

Abstract
Despite the large number of the outstanding researches, pathogenesis of osteonecrosis remains unknown. During the last decades the hypothesis that increased intravascular coagulation may be the pathogenetic mechanism which leads to osteonecrosis is gaining constantly support. Both primary factors of hyper-coagulability, such as resistance to activated protein C, protein C and protein S deficiency, low levels of tissue plasminogen activator, high levels of plasminogen activator inhibitor, von Willebrand factor, lipoprotein (a), and secondary factors of hypercoagulability with factors potentially activating intravascular coagulation, such as pregnancy, antiphospholipid antibodies, systemic lupus erythematosus, hemoglobinopathies and sickle cell disease, and hemato-oncologic diseases are discussed in this article. Although coagulation abnormalities in patients with hip osteonecrosis might represent increased risk factors for the development of bone necrosis by predisposing the patient to thromboembolic phenomena, further investigation is needed to indicate the definite correlation between factors leading to increased intravascular coagulation and pathogenesis of osteonecrosis.
Key words: osteonecrosis, femoral head, intravascular coagulation, hypercoagulability.
Introduction
Osteonecrosis (ON) of the femoral head is a devastating clinical entity characterized primarily by bone ischemia. Necrosis of the subchondral bone gradually results in mechanical deficiency and eventually in degeneration of the hip joint (Figure 1). First publications regarding ON of the femoral head were conducted by Koning and Twynman in 1888.1,2 The following years many investigators studied extensively the pathophysiology of the disease. Despite numerous studies that have been conducted during the years, the pathogenetic mechanism which is responsible for ON of the femoral head still remains obscure.
Figure 1.
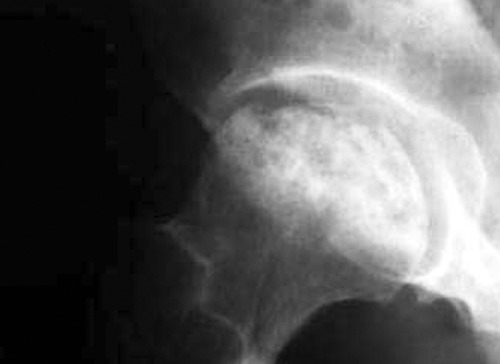
In osteonecrosis of the femoral head the cascade of events leads to collapse of the head and subsequent development of secondary hip osteoarthritis.
The first non traumatic ON of the femoral head, in a patient with coagulation disorders, was reported in 1965 by Hamilton.3 Few years later, both Boettcher et al.4 and Bonfiglio5 published new data which was in agreement with Hamilton's observation. In 1974, Jones et al.6 hypothesized that intravascular coagulation and thrombosis consist the final common mechanism which results in ON (Figure 2). Through the years, this theory has gained support by a number of studies which have indicated increased thrombophilia and hypofibrinolysis in the majority of patients with ON.
Figure 2.
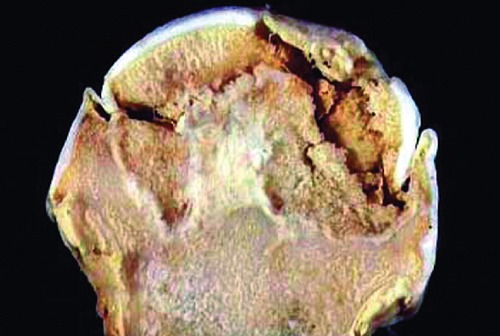
Necrosis of subchondral bone will eventually progress to collapse of the femoral head.
Primary factors of hypercoagulability
Conditions predisposing to increased intravascular coagulation may be either primary, if a subjective pathogenetic factor is not identified, or secondary if are enlisted in the frame of a systematic disorder (Figure 3).
Figure 3.
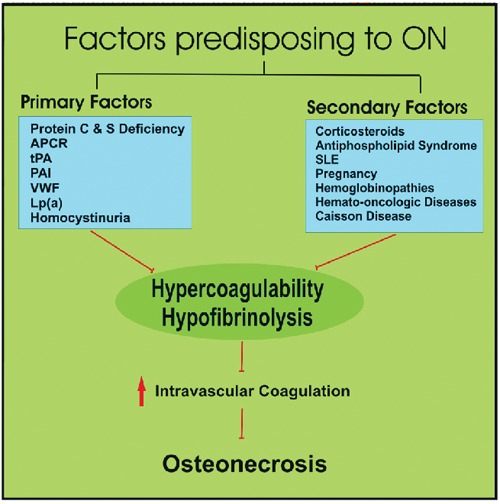
Primary and secondary factors of hypercoagulability/hypofibrinolysis that implicated in the pathogenetic mechanism of osteonecrosis.
