average linkage was performed with the ~24,000 Infinium DNA methylation probes located outside of repeats or known SNPs in HCT116 wild type, DKO8 and DKO1 cell lines. Each row represents a probe; each column represents a sample. The beta value (level of DNA methylation) for each probe is represented with a color scale as shown in the key.
(B) K-means (K=4) clustering of the 566 Infinium DNA methylation probes that maintain DNA methylation in DKO1 sample (a beta value of at least 0.6 and a difference between HCT116 and DKO1 smaller than 0.2) in (A) for 10 TCGA samples (n=4 normal colon and n=6 primary colon adenocarcinoma).
(C) Heatmap of 566 infinium DNA methylation probes in 32 normal tissues retaining the probe order from (B). Primary normal bladder (n=4), sperm (n=1), and primary normal TCGA kidney (n=15), lung (n=4) and ovary (n=8). Whole Genome Amplified DNA (WGA) was used as a negative control for DNA methylation.
(D) Bisulfite sequencing validation of Infinium DNA methylation data from two regions (LDHAL6B and ADAM2) from the somatic-specific DNA methylation cluster and two regions (ARMCX1 and MEOX2) from the cancer-specific DNA methylation cluster. Arrow indicates the position of the Infinium probe. Empty and filled circles denote unmethylated and methylated CpG sites, respectively. Each horizontal row represents one sequenced DNA clone. The number on the right represents the mean DNA methylation score of each region and the number in the parentheses represents the mean DNA methylation score of the specific Infinium CpG site.
Next, we sought to identify whether there was a cancer-specific DNA methylation profile at these regions that maintained DNA methylation even in DKO1 cells, which would potentially include important putative targets for epigenetic therapy. To accomplish this, we first compared the DNA methylation levels of the 566 CpG sites that retained DNA methylation in DKO1 cells to the DNA methylation profile of six primary colon adenocarcinoma tissue samples and four normal colon tissue samples obtained from The Cancer Genome Atlas (TCGA) database. Using k-means clustering, we identified 92 CpG sites, spanning 77 genes that were unmethylated in normal colon and became hypermethylated in colon adenocarcinoma (Figure 1B and Table S1) consistent with a cancer specific methylation profile.
We further compared these data to DNA methylation data of several normal tissues including sperm, bladder, kidney, lung and ovary, which allowed us to identify clusters of gene regions highly enriched for somatic tissue specific DNA methylation. Such genes were methylated in the somatic tissues analyzed and unmethylated in germ cells. This somatic tissue specific cluster comprised 99 CpG sites, spanning 83 genes (Figure 1C and Table S1). Furthermore, we also identified genes that exhibit cell culture-specific DNA methylation, such that these regions are methylated in all cell lines analyzed but unmethylated in primary tissues (Figure 1C and Table S1). This cell culture specific cluster comprised 29 CpG sites, spanning 25 genes. We focused only on these three groups because of their differential DNA methylation profiles. We speculate that the remaining 346 CpG sites might be regions that are more prone to methylation, remaining a good target for residual DNA methylation activity without functional relevance or, alternatively, may have a tissue-specific expression profile, being unmethylated only in specific cell types that were not surveyed in this study. Whole Genome Amplified (WGA) DNA served as a negative control (Figure 1C) to confirm that the regions identified as being methylated in DKO1 cells were not false-positives due to technical problems with the specific Infinium probes.
The distribution of probes, relative to transcription start sites (TSSs), in the cancer specific and somatic tissue specific clusters were found to be very similar to the distribution of the array itself (Figure S1A), while the distribution in the cell culture-specific cluster tended to be slightly more concentrated at the TSS (Figure S1A). However, it should be noted that there was no association between distance to TSS and methylation cluster (somatic, cancer, cell line) as assessed by one-way ANOVA (p>0.05).
We selected genomic regions from each cluster to validate the Infinium-based DNA methylation data using bisulfite sequencing. All of the sequences analyzed showed high levels of DNA methylation in HCT116 wild type and DKO1 cells (Figure 1D), with the CpG site surveyed by the Infinium platform presenting a maximum difference between their beta values of 0.17 in DKO1 when compared to HCT116 wild type. These results demonstrate that even though DKO cells are globally DNA hypomethylated (Rhee et al., 2002; Sharma et al., 2011), the residual DNA methylation is focal and site specific, supporting the hypothesis that there is a functional role for some of the retained DNA methylation.
To further demonstrate the importance of the DNA methylation that is retained in the three identified clusters, we treated DKO1 cells with 1 μM of 5-Aza-2’-deoxycytidine or PBS for 24 hours. After treatment, we allowed at least two population doublings (5 days) for demethylation to occur and then analyzed changes in the DNA methylation profile by the Illumina Infinium array. As expected, most of the regions in the three previously identified clusters were resistant to demethylation, with only 8 regions from the somatic cluster, 5 regions from the cancer cluster and 1 region from the cell culture cluster presenting a difference in the beta value greater than 0.2 (Figure S1B). These regions we considered false-positives, and excluded from subsequent analysis.
Residual methylation in DKO1 can not be explained by an inherent susceptibility to DNA methylation
Our working hypothesis is that the artificial impairment of DNA methyltransferase machinery in DKO1 cells will induce a strong selective pressure for any remaining DNA methylation to be maintained at the regions necessary for cancer cell survival. An equally plausible hypothesis is that the residual methylation reflects an inherent tendency for some genes to remain methylated. Indeed, previous studies suggest that certain genomic regions are more prone to DNA methylation (Estecio et al., 2010; Ohm et al., 2007; Schlesinger et al., 2007; Widschwendter et al., 2007). Therefore, these regions may remain better targets for residual DNA methylation activity. To directly test this alternative hypothesis, we used two known approaches to predict whether a gene is more prone to DNA methyltransferase activity in cancer cells, one based on its chromatin structure (Ohm et al., 2007; Schlesinger et al., 2007; Widschwendter et al., 2007) and another based on its genomic architecture (Estecio et al., 2010).
Genes marked by H3K27me3 in ES cells (ESC) are known to be predisposed to DNA methylation in cancer cells (Ohm et al., 2007; Schlesinger et al., 2007; Widschwendter et al., 2007). Indeed, we found that the H3K27me3 status in ESC can accurately predict the methylation levels in the wild type HCT116 cells (Figure 2A). Then, we tested whether methylation-prone regions (H3K27me3 positive in ESC) would preferentially retain methylation in DKO1 cells, compared to HCT116 cells. If this hypothesis was correct, we should observe an enrichment of methylation-prone genes in the cohort of genes that are methylated in DKO1 cells, since they would be better targets for residual DNA methyltransferase activity. Yet, there was no such enrichment (Figure 2B). Rather, we observed a slight decrease in the proportion of methylation-prone genes that retain methylation in DKO1 cells. These data suggest that genes found to retain DNA methylation in DKO1 cells are not simply predisposed to DNA methylation in cancer cells.
