

Abstract
Lung cancer is the leading cause of cancer death worldwide, and adenocarcinoma is its most common histological subtype. Clinical and molecular evidence indicates that lung adenocarcinoma is a heterogeneous disease, which has important implications for treatment. Here we performed genome-scale DNA methylation profiling using the Illumina Infinium HumanMethylation27 platform on 59 matched lung adenocarcinoma/non-tumor lung pairs, with genome-scale verification on an independent set of tissues. We identified 766 genes showing altered DNA methylation between tumors and non-tumor lung. By integrating DNA methylation and mRNA expression data, we identified 164 hypermethylated genes showing concurrent down-regulation, and 57 hypomethylated genes showing increased expression. Integrated pathways analysis indicates that these genes are involved in cell differentiation, epithelial to mesenchymal transition, RAS and WNT signaling pathways, and cell cycle regulation, among others. Comparison of DNA methylation profiles between lung adenocarcinomas of current and never-smokers showed modest differences, identifying only LGALS4 as significantly hypermethylated and down-regulated in smokers. LGALS4, encoding a galactoside-binding protein involved in cell–cell and cell–matrix interactions, was recently shown to be a tumor suppressor in colorectal cancer. Unsupervised analysis of the DNA methylation data identified two tumor subgroups, one of which showed increased DNA methylation and was significantly associated with KRAS mutation and to a lesser extent, with smoking. Our analysis lays the groundwork for further molecular studies of lung adenocarcinoma by identifying novel epigenetically deregulated genes potentially involved in lung adenocarcinoma development/progression, and by describing an epigenetic subgroup of lung adenocarcinoma associated with characteristic molecular alterations.
Lung cancer is the leading cause of cancer-related death worldwide (Jemal et al. 2011). In many countries, adenocarcinoma has surpassed squamous carcinoma as the most common histological subtype of lung cancer, and it is also the most common histological subtype in women, Asians, and never-smokers (Toh et al. 2006). Lung adenocarcinoma is increasingly recognized as a clinically and molecularly heterogeneous disease. This is exemplified by recent reclassifications based on pathology and patient survival (Travis et al. 2011), the increasing number of clinical trials demonstrating targeted treatments that specifically benefit patients defined by molecular subtypes such as EGFR, KRAS, BRAF, and ERBB2 mutations and EML4-ALK fusions (Pao et al. 2004, 2005a,b; Pao and Girard 2011), as well as observed prognostic gene expression signature profiles (Bhattacharjee et al. 2001; Beer et al. 2002; Larsen et al. 2007). DNA methylation-based profiling has also confirmed the existence of epigenetic subtypes in several cancers (Issa 2004; Li et al. 2010; Noushmehr et al. 2010; Hinoue et al. 2012). Promoter DNA methylation, which is associated with gene silencing, can regulate gene expression in a myriad of biological and pathological processes, including lung cancer (Jones 2002; Belinsky 2004; Kerr et al. 2007; Brock et al. 2008; Risch and Plass 2008). Unlike genetic mutations, DNA methylation is an inherently reversible change, and therefore is of great interest as an active target of drug development (Esteller 2003; Rodriguez-Paredes and Esteller 2011).
While previous studies have profiled DNA methylation in lung adenocarcinoma (Shiraishi et al. 2002; Divine et al. 2005; Tsou et al. 2005, 2007; Toyooka et al. 2006; Tessema and Belinsky 2008; Christensen et al. 2009; Goto et al. 2009; Kubo et al. 2009), they have either been limited in the number of samples or genes assayed, focused on a mix of lung cancer histologies, thereby limiting the ability to identify subtypes, or lacked expression studies that allow the potential function of DNA methylation alterations to be determined. To address these issues, here we analyzed 59 lung adenocarcinoma tumors and matched adjacent non-tumor lung (NTL) tissues. Because adenocarcinoma is the most common lung cancer subtype found in never-smokers, it was important to ensure that cancers from smokers and never-smokers would both be included. Thus, we chose the cases so that approximately half of the tumors were from patients who were never-smokers. Using the Illumina Infinium HumanMethylation27 platform, we interrogated the DNA methylation status of 27,578 CpG dinucleotides spanning 14,475 genes. Focusing on genes differentially methylated in tumor vs. non-tumor lung, we integrated mRNA expression data to identify DNA methylation events with potential functional significance. We verified our findings using an independent set of 28 lung adenocarcinomas and matched adjacent NTL, as well as validated select DNA methylation data using an alternative assay, MethyLight. Lastly, we used both supervised and unsupervised analyses of the DNA methylation data to identify subgroups within the tumors.