Protein C and protein S deficiency, activated protein C resistance
Protein C is synthesized in the liver and binds to protein thrombomodulin, an endothelial cell surface protein. Being converted to an active protease by thrombin, activated protein C, in conjunction with protein S, proteolyses factors Va and VIIIa, inhibiting fibrin formation. Protein C and protein S deficiency is an inherited disorder with autosomal dominant trait.7 Heterozygous protein C deficiency has been associated with recurrent episodes of thrombophlebitis or/and pulmonary thromboembolism. Homozygous protein C deficiency causes extended thromboembolic events. Hereditary partial or complete protein S deficiency has also been associated with increased tendency for venous thrombosis.8
Activated protein C resistance (APCR) was first reported by Dahlback et al. in 1993.9 This type of disorder is caused by a mutation of the gene encoding factor V. An arginine to glutamine substitution in one of the protein's cleavage sites leads to synthesis of a structurally abnormal factor V, known as factor V Leiden. Factor V Leiden is resistant to inactivation by activated protein C.10 APCR is now recognized as the most common cause of venous thrombosis.11 In a study of 216 patients with ON of the femoral head, Korompilias et al.12 reported presence of APCR in 50% of patients, increased levels of Lipoprotein (a) [Lp(a)] in 27.3% of patients, and protein C deficiency in 4.6% of patients. In the same study, anticardiolipin antibodies (aCLA) and lupus anticoagulants (LA) were present in 26% and 2.3%, respectively. Hereditary protein C and protein S deficiency, as well as APCR may lead to hypercoagulability. The presence of these disorders in a relatively high number of patients with ON of the femoral head enforces the hypothesis that increased intravascular coagulation may be the pathogenetic mechanism that results to bone necrosis.
It has been showed that mutation of factor V Leiden is important factor in the pathogenesis of osteonecrosis of the femoral head in childhood (Legg-Calvé-Perthes disease), a clinical entity of unknown origin which is considered as idiopathic form of osteonecrosis.13 In Brazilian population, hetrozygosity for factor V Leiden was the only inherited risk factor for the development of Perthes disease.14 Clinical course of the disease is related to the factor V Leiden mutation. However, this genetic defect is not necessarily enough to cause the disease. It seems that other precipitating factors are required to provoke the entire process. Furthermore, Kechli et al.15 found no association between osteonecrosis of the femoral head in childhood and for factor V Leiden or other hypercoagulable state mutations during or after treatment for paediatric malignancy.
Low levels of tissue plasminogen activator and high levels of plas-minogen activator inhibitor
Plasminogen activator inhibitor (PAI) and tissue plasminogen activator (tPA) are two vital enzymes of the fibrinolytic system which are secreted by endothelial cells. tPA activates plasminogen, which degenerates fibrin. PAI binds to tPA, inhibiting its activity. High levels of PAI and low levels of tPA may cause a destruction of the balance between intravascular coagulation and fibrinolysis leading to hypofibrinolysis.16
The relationship between factors participating in the coagulation cascade and pathogenesis of ON has been extensively investigated. Glueck et al.17 reported disorders in coagulation factors in 87% of patients with ON of the femoral head (12 patients with idiopathic ON, 18 patients with secondary ON). Nine of 12 patients (75%) with idiopathic ON had high levels of PAI and low levels of tPA, whereas 3 of the 12 patients (25%) with idiopathic ON and 4 of the 18 patients (22%) with secondary ON had high levels of Lp(a). In another study, in 74% of patients with ON one or more disorders in coagulation factors were found, such as protein C deficiency, APCR, low levels of tPA, and high levels Lp(a).18 Coagulation disorders, including high levels of PAI, were also reported in a study of 59 patients with ON.19 In a recently published study of Tan et al.,20 low levels of tPA and high levels of PAI were found in 10 patients with osteonecrosis of the femoral head, indicating hypofibrinolysis as a cause for osteonecrosis.
Von Willebrand Factor
Von Willebrand Factor (VWF) is a protein, which is synthesized and stored in endothelial cells. The role of VWF is to facilitate platelet adhesion and to transport factor VIII. Under pathological high levels of VWF platelets aggregation may increase unconscionably leading to thrombotic events.21,22
Zalavras et al.8 studied 68 patients with non-traumatic ON of the femoral head. In their series there were included 17 patients with idiopathic ON and 51 patients with secondary ON. Coagulation factors disorders were present in 58.9% of patients with idiopathic ON and in 62.7% patients with secondary ON. Interestingly, this was the first reported association of high levels of VWF with ON, as 23.5% of idiopathic and 23.5% of secondary ON patients had high levels of VWF.
In another study of patients with osteonecrosis or Legg-Calvé-Perthes disease haemostatic disturbances were retrospectively analyzed.13 Among other findings, in Legg-Calve'-Perthes patients a lower plasmatic VWF antigen level was detected compared with healthy controls.
Lipoprotein a
Lp(a) is a low-density lipoprotein, which consists of a lipid core, an apolipoprotein b [apo(b)], and an apolipoprotein a [apo(a)].23 Apo(a) has a strong structural resemblance with plasminogen. Due to its extensive homology with plasminogen, apo(a) binds to the lysine sites available for plasminogen at the surface of fibrin. By this mechanism apo(a) inhibits the conjunction of plasminogen and tPA at the surface of fibrin disordering the fibrinolytic system. In a recent study, Hirata et al.24 revealed a significant relationship between ON and the low molecular weight form of apo(a).