Figure 2. Residual methylation in DKO1 is not caused by an inherent susceptibility to DNA methylation.
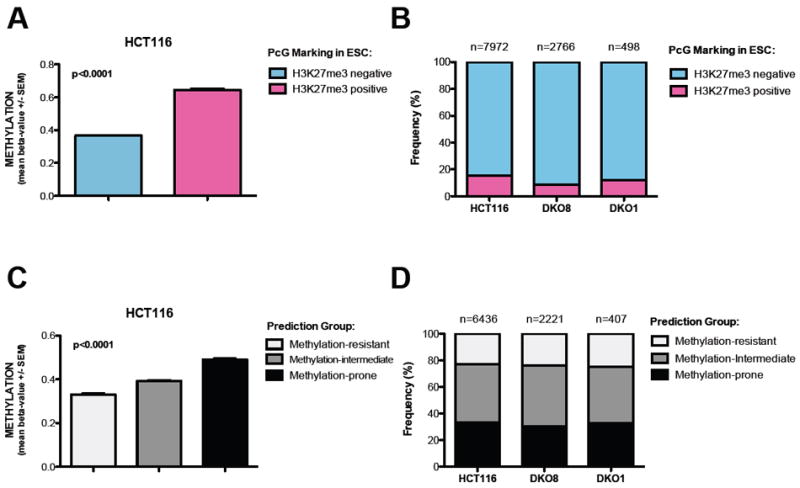
(A) Validation of H3K27me3 status in ESC as a predictive method for DNA methylation in HCT116 cells. Methylation status of ~27,000 CpG sites was determined by Infinium. T-test with Mann Whitney post-test. Data represent the mean ± SEM.
(B) Frequency of probes marked by H3K27me3 in ESC in the cohort of DNA methylated probes (Beta value >0.6) in HCT116, DKO8 and DKO1 cells.
(C) Validation of the predictive method based on genomic architecture (Estecio et al., 2010) in HCT116 cells. Methylation status of ~27,000 CpG sites was determined by Infinium. One way ANOVA with Kruskal-Wallis test. Data represent the mean ± SEM.
(D) Frequency of methylation-prone genes in the cohort of DNA methylated genes (Beta value >0.6) in HCT116, DKO8 and DKO1 cells.
To further test this hypothesis, we performed a similar analysis using a previously published algorithm to predict whether a genomic region is prone, intermediate or resistant to DNA methylation in cancer cells, based on its genomic architecture (Estecio et al., 2010). In our test, this algorithm accurately predicted methylation levels in HCT116 cells (Figure 2C). Similar to the previous analysis, if the genes that maintain methylation in DKO1 cells were simply more prone to DNA methyltransferase activity, one would expect an enrichment of methylation-prone genes in the pool of genes that retain methylation in DKO1 cells. Again, we could not find such enrichment (Figure 2D).
Taken together, these data suggest that the targets of residual DNA methylation in DKO1 cells are not dictated by an inherent predisposition to DNA methylation based on either the chromatin structure or the genomic architecture. These findings further support our original hypothesis that these loci retain methylation due to a functional selection pressure.
Validation of the findings in other types of cancer and association with gene expression
We next validated our findings in a larger test set of colon adenocarcinoma and normal colon samples. Using DNA methylation data available from TCGA we observed a significant increase in DNA methylation in the majority of the CpG sites identified with cancer specific DNA methylation in 168 primary colon adenocarcinoma samples relative to 16 normal colon samples (Figure 3A, Wilcoxon rank sum test followed by a FDR correction, p value < 0.05). We next extended our findings to determine whether this cancer-specific DNA methylation profile was unique to colon adenocarcinoma or if it could also be observed in other tumor types. Using DNA methylation data available from TCGA, we analyzed DNA methylation from 19 lung adenocarcinoma samples against four normal samples. Again, the same pattern emerged, where the identified CpG sites presented an overall significant gain of DNA methylation in the tumor samples (Figure 3C, Wilcoxon rank sum test followed by a FDR correction, p value < 0.05). Indeed, most of the genes statistically determined to be hypermethylated in lung adenocarcinoma were also hypermethylated in colon adenocarcinoma (Figure 3E). These data indicate that the CpG sites we identified as cancer-specific are frequently hypermethylated in other type of human cancer relative to the normal cell counterparts, suggesting these regions might have a more fundamental role in tumorigenesis, such as cell survival.
Figure 3. Validation of CpG sites identified with cancer-specific DNA methylation using independent datasets and association with gene repression.
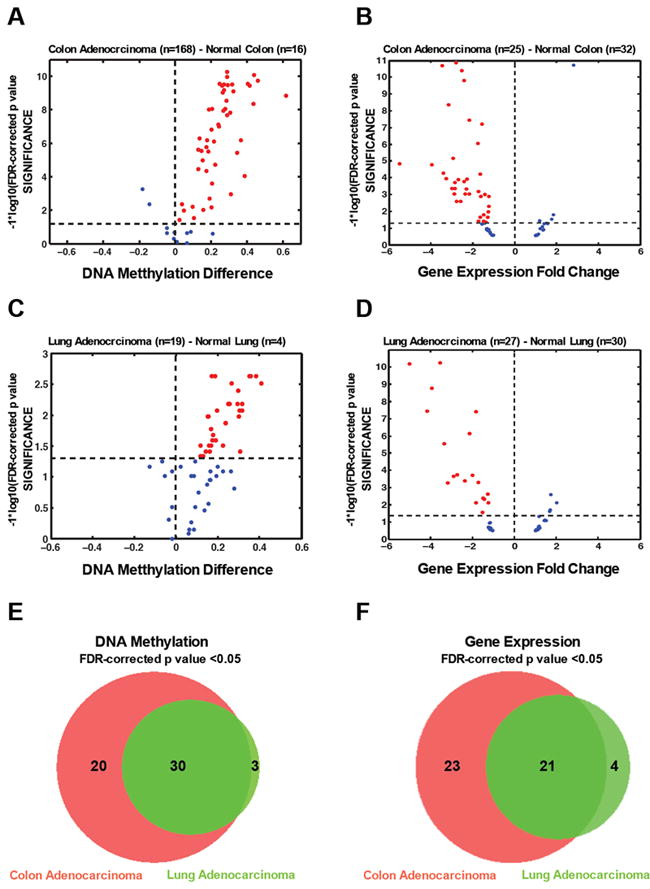
(A) Volcano plot of the CpG loci identified as cancer-specifically methylated in colon adenocarcinoma (Normal n=16, Cancer n=168) from the TCGA Data Portal. The beta value difference in DNA methylation between the tumor samples and the correspondent normal samples is plotted on the x-axis, and the p value for a FDR-corrected Wilcoxon rank sum test of differences between the tumor and correspondent normal samples (-1* log10 scale) is plotted on the y-axis. Probes that are significantly hypermethylated (FDR adjusted p<0.05) in tumors are shown in red.