Results
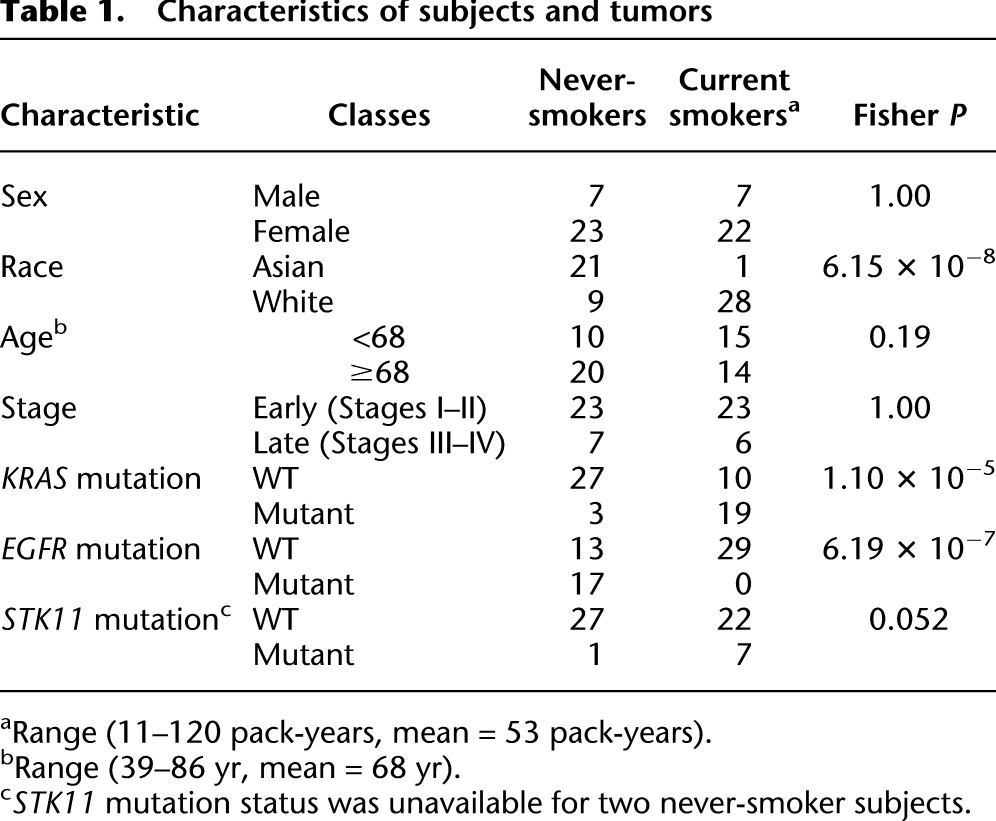
Genome-scale DNA methylation profiles were obtained for 59 lung adenocarcinomas and matched adjacent NTL tissue (Table 1). Thirty tumors were from never-smokers (defined here as less than 100 cigarettes in a lifetime), while 29 were from current smokers. Before any statistical tests were conducted, we inspected the data for the presence of substantial confounding batch effects due to the separate plates or chips (Leek et al. 2010). We did not observe any such effects (see Supplemental Fig. 1; Methods). One NTL sample was eliminated for quality-control reasons (see Methods); 117 samples were thus further analyzed (as outlined in Supplemental Fig. 2).
Table 1.
Characteristics of subjects and tumors
Identification of differentially methylated regions in lung adenocarcinoma
We first performed an exploratory two-dimensional (2D) hierarchical clustering of the top 5000 probes that varied most across the 117 samples (Fig. 1A). The DNA methylation profiles of tumors and NTL resulted in separate clusters, with the exception of one NTL sample (3022_N), indicating a substantial difference in DNA methylation profiles between the tumor and non-tumor samples. We next performed a locus-by-locus differential DNA methylation analysis of tumors vs. NTL to identify differentially methylated probes. Using our criteria of Q < 0.05 and a minimum median β-value difference of 20%, we identified 681 probes (520 genes) that were significantly hypermethylated in tumors, and 275 probes (247 genes) that were significantly hypomethylated (Fig. 1B; Supplemental Table 1). Some of our most hypermethylated loci include HOX genes, specifically HOXB4, HOXA9, and HOXA7. Seventeen different HOX genes passed our strict cutoffs, many passing with multiple probes, supporting previous observations of widespread DNA methylation of the Polycomb complex–targeted HOX genes (Shiraishi et al. 2002; Rauch et al. 2007). Some of our most hypomethylated loci include CASP8 and TNFRSF10A, both involved in TNF-receptor-mediated apoptosis (Boldin et al. 1996; Wang and El-Deiry 2003). To investigate the categories of genes exhibiting altered DNA methylation, we performed a DAVID functional annotation analysis (Huang da et al. 2009). The differentially hypermethylated set of genes was significantly enriched in GO biological processes including regulation of transcription, embryonic morphogenesis, cell–cell signaling, and cell surface receptor–linked signal transduction, while the differentially hypomethylated set was significantly enriched in processes including epidermal cell differentiation, epithelial cell differentiation, and defense response (Benjamini-Hochberg [BH] adjusted p < 0.05).