Published data suggest that high levels of Lp(a) may cause hypofibrinolysis, thrombogenesis, and atherogenesis,25 which may play a distinct role in the pathogenesis of bone necrosis. High levels of Lp(a) may cause thrombotic venous occlusion, which in turns may result in intramedullary hypertension, reduced arterial perfusion, and finally ON.17 On the other hand, according to Mont and colleagues,26 difference between elevated Lp(a) levels in 31 lupus patients that developed ON and 72 healthy individuals was statistically insignificant. This is in agreement with Jone's et al.27 observations who studied the presence of thrombophilia and decreased fibrinolysis in 45 patients with ON. When these patients compared to 40 control patients, the authors showed statistically insignificant difference in serum levels of Lp(a) between the 2 groups. However, Hirata et al.24 did not find relationship between plasma levels of Lp(a) and the development of ON. The same authors showed that apo(a) phenotype is a risk factor of corticosteroid-induced ON of the femoral head after renal transplant.24 Thus, preoperative analysis of apo(a) phenotype may be an important predictor for the development of ON after treatment, while Lp(a) may be used as an useful marker for the disease.28
Homocystinuria
Homocystinuria is the result of an enzymatic deficiency in methionine metabolism. Enzymatic defects can be either primary or a result of defects in the cytosolic metabolism of cobolamin.29,30 Patients homozygous for homocystinuria may develop life-threatening thromboembolic events before the age of 30. Thrombotic episodes associate with homocystinuria include deep-vein thrombosis, pulmonary embolism, and arterial thrombosis.29,31–33 Osteonecrosis can also develop as a result of homocystinuria associated hypercoagulability.
Thrombosis, venous, arterial, or both, is not always present in patients homozygous for homocystinuria. It has been shown that one reason for the variability of thrombosis in patients with homocystinuria is the presence or absence of factor V Leiden.34 Therefore, it can be suggested that in patients with homocystinuria additional contributing factors may be needed for osteonecrosis to occur.
Secondary factors of hypercoagulability
Antiphospholipid antibodies - antiphospholipid syndrome
LA and aCLA antibodies, as well as antibodies causing false positive tests for syphilis, belong to a family known as antiphospholipid antibodies. They are active against negatively charged phospholipids and have been associated with an increased tendency for thrombotic phenomena.35,36 Platelet membranes, endothelial cells, and proteins of the coagulation cascade, such as prothrombin, protein C, and protein S are considered to be possible targets of antiphospholipid antibodies.35,37
The morbid condition, which is characterized by arterial and venous thrombosis or morbidity during pregnancy in the presence of antiphospholipid antibodies is defined as antiphospholipid syndrome.38 Antiphospholipid syndrome is classified as primary, when occurs in the absence of any other underlying disease and secondary, when occurs in the presence of systemic lupus erythematosus (SLE) or other lupuslike disease.39 Antiphospholipid antibodies are active against vessels of all sizes, such as the aortic arch, coronary, retinal and peripheral arteries, pulmonary, cerebral and small skin vessels, leading to thromboembolic events.39,40 In such cases, ON may be explained by bone ischemia due to arterial or venous microthrombosis caused by the presence of antiphospholipid antibodies.
The association of aCLA with osteonecrosis in SLE patients have been demonstrated in several studies.26,41,42 Mont et al.26 using logistic regression analysis, proved that in patients with SLE and osteonecrosis IgG aCLA levels were independently and positively associated with ON. Moreover, osteonecrosis has been reported as the first manifestation in 3 patients with antiphospholipid syndrome,43 while in others has been associated with aCLA without any features of antiphospholipid syndrome.41
Asherson et al.44 described 5 patients with secondary antiphospholipid syndrome and corticosteroid administration who presented ON. Few years later, it was the same investigator who documented 2 patients with primary antiphospholipid syndrome (APLS) who developed ON of the femoral head in the absence of previous corticosteroid therapy.43 The hypothesis that antiphospholipid syndrome is associated with the development of ON was established after numerous studies.45–47
In 1997, a study was conducted in 40 patients (25 men, 15 women) with non traumatic ON of the femoral head in order to investigate pathologic levels of anticardiolipin antibodies.41 Fifteen of 40 patients (37.5%) had low or mediate levels of aCLA. More specifically, 6 patients had elevated levels of IgM, 6 patients had elevated levels of IgA, whereas 3 patients had elevated levels of both IgM and IgA. The correlation between aCLA and ON of the femoral head was also demonstrated in patients with HIV disease.48
Corticosteroids
Corticosteroids are one of the most common and best studied predisposing factors of ON. Dosages associated with ON are >2 gr prednisone or its equivalents, within a period of 2–3 months. After corticosteroid therapy the risk period for the development of ON of the femoral head ranges from 3 to 12 months.49
The reported incidence of ON after kidney transplantation ranges from 3–16%.50,51 Corticosteroids play an important role in ON of femoral head in kidney allograft recipients. However, it has been found that factor V Leiden and prothrombin gene mutations are also of paramount importance for the development of ON in these patients.52 Moreover, lower steroid administration under ciclosporin A treatment may be the reason of the decreased incidence of ON in this group of patients.53 In a study of 287 renal transplant recipients, Inoue et al.54 found no relation between ON of the femoral head and total oral dosage, maximum oral dosage, and pulsed dosage of steroids. However, they showed that lower average daily oral dosage is an important factor for reducing ON development in renal transplant patients. Thus, reducing the oral steroid dosages after renal transplantation may reduce the occurrence of ON. On the other hand, the use of other immunosuppressive medication other than steroids in order to avoid ON is still controversial due to a higher risk of ON in patients under ciclosporin A or tacrolimus.55,56
Systemic lupus erythematosus
The development of ON in patients with SLE was first reported by Dubois in 1960.57 Numerous studies indicating the correlation between ON and SLE followed ever since.58–60 However, it has not been clarified yet if ON is a direct result of the disease or is caused by underlying factors, such as corticosteroid therapy57,58,61 or presence of antiphospholipid antibodies.45,62
The incidence of corticosteroid usage and SLE on the development of ON of the femoral head was studied in 30 rabbits.49 For this reason, lupus-like disease was caused in 20 rabbits. Ten of them were treated with high dosages of prednisolone. Ten healthy rabbits were also treated with high dosages of corticosteroids. The study indicated the presence of characteristic changes of ON in the femoral head of the rabbits, which had a lupuslike disease and were treated with high dosages of prednisolone. The most common features were the extravasation of red blood cells into the marrow, the bone marrow edema, the marrow necrosis and the thrombus formation in veins, small arteries, and arterioles of the metaphysis and diaphysis and in extraosseus vessels of the metaphysis of rabbits' femur. The immunologic reaction in patients with SLE in combination with the effects of corti-costeroids may be predisposing factors, which potentiate these thrombotic events.