(B) Volcano plot gene expression data of cancer-specific DNA methylated genes. Gene expression data was obtained from GEO (GSE 8671) from primary normal colon (n=32) and primary colon cancer (n=25). For the volcano plots, gene expression fold change between the normal tissues and the tumor tissues is plotted on the x-axis, and the p value for a FDR-corrected t-test of differences between the normal and the tumor tissues (-1* log10 scale) is plotted on the y-axis. Probes that are significantly (p<0.05) down-regulated in tumor tissues are shown in red.
(C) Volcano plot of the CpG loci identified as cancer-specifically methylated in lung adenocarcinoma (Normal n=4, Cancer n=19) from the TCGA Data Portal. The beta value difference in DNA methylation between the tumor samples and the correspondent normal samples is plotted on the x-axis, and the p value for a FDR-corrected Wilcoxon rank sum test of differences between the tumor and correspondent normal samples (-1* log10 scale) is plotted on the y-axis. Probes that are significantly hypermethylated (FDR adjusted p<0.05) in tumors are shown in red.
(D) Volcano plot gene expression data of cancer-specific DNA methylated genes. Gene expression data was obtained from GEO (GSE7670) from primary lung adenocarcinoma (n=27) and primary lung (n=30). For the volcano plots, gene expression fold change between the normal tissues and the tumor tissues is plotted on the x-axis, and the p value for a FDR-corrected t-test of differences between the normal and the tumor tissues (-1* log10 scale) is plotted on the y-axis. Probes that are significantly (p<0.05) down-regulated in tumor tissues are shown in red.
(E) Venn diagram showing the overlap between the genes statistically hypermethylated in colon adenocarcinoma (n=50, FDR adjusted p<0.05) and lung agenocarcinoma (n=33, FDR adjusted p<0.05).
(F) Venn diagram showing the overlap between the genes statistically repressed in colon adenocarcinoma (n=44, FDR adjusted p<0.05) and lung agenocarcinoma (n=25, FDR adjusted p<0.05).
Since DNA methylation of CpG islands located in promoter regions is well known to be correlated with gene silencing (Cedar and Bergman, 2009; Jones and Baylin, 2007; Portela and Esteller, 2010), we investigated the expression state of the genes identified using independent data sets. We selected two microarray datasets from the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/): colon adenocarcinoma against normal colon (GSE 8671) (Sabates-Bellver et al., 2007), and lung adenocarcinoma against normal lung (GSE7670) (Su et al., 2007).
We found an inverse correlation between DNA methylation and gene expression when we analyzed the gene expression data of 32 normal colon samples and 25 colon tumor samples. The majority of the genes subject to cancer-specific DNA methylation displayed decreased gene expression in colon cancer samples compared to normal colon (Figure 3B, t-test followed by a FDR correction, p value < 0.05). We also observed that some genes showed a similarly low level of expression in both samples, probably due to an epigenetic switch in the silencing mechanism where the gene was already silenced in the normal sample by another epigenetic mechanism and became de-novo DNA methylated in cancer (Gal-Yam et al., 2008).
Moreover, we found a similar gene expression pattern in lung adenocarcinoma, where most of the cancer-specific DNA methylation genes displayed decreased gene expression in the tumor when compared to the correspondent normal tissue (Figure 3D, t-test followed by a FDR correction, p value < 0.05). Again, most of the genes statistically repressed in lung adenocarcinoma were also repressed in colon adenocarcinoma (Figure 3F). This data further suggests the there is a functional relevance of identified DNA methylation.
Altogether, by combining gene expression with DNA methylation data we identified regions that are candidates for DNA methylation-mediated gene silencing. Moreover, the gene expression data corroborate our cluster analysis by using a different method and independent datasets to demonstrate biological differences in the gene clusters we identified.
Spontaneous loss of DNA methylation at the identified genomic regions is associated with cell death
DKO1 cells have highly impaired DNA methyltransferase machinery due to the absence of DNMT3B, very low protein levels of DNMT3A and low levels of a truncated DNMT1 (Egger et al., 2006; Sharma et al., 2011). As a consequence of the impaired DNA methyltransferase machinery, the global DNA methylation level in this cell line is very low, with most of the genes that were methylated in the parental HCT116 cells losing this methylation in DKO1 cells. Therefore, we hypothesize that DKO1 cells would be under a constant selective pressure to maintain the residual DNA methylation at key regions necessary for this cancer cell to survive.
We next investigated whether DKO1 cells exhibit a higher basal level of cell death than HCT116 wild type cells. When quantifying cell death by measuring the externalization of phosphatidylserine (PS) using Annexin-V by flow cytometry, we observed at least four times more spontaneous cell death in DKO1 than in the parental HCT116 cells (Figure 4A). This suggests that DKO1 cells are indeed under constant selective pressure, probably because during cell division, some daughter cells lose DNA methylation at key regions due to the impaired DNA methyltransferase activity in DKO1 cells (Egger et al., 2006; Spada et al., 2007) and consequently, they cannot survive.
Figure 4. Apoptosis analysis of HCT116 and DKO1 cells. See also Figure S2.
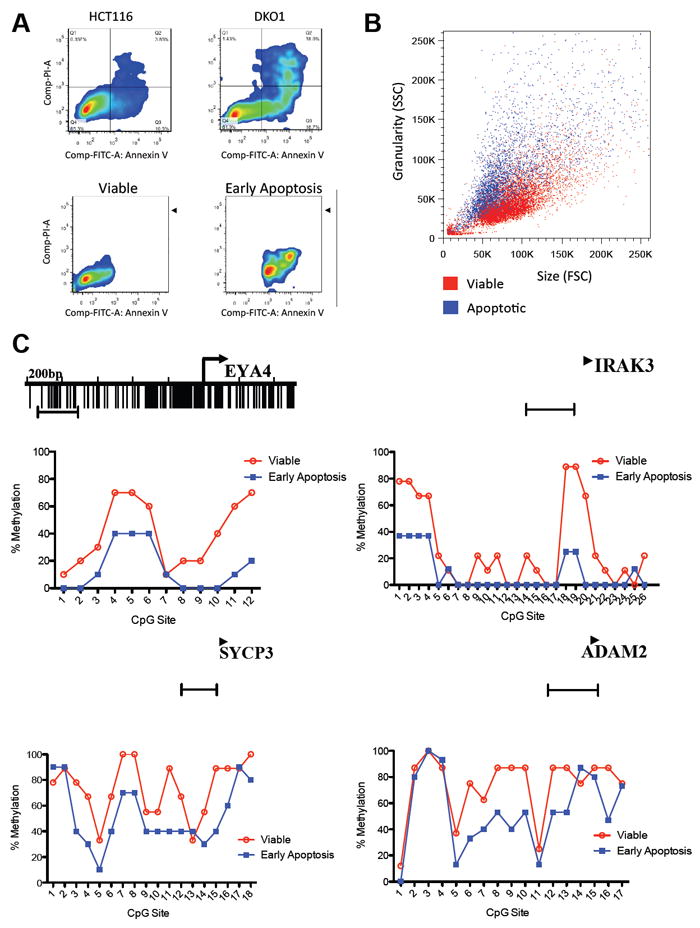
(A) HCT116 wild type and HCT116 DKO1 cells were stained with annexin V-FITC and Propidium Iodide (PI) and analyzed by FACS, showing an increased level of basal apoptotic cell death in the HCT116 DKO1 cell line compared to HCT116 wild type. HCT116 DKO1 cells were then sorted in viable (annexin V and PI negative) and early apoptosis (annexin V positive and PI negative).
(B) The morphology of viable DKO1 and early apoptosis is clearly distinct. The apoptotic cells (blue) show a characteristic phenotype of higher SSC and lower FSC than the viable cells (red).
(C) Bisulfite sequencing analysis of CpG methylation status of four regions, from cancer-specific methylated cluster (EYA4 and IRAK3) and from somatic tissue-specific DNA methylation cluster (SYCP3 and ADAM2). The mean percent methylation at each CpG site is derived from clones showed on Figure S2A. The capped line represents the region analyzed.
We took advantage of the increased rates of spontaneous cell death in DKO1 cells to further test our hypothesis that cancer cells depend on constant DNA methylation of these regions in order to survive. Using cell sorting, we first separated DKO1 cells into two populations, Annexin-V positive (early spontaneous apoptosis) and Annexin-V negative (viable cells) (Figure 4A). These two populations have distinct morphologies, with Annexin-V positive cells in the range of lower Forward Scatter (FSC) and higher Side Scatter (SSC), a characteristic feature of apoptotic cells (Darzynkiewicz et al., 1992), compared to Annexin-V negative cells (Figure 4B).
We then compared the DNA methylation levels of EYA4 and IRAK3 gene promoter regions in early apoptotic and viable cells. We have previously defined these genes as harboring cancer-specific DNA methylation and differential expression in cancer versus normal cells. Furthermore, these genes were in the top-tier for significantly hypermethylated genes, and for gene repression, in colon and lung adenocarcinoma. In addition, we also compared the DNA methylation levels of SYCP3 and ADAM2 gene promoter regions between early apoptotic and viable cells. These genes were identified as having somatic cell-specific DNA methylation and differential gene expression between somatic and germ cells (data not shown). In agreement with our hypothesis that DNA methylation-induced silencing of these regions is required for survival, early apoptotic cells showed at least a 27% reduction in DNA methylation in all four regions analyzed, with some specific CpG sites having as much as 80% reduction in DNA methylation (Figure 4C and Figure S2A). Since degradation of cellular mRNA is an early apoptosis-induced event (Del Prete et al., 2002), we could not reliably measure whether this demethylation was associated with re-expression of these genes in the dying cells. In contrast, DNA degradation is a late apoptotic event, which allowed us to study the DNA methylation status during the first steps of apoptosis.
An alternative hypothesis is that global demethylation in DKO1 cells, due to impaired DNA methyltransferase activity, leads to genomic instability and cell death. To test this hypothesis, we measured the global DNA methylation levels of the early spontaneous apoptotic and viable cells, and did not find a global reduction in DNA methylation (Figure S2B), further suggesting that demethylation of these specific genes lead to cell death. In addition, to test whether apoptosis itself could cause demethylation of these regions, we treated HCT116 cells with 0.2uM of Staurosporine (STS), a drug known to induce cell death by blocking protein kinases (Manns et al., 2011). Next, we sorted viable and STS-induced dead cells and did not observe any difference in DNA methylation of these candidate regions (Figure S2C). Altogether, this strongly suggests that demethylation of these regions is causing cell death rather that the other way around.
This data, together with our previous data showing that DKO1 cells have reduced cell viability when this low level of DNMT1, and consequently the DNA methylation level, is further reduced by RNAi (Egger et al., 2006; Spada et al., 2007), and that complete knock-out of the maintenance DNMT1 leads to massive cell death (Chen et al., 2007), demonstrate that these cells are under constant selective pressure to retain DNA methylation at these key regions that we identified here in order to survive.
Functional Validation
We further sought to further demonstrate that re-expression of genes whose DNA methylation is critical for cancer cell survival leads to increased cell death. We cloned the cDNA of six genes from the cancer cluster (IRAK3, P2RY14, CDO1, BCHE, ESX1 and ARMCX1), two from the somatic cluster (ADAM2 and SYCP3), and one from the cell line cluster (STEAP4) into the pLJM1 lentiviral vector to individually re-express these genes in HCT116 and RKO colon carcinoma cell lines (Figure S3A and S3B). We observed that expression of each of these genes decreased cell viability in both HCT116 and RKO cells (Figure 5A and 5B). We also re-expressed NOX4 as a control gene (Figure 5A and 5B). NOX4 was heavily methylated in HCT116 (beta value of 0.95) and completely demethylated in DKO1 (beta value of 0.007), suggesting that DNA methylation-mediated repression of this gene is not necessary for DKO1 survival. It is important to note that these 10 genes have a low relative expression in RKO (Figure S3B) and a very high basal DNA methylation level in this cell line (Figure S3E).
Figure 5. Functional validations. See also Figure S3.