Pregnancy
Osteonecrosis of the femoral head is an uncommon complication of pregnancy.63,64 The symptoms usually begin during the third trimester or immediate postpartum period. Increased intramedullary pressure due to impaired venous drainage has been proposed as a potential mechanism.65 Hypercoagulability occurs during pregnancy because of depression of fibrinolytic system, proteins C, S and antithrombin III deficiencies, high levels of PAI and hyperlipemia. These disorders can be potentiated by a bacterial or viral infection leading to thrombotic phenomena. Furthermore, complications of pregnancy, such as preeclampsia, eclampsia, and fatty liver of pregnancy may cause the entrance of placental tissue factor, amniotic fluid or embolic lipids in the maternal circulation, triggering the coagulation system and leading to hypercoagulability,66 which in turn may result in ON of the femoral head.
Hemoglobinopathies
Hemoglobinopathies are also considered as a predisposing factor of ON of the femoral head. Patients with hemoglobin SS genotype and a-thalassemia are at highest risk to develop ON.67,68 Francis69 reported increased intravascular coagulation in patients with sickle cell disease, because of increased platelet activity, thrombin synthesis and fibrin formation, and decreased activity of fibrinolytic system. Kucuk et al.70 reported the presence of antiphospholipid antibodies in patients with sickle cell disease. In these patients bilateral hip involvement is a common finding. Thus, screening of the asymptomatic hip should be performed on a regular basis. Hernigou et al.71 recommended screening of the asymptomatic hip at 6 months intervals especially in patients with large volume of ON and collapse of the contralateral hip.
In a recent study, evaluation of the physical history of asymptomatic ON of the femoral head in 121 patients with sickle cell disease and symptomatic ON to the contralateral hip was performed.71 One hundred and ten (91%) previously asymptomatic patients developed hip pain, while collapse occurred in 93 hips (77%). According to the data of this study, clinical progression of ON in patients with sickle cell disease is more frequent and more rapid than in ON due to other causes. Moreover, considering the fact that most of the asymptomatic hips collapsed three years, the authors suggested that a prophylactic surgical procedure should be considered in order to retard the progression of the disease.71
Hemato-oncologic diseases
ON is an uncommon, long-term complication of hemato-oncologic diseases, such as multiple myeloma,72,73 acute lympoblastic leukemia (ALL),74–76 and chronic myelogenous leukaemia (CML).76,77
Recently, Talamo et al.72 found high incidence of ON in patients treated for multiple myeloma. Cumulative dexomethasone dose, male sex, and younger age were found to be predisposing factors of ON among these patients. In another study ON was presented as a complication during treatment of ALL with corticosteroids.75 The overall 5-year cumulative incidence for ON was 1.8%.
Hypercoagulability is considered to play a main role in CML-induced ON.78 CML leads to leukostasis, which is characterised by occlusion of microcirculation by aggregation of leukemic cells and thrombi.79 Microcirculatory obstruction of the femoral head is probably the underlying pathology for the development of ON in these patients.
Caisson disease
Caisson disease is considered as another predisposing factor of ON of the femoral head.80 Hypercoagulability has been reported in patients with hyperbaric exposure, as fibrinogen, platelets, and lipid aggregate at the blood-bubble interface. This aggregation in combination with low antithrombin III activity and increased synthesis of fibrin leads to increased intravascular coagulation, which in turn may result in ON.
The incidence of ON in 4980 British professional divers was found to be as high as 4.2%.81 However, in that study only conventional radiographs were used. In a recent controlled study, Bolte et al.82 using magnetic resonance imaging examined 32 military divers and 28 non-divers. They found no higher prevalence of ON in military divers than in non-divers. This finding was attributed to increased medical controls, improved physical fitness, and decompression procedures followed by experience military divers.
Conclusions
Although many questions regarding the pathogenesis of ON are still unanswered, a great number of published series are in favour of the theory of increased intravascular coagulation. The systematic study of the above predisposing factors regarding the increased intravascular coagulation and the consequent ischemic damages within the femoral head may help to understand pathogenesis of ON.