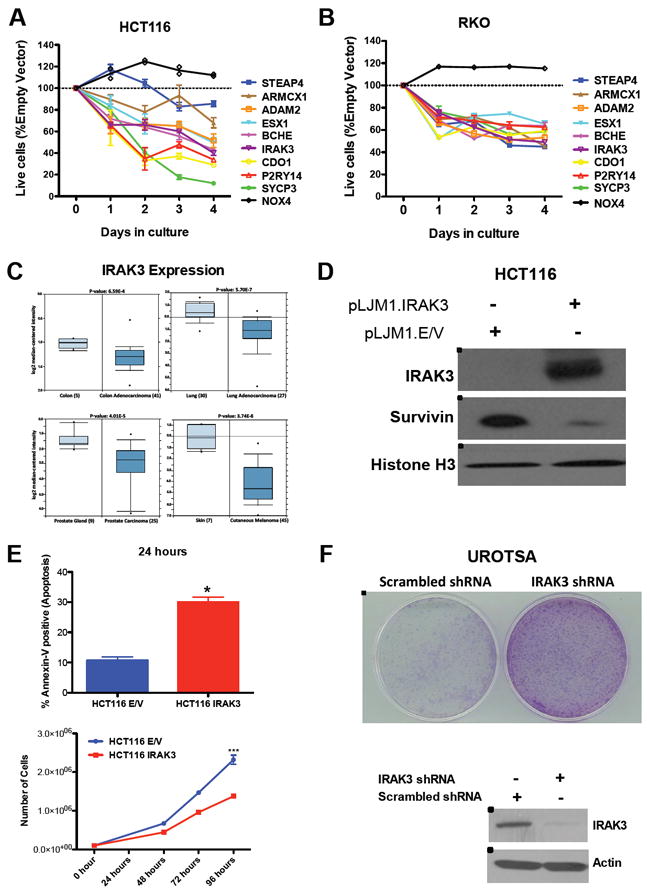
(A) Overexpression of nine candidate genes from the cancer cluster (P2RY14, IRAK3, CDO1, ESX1, ARMCX1, BCHE), somatic cluster (SYCP3 and ADAM2), cell culture cluster (STEAP4). Shown is the fraction of Empty-Vector at the indicated times, normalized to the day 0 values. NOX4 was used as a control gene, since it is hypermethylated in HCT116 cells and completely demethylated in DKO1 cells.
(B) Overexpression of the same nine candidate genes reduces viability of RKO cancer cells. Shown is the fraction of Empty-Vector at the indicated times, normalized to the day 0 values. NOX4 was used as a control gene.
(C) Meta-analysis using the oncomine (www.oncomine.org) for IRAK3 expression. Box plots showing decreased expression of IRAK3 during tumorigenesis on datasets performed in colon adenocarcinoma (Kaiser et al., 2007); lung adenocarcinoma (Su et al., 2007); prostate carcinoma (Welsh et al., 2001) and cutaneous melanoma (Talantov et al., 2005). The y-axis represents log2 median-centered intensity (normalized expression). Shaded boxes represent the interquartile range (25th–75th percentile). Whiskers represent the 10th–90th percentile. The bars denote the median.
(D) Overexpression of IRAK3 in HCT116 cells induces a reduction in the Survivin levels. Western-Blot analyzes of IRAK3 and Survivin after lentiviral infection with pLJM1 empty vector (E/V) or pLJM1-IRAK3. Histone H3 was used as a loading control.
(E) IRAK3 expression induces cell death of cancer cells. HCT116 infected with pLJM1 empty vector or pLJM1 IRAK3 were stained with annexin V-FITC and Propidium Iodide (PI) and analyzed by FACS, showing and increased level of cell death in the cell overexpressing IRAK3 (upper panel). Re-expression of IRAK3 in HCT116 wild type cells showed a reduced cell number in culture than HCT116 empty vector (lower panel). *P<0.05, ***P<0.0001. Data represent the mean ± SEM.
(F) IRAK3 knock-down induces colony formation in a non-tumorigenic cell. UROTSA infected with a shRNA against IRAK3 presented a higher colony formation activity than UROTSA infected with a scrambled shRNA. Western-Blot analysis of IRAK3 after lentiviral infection with shRNA against IRAK3 or a scrambled shRNA. Actin was used as a loading control.
To gain more detailed information of how cancer cells become dependent on DNA methylation of these genes, we investigated how the silencing of one candidate, Interleukin-1 Receptor-Associated Kinase 3 (IRAK3), affects cancer cell survival in more detail. IRAK3 has a cancer-specific DNA methylation pattern, a reduced expression in colon adenocarcinoma compared to normal colon, and a decreased DNA methylation in spontaneously dying DKO1 cells when compared to viable DKO1 cells. In addition, IRAK3 was a promising candidate because, through IRAK1 (Kobayashi et al., 2002), it indirectly inhibits three essentials pathways that cancer cells rely on to survive: STAT3, NFkB and MAPK (Figure S3F) (Ngo et al., 2011; Su et al., 2009; Turnis et al., 2010). These pathways, in turn, regulate the expression of the anti-apoptotic gene SURVIVIN (Jiang Sr et al., 2011; Zhou et al., 2009). Consistent with our hypothesis, using the Oncomine platform (www.oncomine.org), we found reduced IRAK3 expression in several types of cancer when compared to normal tissue (Figure 5C) including colon adenocarcinoma when compared to normal colon (Student’s t-test p value = 6.59E-4, top 15% under-expressed gene rank) (Kaiser et al., 2007), in lung adenocarcinoma when compared to normal lung (Student’s t-test p value = 5.70E-7, top 6% under-expressed gene rank) (Su et al., 2007), in prostate carcinoma when compared to normal prostate (Student’s t-test p value = 4.01E-5, top 3% under-expressed gene rank) (Welsh et al., 2001) and in cutaneous melanoma when compared to normal skin (Student’s t-test p value = 3.74E-8, top 3% under-expressed gene rank) (Talantov et al., 2005) (Figure 5C). The consistently reduced expression level of IRAK3 in a variety of cancers suggests that its silencing plays a role in the tumorigenic process. In addition, the down-regulation of IRAK3 was correlated with a statistically significant up-regulation of IRAK1 and SURVIVIN in the same studies (Figure S3C and S3D).