References
- 1.König F. Beiträge zur ätiologie der corpora mobilis. Entstehung derselben durch osteochondritis dissecans. Dtsch Z Chir. 1888;27:99–109. [Google Scholar]
- 2.Twynham GE. A case of Caisson disease. Br Med J. 1888;1:190–1. [Google Scholar]
- 3.Hamilton HH, Bonfiglio M, Sheets RF, Connor WE. Relation of altered hemostasis to idiopathic aseptic necrosis of the femoral head. J Clin Invest. 1965;44:1058–65. [Google Scholar]
- 4.Boettcher WG, Bonfiglio M, Hamilton HH, et al. Non-traumatic necrosis of the femoral head: Part I. Relation of altered hemostasis to etiology. J Bone Joint Surg Am. 1970;52:312–21. [PubMed] [Google Scholar]
- 5.Bonfiglio M. Lambertsen CJ. Underwater Physiology V. Proc. Fifth Symposium Underwater Physiology. Bethesda: MD: FASEB; 1976. Development of bone necrosis lesions; pp. 117–132. [Google Scholar]
- 6.Jones JP, Jr, Sakovich L, Anderson CE. Beckman EL, Elliot D, Smith EM. Dysbarism-Related Osteonecrosis. Washington, DC: US Government Printing Office; 1974. Experimentally produced osteonecrosis as a result of fat embolism; pp. 117–117. [Google Scholar]
- 7.Zalavras CH, Dailiana Z, Elisaf M, et al. Potential aetiological factors concerning the development of osteonecrosis of the femoral head. Eur J Clin Invest. 2000;30:215–21. doi: 10.1046/j.1365-2362.2000.00621.x. [DOI] [PubMed] [Google Scholar]
- 8.Francis R. The protein C anticoagulant pathway and thrombosis. West J Med. 1985;143:95–6. [PMC free article] [PubMed] [Google Scholar]
- 9.Dahlback B, Carlson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA. 1993;90:1004–8. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murin S, Marelich GP, Arroliga AC, Matthay RA. Hereditary thrombophilia and venous thromboembolism. Am J Respir Crit Care Med. 1998;158:1369–73. doi: 10.1164/ajrccm.158.5.9712022. [DOI] [PubMed] [Google Scholar]
- 11.Svensson PJ, Dahlback B. Resistance to activated protein C as a basis for venous thrombosis. N Engl J Med. 1994;330:517–22. doi: 10.1056/NEJM199402243300801. [DOI] [PubMed] [Google Scholar]
- 12.Korompilias VA, Ortel LT, Urbaniak RJ. Coagulation abnormalities in patients with hip osteonecrosis. Orthop Clin N Am. 2004;35:265–71. doi: 10.1016/j.ocl.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Pósán E, Szepesi K, Gáspár L, et al. Thrombotic and fibrinolytic alterations in the aseptic necrosis of femoral head. Blood Coagul Fibrinolysis. 2003;14:243–8. doi: 10.1097/01.mbc.0000061299.28953.34. [DOI] [PubMed] [Google Scholar]
- 14.Arruda VR, Belangero WD, Ozelo MC, et al. Inherited risk factor for thrombophilia among children with Legg-Calve'-Perthes Disease. J Ped Orthop. 1999;19:84–7. [PubMed] [Google Scholar]
- 15.Kechli AM, Wilimas J, Pui CH, et al. Factor V Leiden and other hypercoagulable state mutations are not associated with osteonecrosis during or after treatment for pediatric maliganancy. J Pediatr. 1999;134:310–4. doi: 10.1016/s0022-3476(99)70455-5. [DOI] [PubMed] [Google Scholar]
- 16.Molino D, De Santo NG, Marotta R, et al. Plasma levels of plasminogen activator inhibitor type 1, factor VIII, prothrombin activation fragment 1+2, anticardiolipin, and antiprothrombin antibodies are risk factors for thrombosis in hemodialysis patients. Semin Nephrol. 2004;24:495–501. doi: 10.1016/j.semnephrol.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Glueck CJ, Freiberg R, Glueck HI, et al. Hybrofibrinolysis: A common, major cause of osteonecrosis. Am J Hematol. 1994;45:156–66. doi: 10.1002/ajh.2830450212. [DOI] [PubMed] [Google Scholar]
- 18.Glueck CJ, Freiberg R, Tracy T, et al. Thrombophilia and Hypofibrinolysis. Pathophysiologies of Osteonecrosis. Clin Orthop Relat Res. 1997;334:43–56. [PubMed] [Google Scholar]
- 19.Glueck CJ, Fontaine RN, Gruppo R, et al. The plasminogen activator inhibitor-1 gene, hypofibrinolysis, and osteonecrosis. Clin Orthop Relat Res. 1999;366:133–46. doi: 10.1097/00003086-199909000-00017. [DOI] [PubMed] [Google Scholar]
- 20.Tan X, Cai D, Wu Y, et al. Comparative analysis of serum proteomes: discovery of proteins associated with osteonecrosis of the femoral head. Translational Research. 2006;148:114–9. doi: 10.1016/j.trsl.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell R, Kumar V. Kumar V, Cotran T, Robbins K. Basic pathology. Philadelphia: Saunders; 1997. Hemodynamic disorders, thrombosis and shock; pp. 60–80. [Google Scholar]
- 22.Zwaginga JJ, Ijsseldijk MJ, Beeser-Visser N, et al. High von Willebrand factor concentration compensates a relative adhesion defect in uremic blood. Blood. 1990;75:1498–08. [PubMed] [Google Scholar]
- 23.Berg K. Scanu AM. Lp(a) San Diego (CA): Academic Press; 1990. Lp(a) lipoprotein: an overview; pp. 1–23. [Google Scholar]
- 24.Hirata T, Fujioka M, Takahashi K, et al. Low molecular weight phenotype of apolipoprotein(a) is a risk factor of corticosteroid-induced osteonecrosis of the femoral head after renal transplant. J Rheumatol. 2007;34:516–22. [PubMed] [Google Scholar]
- 25.Fortmann SP, Marcovina SM. Lipoprotein(a), a clinically elusive lipoprotein particle. Circulation. 1997;95:295–6. doi: 10.1161/01.cir.95.2.295. [DOI] [PubMed] [Google Scholar]
- 26.Mont MA, Glueck CJ, Pacheco IH, et al. Risk factors for osteonecrosis in systemic lupus erythematosus. J Rheumatol. 1997;24:654–62. [PubMed] [Google Scholar]
- 27.Jones LC, Mont MA, Le TB, et al. Procoagulants and osteonecrosis. J Rheumatol. 2003;30:783–91. [PubMed] [Google Scholar]
- 28.Mont MA, Ulrich SD, Seyler TM. Role of thrombotic and fibrinolytic alterations in the pathogenesis and treatment of osteonecrosis. J Rheumatol. 2007;34:466–8. [PubMed] [Google Scholar]
- 29.Mudd SH, Levy HL, Skovby F. Scriver CR, Beaudet AL, Sly WS, Valle DL. The metabolic and molecular bases of inherited disease. 7th ed. Vol. 1. New York: McGraw-Hill; 1995. Disorders of transsulfuration; pp. 1279–1327. [Google Scholar]
- 30.Mudd SH, Skovby F, Levy HL, et al. The natural history of homocystinuria due to cystathionine b-synthase deficiency. Am J Hum Genet. 1985;37:1–31. [PMC free article] [PubMed] [Google Scholar]
- 31.Holme E, Kjellman B, Ronge E. Betaine for treatment of homocystinuria caused by methylenetetrahydrofolate reductase deficiency. Arch Dis Child. 1989;64:1061–4. doi: 10.1136/adc.64.7.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosenblatt DS. Scriver CR, Beaudet AL, Sly WS, Valle DL. The metabolic and molecular bases of inherited disease. 7th ed. Vol. 2. New York: McGraw-Hill; 1995. Inherited disorders of folate transport and metabolism; pp. 3111–3128. [Google Scholar]
- 33.Fenton WA, Rosenberg LE. Scriver CR, Beaudet AL, Sly WS, Valle DL. The metabolic and molecular bases of inherited disease. 7th ed. Vol. 2. New York: McGraw-Hill; 1995. Inherited disorders of cobalamin transport and metabolism; pp. 3129–3149. [Google Scholar]
- 34.Mandel H, Brenner B, Berant M, et al. Coexistence of hereditary homocystinuria and factor V Leiden-effect on thrombosis. N Engl J Med. 1996;334:763–8. doi: 10.1056/NEJM199603213341204. [DOI] [PubMed] [Google Scholar]
- 35.Hughes GRV. The antiphospholipid syndrome: ten years on. Lancet. 1993;342:341–4. doi: 10.1016/0140-6736(93)91477-4. [DOI] [PubMed] [Google Scholar]
- 36.Khamashta MA, Asherson RA. Hughes syndrome: Antiphospholipid antibodies move closer to thrombosis in 1994. Br J Rheumatol. 1995;34:493–4. doi: 10.1093/rheumatology/34.6.493. [DOI] [PubMed] [Google Scholar]
- 37.Triplett DA. Antiphospholipid antibodies and thrombosis. A consequence, coincidence, or cause. Arch Pathol Lab Med. 1993;117:78–88. [PubMed] [Google Scholar]
- 38.Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthitis Rheum. 1999;42:1309–11. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 39.Lockshin MD. Answers to the antiphospho-lipid syndrome. N Engl J Med. 1995;332:1025–7. doi: 10.1056/NEJM199504133321510. [DOI] [PubMed] [Google Scholar]
- 40.Bick RL, Pegram M. Syndromes of hypercoagulability and thrombosis. Semin Thromb Hemost. 1994;20:109–32. doi: 10.1055/s-2007-1001895. [DOI] [PubMed] [Google Scholar]
- 41.Korompilias VA, Gilkeson SG, Ortel LT, et al. Anticardiolipin antibodies and osteonecrosis of the femoral head. Clin Orthop Relat Res. 1997;345:174–80. [PubMed] [Google Scholar]
- 42.Tektonidou GM, Malagari K, Vlachoyian-nopoulos GP, et al. Asymptomatic avascular necrosis in patients with primary antiphospholipid syndrome in the absence of corticosteroid use. Arthritis Rheum. 2003;48:732–6. doi: 10.1002/art.10835. [DOI] [PubMed] [Google Scholar]