To formally test whether the decreased expression of IRAK3 was directly responsible for the increased expression of SURVIVIN and, consequently, increased cell survival, we re-expressed IRAK3 in HCT116 cells. Re-expression of IRAK3 caused a striking reduction in Survivin protein levels (Figure 5D) and caused a significant increase in cell death (Student’s t-test p=0.0219, Figure 6E) and decrease in cell viability (p<0.0001, Figure 5E), confirming that cancer cells require DNA methylation induced silencing of IRAK3, and thus become dependent on the aberrant DNA methylation. Moreover, we sought to determine whether the silencing of IRAK3 has any effect in a non-transformed cell. We performed a colony formation assay in a non-tumorigenic cell line, UROTSA, infected with IRAK3 shRNA or a scrambled shRNA (Figure 5F). We observed that IRAK3 knockdown was sufficient to induce a striking increase in colony formation (Figure 5F). Thus, demonstrating that the criteria we used was successful to identify functionally relevant genes and demonstrate that cancer cells become addicted to their epigenetic silencing.
DISCUSSION
Several genome-wide studies have revealed that a large number of promoter regions become de-novo methylated in cancer (Noushmehr et al., 2010; Portela and Esteller, 2010). However, defining the specific ‘driver’ gene regions that cancer cells depend on for survival has proven extremely difficult (Kalari and Pfeifer, 2010). In this study, we have defined the gene promoters whose DNA methylation is required for survival of somatic cancer cells in culture. This group of genes could be further subdivided into at least three sub-groups: those necessary to be methylated for the survival of 1) somatic cells, 2) cancer cells and 3) cells in culture. These sets of genes retain DNA hypermethylation even after strong depletion of DNA methyltransferase activity, suggesting that DNA methylation is the main epigenetic mechanism used to maintain silencing, as these cells do not seem able to switch to other repression mechanisms such as histone modifications alone.
Genes with germ line specific expression need to be tightly regulated in somatic tissues, as their aberrant expression could be lethal for somatic cells. For example, the gene ‘Stil’, in drosophila, is only expressed in germ cells and is necessary for germ cell survival. When ‘Stil’ is transiently expressed in somatic tissues it results in lethality (Sahut-Barnola and Pauli, 1999). The genes we identified in the somatic-specific DNA methylation group are mainly germ cell-specific genes and their demethylation, and resulting re-expression in somatic cells can trigger apoptosis, as we showed for SYCP3 and ADAM2. This suggests a primary role for DNA methylation as a mechanism for repression of testes specific genes in somatic cells (which includes cancer cells). It also lends confidence to our analysis, as it is known that several CpG island genes are normally DNA methylated in somatic tissue and unmethylated in germ cells (Shen et al., 2007).
Intriguingly, none of the genes identified in the cancer specific group, whose DNA methylation is necessary for the survival of cancer cells, are classic known Tumor Suppressor Genes (TSGs). This suggests that the genes we identified here are previously unknown tumor suppressors whose silencing is necessary for cancer cell survival. Interestingly, this group encompasses several cell signaling molecules, such as those with nucleotide receptor activity and G-protein coupled receptors (GPCRs). GPCRs have recently been described to be significantly mutated in several kinds of cancer and were found as a top category in a systematic search for tumor suppressor genes by exome and transcriptome sequencing (Kan et al., 2010; Zhao et al., 2010). Taken together, these results indicate a more significant role of GPCRs, as TSGs, in cancer than previously thought.
TSGs can be silenced in cancer cells by several epigenetic mechanisms including DNA methylation, histone modifications and nucleosome positioning (Jones and Baylin, 2007). It is also known that cancer cells depend on the silencing of TSGs and, consequently, are hypersensitive to the re-expression of these genes (Luo et al., 2009; Martins et al., 2006; Ventura et al., 2007; Xue et al., 2007). We propose here that because of this TSG hypersensitivity, and because some of these key TSGs are silenced by epigenetic mechanisms, cancer cells become addicted to this aberrant epigenetic silencing.
Indeed, we demonstrated that re-expression of the genes we identified here decreases cell viability, highlighting the need for their constant repression. Furthermore, we showed that one of these genes, IRAK3, negatively regulates expression of the anti-apoptotic gene SURVIVIN. It was recently shown that oncogenic activation of MYD88 lead to IRAK1 phosphorylation and, consequently, NFkB activation, promoting cell survival (Ngo et al., 2011). IRAK3 is a negative regulator of this signaling pathway, inhibiting IRAK1 phosphorylation (Janssens and Beyaert, 2003) thus supporting our findings and suggesting a tumor suppressor role for IRAK3. IRAK3 was also previously identified, in an RNAi-based genetic screen, as able to suppress transformation of human mammary epithelial cells (Westbrook et al., 2005), consistent with our finding that IRAK3 knock-down in non-tumorigenic cells increased colony formation and suggesting that the genes found in this cancer group are excellent targets for therapy. In addition, because these genes depend on DNA methylation-mediated gene silencing, these may be especially good targets for epigenetic therapy (Kelly et al., 2010).
It has been known for many years that de-novo DNA methylation occurs during the cell culturing process (Antequera et al., 1990; Jones et al., 1990; Wilson and Jones, 1983). Here, we identified a group of genes whose silencing by DNA methylation is required for cells to survive in culture. This group of genes was highly methylated in colon, bladder and breast cell lines and unmethylated in primary matched tissue analyzed, independent of the tumorigenic state (data not shown). Intriguingly, this group encompasses many nucleosome assembly genes, including several histone variants about which little is known. These results suggest that during the cell culturing process, extensive changes in expression of nucleosome constituents are necessary for cell survival. It also highlights the importance of careful interpretation of epigenetics results obtained from cell culture experiments.
Taken together, by identifying the minimal DNA methylation profile necessary for the survival of cancer cells and comparing this profile in several primary normal tissues and cancer types we were able to find, and experimentally validate, a group of genes whose de-novo methylation in cancer is functionally relevant for the survival of cancer cells. We also found that, despite the complex nature of tumorigenesis, cancer cells become dependent on the DNA methylation-mediated epigenetic silencing of these genes. These driver epigenetic events associated with cancer cell survival are potentially good candidates for the development of new, target specific, therapies.
Experimental Procedures
Cell lines, DNA and RNA preparations, Antibodies, and Primers
These are described in the Supplemental Experimental Procedures.
DNA methylation assay
Genomic DNA samples (1 μg each) were bisulfite converted using the Zymo EZ DNA methylation kit (Zymo Research, Orange, CA; cat # D5002) according to the manufacturer’s instructions. Bisulfite-converted DNA was eluted in an 18 μl volume, and 3 μl were removed for post-bisulfite quality control tests as described previously (Campan et al., 2009). All cell lines and clinical samples passed bisulfite conversion quality control and were subsequently processed for the Illumina Infinium DNA methylation platform (HumanMethylation27 BeadChip). A beta (β) value of 0 to 1.0 was reported for each CpG site (methylation from 0% to 100%, respectively). β values were calculated as described previously (Wolff et al., 2010). The Infinium methylation assays were performed by the USC Epigenome Center in accordance with the manufacturer’s instructions. The assay information is available at www.illumina.com. Heat-maps were generated for the beta-values.