- 43.Asherson RA, Khamashta MA, Ordi-Ros J, et al. The primary antiphospholipid syndrome: Major clinical and serological features. Medicine (Baltimore) 1989;68:366–74. [PubMed] [Google Scholar]
- 44.Asherson RA, Jungers P, Liote F. XVIIth International Congress of Rheumatology. Sydney, Australia: 1985. Ischaemic necrosis of bone associated with the lupus anticoagulant and antibodies to cardiolipin; pp. 282–282. [Google Scholar]
- 45.Asherson RA, Liote F, Page B, et al. Avascular necrosis of bone and antiphospolipid antibodies in systemic lupus erythematosus. J Rheumatol. 1993;20:284–8. [PubMed] [Google Scholar]
- 46.Vela P, Battle E, Salas E, Marco P. Primary antiphospholipid syndrome and osteonecrosis. Clin Exp Rheumatol. 1991;9:545–6. [PubMed] [Google Scholar]
- 47.Seleznick MJ, Silveira LH, Espinosa LR. Avascular necrosis associated with anticardiolipin antibodies. J Rheumatol. 1991;18:1416–7. [PubMed] [Google Scholar]
- 48.Miller KD, Masur H, Jones EC, et al. High prevalence of osteonecrosis of the femoral head in HIV-infected adults. Ann Intern Med. 2002;137:17–25. doi: 10.7326/0003-4819-137-1-200207020-00008. [DOI] [PubMed] [Google Scholar]
- 49.Mont M, Jones L, Hungerford D. Nontraumatic osteonecrosis of the femoral head: Ten years later. J Bone Joint Surg Am. 2006;88:1117–32. doi: 10.2106/JBJS.E.01041. [DOI] [PubMed] [Google Scholar]
- 50.Elmstedt E, Svahn T. Skeletal complications following renal transplantation. Acta Orthop Scand. 1981;52:279–86. doi: 10.3109/17453678109050104. [DOI] [PubMed] [Google Scholar]
- 51.Nielson HE, Melson F, Christensen MS. Aseptic necrosis of bone following renal transplantation. Acta Med Scand. 1977;202:27–32. doi: 10.1111/j.0954-6820.1977.tb16777.x. [DOI] [PubMed] [Google Scholar]
- 52.Ekmekci Y, Keven K, Akar N, et al. Thrombophilia and avascular necrosis of femoral head in kidney allograft recipients. Nephrol Dial Transplant. 2006;21:3555–8. doi: 10.1093/ndt/gfl400. [DOI] [PubMed] [Google Scholar]
- 53.Lausten GS, Lemser T, Jensen PK, Egfjord M. Necrosis of the femoral head after kidney transplantation. Clin Transplantation. 1998;12:572–4. [PubMed] [Google Scholar]
- 54.Inoue S, Horii M, Asano T, et al. Risk factors for nontraumatic osteonecrosis of the femoral head after renal transplantation. J Orthop Sci. 2003;8:751–6. doi: 10.1007/s00776-003-0716-9. [DOI] [PubMed] [Google Scholar]
- 55.Sakai T, Sugano N, Kakado Y, et al. Tacrolimus may be associated with lower osteonecrosis rates after renal transplantation. Clin Orthop Relat Res. 2003;415:163–70. doi: 10.1097/01.blo.0000093908.26658.df. [DOI] [PubMed] [Google Scholar]
- 56.Abbott KC, Koff J, Bohen EM, et al. Maintenance immunosupression use and the associated risk of avascular necrosis after kidney transplantation in the United States. Transplantation. 2005;79:330–6. doi: 10.1097/01.tp.0000149894.95435.7f. [DOI] [PubMed] [Google Scholar]
- 57.Dubois EL, Cozen L. Avascular necrosis in SLE. JAMA. 1960;174:966–71. doi: 10.1001/jama.1960.03030080028005. [DOI] [PubMed] [Google Scholar]
- 58.Goldie I, Tibblin G, Scheller S. Systemic lupus erythematosus and aseptic bone necrosis. Acta Med Scand. 1967;182:55–63. [PubMed] [Google Scholar]
- 59.Hurley RM, Steinberg RH, Patriquin H, Drummond KN. Avascular necrosis of the femoral head in childhood systemic lupus erythematosus. Can Med Assoc J. 1974;111:781–4. [PMC free article] [PubMed] [Google Scholar]
- 60.Nilsen KH. Systemic lupus erythematosus and avascular bone necrosis. N Z Med J. 1977;85:472–5. [PubMed] [Google Scholar]
- 61.Zizic TM, Mafcoux C, Hungerford DS, et al. Corticosteroid therapy associated with ischemic necrosis of bone in systemic lupus erythematosus. Am J Med. 1985;79:596–604. doi: 10.1016/0002-9343(85)90057-9. [DOI] [PubMed] [Google Scholar]
- 62.Abeles M, Urman JD, Rothfield NF. Aseptic necrosis of bone in systemic lupus erythematosus. Relation to corticosteroid therapy. Arch Intern Med. 1978;138:750–4. [PubMed] [Google Scholar]
- 63.Hasegawa Y, Iwase T, Iwasada S, et al. Osteonecrosis of the femoral head associated with pregnancy. Arch Orthop Trauma Surg. 1999;119:112–4. doi: 10.1007/s004020050370. [DOI] [PubMed] [Google Scholar]