All the DNA methylation data from primary tissue was obtained from TCGA (http://cancergenome.nih.gov/).
To analyze the DNA methylation status of individual DNA molecules, we cloned bisulfite converted PCR fragments into the pCR2.1 vector using the TOPO-TA cloning kit (Invitrogen, Carlsbad, CA). Individual colonies were screened for the insert and the region of interest was sequenced using M13 primers as previously described (Wolff et al., 2010).
H3K27me3 status in ESCs as a predictive method for DNA methylation in cancer
H3K27me3 profile in H9 Embryonic Stem Cells was obtained from previously published data (Lee et al., 2006). Next, we intersected the genomic position of the infinium probes with the H3K27me3 status in H9 ESCs to define which probes are H3K27me3 positives in ESCs and which probes are H3K27me3 negatives in ESCs. From the approximately 24000 probes used in Fig.1, we found approximately 7.7% as H3K27me3 positives in ESCs and 79.7% as H3K27me3 negative in ESCs and we could not determine the H3K27me3 status in ESCs of 12.6% of the probes.
Genomic architecture as a predictive method for DNA methylation in cancer
We used previously published data (Estecio et al., 2010) that predict the inherent susceptibility to DNA methylation in cancer based on SINE and LINE retrotransposons density in a 20-kb window around the TSS of each gene. Briefly, they calculate the log-odds ratio of SINE and LINE retrotransposons per 1-kb window and the sum of log-odds scores in the 20-kb region allowed the classification of each gene as methylation-prone, methylation-intermediate, and methylation-resistant.
Apoptosis Assay
Cellular apoptosis was measured by Annexin-V and Propidium Iodide (PI) staining using Annexin V-FITC Apoptosis Detection Kit (MBL), according to the manufacturer’s protocol. Following staining, the cells were analyzed and sorted by FACS analysis as described previously (De Carvalho et al., 2011).
Gene Expression Analysis
All gene expression data from primary tissue was obtained from GEO (GSE8671 and GSE7670). The data was median-normalized and log2 transformed.
Ectopic gene expression
These are described in the Supplemental Experimental Procedures.
Statistical Analysis
These are described in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
New approach to discriminate between ‘driver’ and ‘passenger’ epigenetic events
Cancer cells become dependent on DNA methylation of a few key target genes
Somatic cells depend on DNA methylation to silence some germline-specific genes
Cells in culture rely on aberrant DNA methylation for survival
Significance.
Epigenetic modifications are potentially reversible, making them good ’druggable’ targets. Here we show that cancer cells cannot survive in the absence of aberrant DNA methylation of specific promoter regions. This process may render cancer cells more susceptible to epigenetic therapy. We also found that physiological DNA methylation of germline-specific genes is necessary for somatic cell survival, suggesting a physiological dependence on continuous DNA methylation of these regions in somatic tissues. Moreover, by defining the promoter regions that must be methylated in order for cells to survive in culture, we found several genes that acquire de-novo DNA methylation in cell lines, highlighting the importance of careful interpretation of epigenetic results obtained from cell culture experiments.
Acknowledgments
This work was supported by R37CA082422 (P.A.J). We would like to thank Dr. Bert Vogelstein and Dr. Stephen B. Baylin for kindly providing DKO cells and also Lora Barsky from the Broad CIRM center, Flow Cytometry Core Facility for technical assistance with FACS analysis. We thank the members of the Jones laboratory and Daniel Weisenberger for helpful discussions and for careful reading of the manuscript. We thank Kimberly Siegmund for the helpful discussions about our statistical approaches. We also thank Peter Laird for useful discussions and The Cancer Genome Atlas (TCGA).
Footnotes
Accession Numbers
Infinium methylation data were deposited in NCBI’s Gene Expression Omnibus; series accession number GSE36534.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antequera F, Boyes J, Bird A. High levels of de novo methylation and altered chromatin structure at CpG islands in cell lines. Cell. 1990;6:503–514. doi: 10.1016/0092-8674(90)90015-7. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- Campan M, Weisenberger DJ, Trinh B, Laird PW. MethyLight. Methods Mol Biol. 2009;507:325–337. doi: 10.1007/978-1-59745-522-0_23. [DOI] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet. 2009;10:295–304. doi: 10.1038/nrg2540. [DOI] [PubMed] [Google Scholar]
- Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, Ueda Y, Li E. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39:391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- Darzynkiewicz Z, Bruno S, Del Bino G, Gorczyca W, Hotz MA, Lassota P, Traganos F. Features of apoptotic cells measured by flow cytometry. Cytometry. 1992;13:795–808. doi: 10.1002/cyto.990130802. [DOI] [PubMed] [Google Scholar]
- De Carvalho DD, Binato R, Pereira WO, Leroy JM, Colassanti MD, Proto-Siqueira R, Bueno-Da-Silva AE, Zago MA, Zanichelli MA, Abdelhay E, et al. BCR-ABL-mediated upregulation of PRAME is responsible for knocking down TRAIL in CML patients. Oncogene. 2011;30:223–233. doi: 10.1038/onc.2010.409. [DOI] [PubMed] [Google Scholar]
- De Carvalho DD, You JS, Jones PA. DNA methylation and cellular reprogramming. Trends Cell Biol. 2010;20:609–617. doi: 10.1016/j.tcb.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Prete MJ, Robles MS, Guao A, Martinez AC, Izquierdo M, Garcia-Sanz JA. Degradation of cellular mRNA is a general early apoptosis-induced event. Faseb J. 2002;16:2003–2005. doi: 10.1096/fj.02-0392fje. [DOI] [PubMed] [Google Scholar]