- 64.Vandenbussche E, Madhar M, Nich C, et al. Bilateral osteonecrosis of the femoral head after pregnancy. Arch Orthop Trauma Surg. 2005;125:201–3. doi: 10.1007/s00402-004-0750-x. [DOI] [PubMed] [Google Scholar]
- 65.Sweet DE, Madewell JE. Resnick D, Niwayama G. Diagnosis of bone and joint disorders. Philadelphia: WB Saunders; 1988. Pathogenesis of osteonecrosis; pp. 3189–3237. [Google Scholar]
- 66.Jones JP., Jr Urbaniak JR, Jones JP. Osteonecrosis: etiology, diagnosis, and treatment. Rosemont, IL: American Academy of Orthopaedic Surgeons; 1997. Osteonecrosis and bone marrow edema syndrome: similar etiology but different pathogenesis; pp. 181–187. [Google Scholar]
- 67.Montella BJ, Nunley JA, Urbaniak JR. Osteonecrosis of the femoral head associated with pregnancy. A preliminary report. J Bone Joint Surg Am. 1999;81:790–8. doi: 10.2106/00004623-199906000-00006. [DOI] [PubMed] [Google Scholar]
- 68.Ware HE, BrookS AP, Toye R, Berney SI. Sickle cell disease and silent avascular necrosis of the hip. J Bone Joint Surg Br. 1991;73:947–9. doi: 10.1302/0301-620X.73B6.1955442. [DOI] [PubMed] [Google Scholar]
- 69.Francis RB., Jr Platelets, coagulation, and fibrinolysis in sickle cell disease: their possible role in vascular occlusion. Blood Coagul Fibrinolysis. 1991;2:341–53. doi: 10.1097/00001721-199104000-00018. [DOI] [PubMed] [Google Scholar]
- 70.Kucuk O, Gilman-Sachs A, Beaman K, et al. Antiphospholipid antibodies in sickle cell disease. Am J Hematol. 1993;42:380–3. doi: 10.1002/ajh.2830420409. [DOI] [PubMed] [Google Scholar]
- 71.Hernigou P, Habibi A, Bachir D, Galacteros F. The natural history of asymptomatic osteonecrosis of the femoral head in adults with sickle cell disease. J Bone Joint Surg Am. 2006;88:2565–72. doi: 10.2106/JBJS.E.01455. [DOI] [PubMed] [Google Scholar]
- 72.Ashraf B, Dianna W, Andrew S, et al. Osteonecrosis of the jaw in multiple myeloma patients: clinical features and risk factors. J Clin Oncol. 2006;24:945–52. doi: 10.1200/JCO.2005.04.2465. [DOI] [PubMed] [Google Scholar]
- 73.Talamo G, Angtuaco E, Walker RC, et al. Avascular necrosis of femoral and/or humeral heads in multiple myeloma: results of a prospective study of patients treated with dexamethasone-based regimens and high-dose chemotherapy. J Clin Oncol. 2005;23:5217–23. doi: 10.1200/JCO.2005.11.676. [DOI] [PubMed] [Google Scholar]
- 74.Arico M, Boccalatte MF, Silvestri D, et al. Osteonecrosis: an emerging complication of intensive chemotherapy for childhood acute lymphoblastic leukemia. Haematologica. 2003;88:747–53. [PubMed] [Google Scholar]
- 75.Burger B, Beier R, Zimmermann M, et al. Osteonecrosis: a treatment related toxicity in childhood acute lymphoblastic leukemia (ALL)-experiences from trial ALL-BFM 95. Pediatr Blood Cancer. 2005;44:220–5. doi: 10.1002/pbc.20244. [DOI] [PubMed] [Google Scholar]
- 76.Gibson J, Joshua DE, Collis D, Kronenberg H. Chronic myeloid leukaemia presenting as femoral head necrosis. Scand J Haematol. 1984;32:376–8. doi: 10.1111/j.1600-0609.1984.tb00691.x. [DOI] [PubMed] [Google Scholar]
- 77.Leone J, Vilque JP, Pignon B, et al. Avascular necrosis of the femoral head as a complication of chronic myelogenous leukaemia. Skeletal Radiol. 1996;25:696–8. doi: 10.1007/s002560050163. [DOI] [PubMed] [Google Scholar]
- 78.Joseph DE, Egesie OJ, Alao OO. Osteonecrosis complicating chronic myeloid leukaemia. Niger J Med. 2006;15:337–9. [PubMed] [Google Scholar]
- 79.Moon JY, Kim BS, Yun HR, et al. A case of avascular necrosis of the femoral head as initial presentation of chronic myelogenous leukemia. Korean J Intern Med. 2005;20:255–9. doi: 10.3904/kjim.2005.20.3.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.James CCM. Late bone lesions in Caisson disease: three cases in submarine personnel. Lancet. 1945;2:6–8. [Google Scholar]
- 81.Aseptic bone necrosis in commercial divers. A report from the decompression sickness central registry and radiological panel. Lancet. 1981:384–8. [PubMed] [Google Scholar]
- 82.Bolte H, Koch A, Tetzlaff K, et al. Detection of dysbaric osteonecrosis in military divers using magnetic resonance imaging. Eur Radiol. 2005;15:368–75. doi: 10.1007/s00330-004-2452-8. [DOI] [PubMed] [Google Scholar]