- Egger G, Jeong S, Escobar SG, Cortez CC, Li TW, Saito Y, Yoo CB, Jones PA, Liang G. Identification of DNMT1 (DNA methyltransferase 1) hypomorphs in somatic knockouts suggests an essential role for DNMT1 in cell survival. Proc Natl Acad Sci U S A. 2006;103:14080–14085. doi: 10.1073/pnas.0604602103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estecio MR, Gallegos J, Vallot C, Castoro RJ, Chung W, Maegawa S, Oki Y, Kondo Y, Jelinek J, Shen L, et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. 2010;20:1369–1382. doi: 10.1101/gr.107318.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, Christos PJ, Schifano E, Booth J, van Putten W, Skrabanek L, et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell. 2010;17:13–27. doi: 10.1016/j.ccr.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Yam EN, Egger G, Iniguez L, Holster H, Einarsson S, Zhang X, Lin JC, Liang G, Jones PA, Tanay A. Frequent switching of Polycomb repressive marks and DNA hypermethylation in the PC3 prostate cancer cell line. Proc Natl Acad Sci U S A. 2008;105:12979–12984. doi: 10.1073/pnas.0806437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell. 2003;11:293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Sr, He B, Li NP, Ma J, Gong GZ, Zhang M. The oncogenic role of NS5A of hepatitis C virus is mediated by up-regulation of survivin gene expression in the Hepatocellular cell through p53 and NF-KappaB pathways. Cell Biol Int. 2011 doi: 10.1042/CBI20110102. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA, Wolkowicz MJ, Rideout WM, 3rd, Gonzales FA, Marziasz CM, Coetzee GA, Tapscott SJ. De novo methylation of the MyoD1 CpG island during the establishment of immortal cell lines. Proc Natl Acad Sci U S A. 1990;87:6117–6121. doi: 10.1073/pnas.87.16.6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser S, Park YK, Franklin JL, Halberg RB, Yu M, Jessen WJ, Freudenberg J, Chen X, Haigis K, Jegga AG, et al. Transcriptional recapitulation and subversion of embryonic colon development by mouse colon tumor models and human colon cancer. Genome Biol. 2007;8:R131. doi: 10.1186/gb-2007-8-7-r131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalari S, Pfeifer GP. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv Genet. 2010;70:277–308. doi: 10.1016/B978-0-12-380866-0.60010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Hernandez LD, Galan JE, Janeway CA, Jr, Medzhitov R, Flavell RA. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manns J, Daubrawa M, Driessen S, Paasch F, Hoffmann N, Loffler A, Lauber K, Dieterle A, Alers S, Iftner T, et al. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: activation of caspase-9 in the absence of Apaf-1. Faseb J. 2011 doi: 10.1096/fj.10-177527. [DOI] [PubMed] [Google Scholar]
- Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–1334. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Meissner A. Epigenetic modifications in pluripotent and differentiated cells. Nat Biotechnol. 2010;28:1079–1088. doi: 10.1038/nbt.1684. [DOI] [PubMed] [Google Scholar]
- Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011 doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nature genetics. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Sabates-Bellver J, Van der Flier LG, de Palo M, Cattaneo E, Maake C, Rehrauer H, Laczko E, Kurowski MA, Bujnicki JM, Menigatti M, et al. Transcriptome profile of human colorectal adenomas. Mol Cancer Res. 2007;5:1263–1275. doi: 10.1158/1541-7786.MCR-07-0267. [DOI] [PubMed] [Google Scholar]
- Sahut-Barnola I, Pauli D. The Drosophila gene stand still encodes a germline chromatin-associated protein that controls the transcription of the ovarian tumor gene. Development. 1999;126:1917–1926. doi: 10.1242/dev.126.9.1917. [DOI] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nature genetics. 2007;39:232–236. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- Sharma S, De Carvalho DD, Jeong S, Jones PA, Liang G. Nucleosomes Containing Methylated DNA Stabilize DNA Methyltransferases 3A/3B and Ensure Faithful Epigenetic Inheritance. PLoS Genet. 2011;7:e1001286. doi: 10.1371/journal.pgen.1001286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Kondo Y, Guo Y, Zhang J, Zhang L, Ahmed S, Shu J, Chen X, Waterland RA, Issa JP. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007;3:2023–2036. doi: 10.1371/journal.pgen.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell. 2007;130:986–988. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Spada F, Haemmer A, Kuch D, Rothbauer U, Schermelleh L, Kremmer E, Carell T, Langst G, Leonhardt H. DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol. 2007;176:565–571. doi: 10.1083/jcb.200610062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Zhang T, Tyson J, Li L. The interleukin-1 receptor-associated kinase M selectively inhibits the alternative, instead of the classical NFkappaB pathway. J Innate Immun. 2009;1:164–174. doi: 10.1159/000158541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC, Lin CH, Whang-Peng J, Hsu SL, Chen CH, Huang CY. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics. 2007;8:140. doi: 10.1186/1471-2164-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlay PC, Jones PA. DNA methylation and cancer. Prog Drug Res. 2011;67:1–23. doi: 10.1007/978-3-7643-8989-5_1. [DOI] [PubMed] [Google Scholar]
- Talantov D, Mazumder A, Yu JX, Briggs T, Jiang Y, Backus J, Atkins D, Wang Y. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin Cancer Res. 2005;11:7234–7242. doi: 10.1158/1078-0432.CCR-05-0683. [DOI] [PubMed] [Google Scholar]
- Turnis ME, Song XT, Bear A, Foster AE, Gottschalk S, Brenner MK, Chen SY, Rooney CM. IRAK-M removal counteracts dendritic cell vaccine deficits in migration and longevity. J Immunol. 2010;185:4223–4232. doi: 10.4049/jimmunol.0903507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes--the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Welsh JB, Sapinoso LM, Su AI, Kern SG, Wang-Rodriguez J, Moskaluk CA, Frierson HF, Jr, Hampton GM. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- Westbrook TF, Martin ES, Schlabach MR, Leng Y, Liang AC, Feng B, Zhao JJ, Roberts TM, Mandel G, Hannon GJ, et al. A genetic screen for candidate tumor suppressors identifies REST. Cell. 2005;121:837–848. doi: 10.1016/j.cell.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW. Epigenetic stem cell signature in cancer. Nature genetics. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- Wilson VL, Jones PA. DNA methylation decreases in aging but not in immortal cells. Science. 1983;220:1055–1057. doi: 10.1126/science.6844925. [DOI] [PubMed] [Google Scholar]
- Wolff EM, Chihara Y, Pan F, Weisenberger DJ, Siegmund KD, Sugano K, Kawashima K, Laird PW, Jones PA, Liang G. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010;70:8169–8178. doi: 10.1158/0008-5472.CAN-10-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Kirkness EF, Caballero OL, Galante PA, Parmigiani RB, Edshall L, Kuan S, Ye Z, Levy S, Vasconcelos AT, et al. Systematic detection of putative tumor suppressor genes through the combined use of exome and transcriptome sequencing. Genome Biol. 2010;11:R114. doi: 10.1186/gb-2010-11-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Bi C, Janakakumara JV, Liu SC, Chng WJ, Tay KG, Poon LF, Xie Z, Palaniyandi S, Yu H, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009;113:4052–4062. doi: 10.1182/blood-2008-05-156422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.