Figure 1.

Identification of DNA methylation differences between lung adenocarcinoma and NTL. (A) Two-dimensional hierarchical clustering was performed using the 5000 most variable Infinium DNA methylation probes across all samples (n = 117). Probes are in rows; samples are in columns. Note that both hypermethylation and hypomethylation in tumors compared with NTL is seen, and that hypomethylation largely occurs outside of CpG islands. (B) Volcano plot of the differential DNA methylation analysis. (X-axis) Median β-value difference (median tumor-median NTL); (y-axis) Q-values for each probe (−1 × log10 scale). (Vertical dotted lines) 20% change in β-values; (horizontal dotted line) the significance cutoff. One gene, CDH13, showed both significant hypermethylation and hypomethylation (see text and Supplemental Figs. 3 and 4). (C) Proportions of probes from genes with associated CpG islands (CGI) and probe locations, categorized as promoter (±1 kb from TSS) or nonpromoter regions. (D) Overlap of significant unique gene lists using an independent sample set (see also Supplemental Figs. 2 and 5).
For the differentially methylated probes, we also investigated whether or not they corresponded to genes with associated CpG islands and were located in promoters (Fig. 1C). Sixty-four percent of probes hypermethylated in tumors corresponded to genes with associated CpG islands, vs. only 7% of probes hypomethylated in tumors (Fisher p < 2.2 × 10−16). In addition, 77% of probes hypermethylated in tumors were located in promoter regions (defined as the region 1 kb upstream or downstream of the nearest transcription start site), vs. only 68% of hypomethylated probes (Fisher p < 0.005). Our findings support previous observations showing significant differences between the characteristics of genes that gain DNA methylation during tumorigenesis, vs. those that lose DNA methylation (Ohm et al. 2007; Schlesinger et al. 2007). One gene, CDH13—known to be repressed in lung cancer (Sato et al. 1998; Toyooka et al. 2003; Selamat et al. 2011)—was represented by nine probes on this platform and was present in both the hypermethylated and hypomethylated lists (Supplemental Figs. 3, 4). Three CDH13 probes were included in our set of the 5000 most variable probes. Of these, two were statistically significantly hypermethylated in tumors, while one was hypomethylated. The former were both located in the promoter region of the gene, and four additional probes in this region also showed hypermethylation in tumors, although they did not meet the strict criteria for significance. The hypomethylated probe was not located in or near a CpG island and resided in intron 1 of the gene, >10 kb from the transcription start site. The two additional probes in this region showed trends of hypomethylation, although they did not reach statistical significance in the discovery analysis. Thus, the CDH13 gene appears to be characterized by widespread differential and bidirectional changes in DNA methylation in lung adenocarcinoma vs. the adjacent NTL. This exemplifies the recent observation that methylation gain in the promoter can be coupled with methylation loss in the gene body (Berman et al. 2012).
To ensure that our findings were not dependent on the specific population analyzed, we performed an additional differential DNA methylation analysis on an independent sample set of 28 lung adenocarcinomas and 27 NTL (Supplemental Table 2), using the same Infinium platform. By applying identical statistical criteria, we identified 313 significantly hypermethylated genes and 85 significantly hypomethylated genes in this verification set (Supplemental Fig. 5A,B). While the smaller number of genes that passed our criteria is expected due to the smaller sample size of the verification set, we found that 95% of the hypermethylated genes and 80% of hypomethylated genes in the verification set had been identified in our discovery analysis (Fig. 1D), supporting our initial findings. To provide technical validation of our observations, we assessed the DNA methylation levels of 12 genes using the real-time PCR-based MethyLight technique on DNA from 26 tumor/NTL pairs, of which 10 tumors and 13 NTL samples were also part of the Infinium verification data set (Supplemental Fig. 5C). These 12 genes were chosen based on significance, functional relevance, and assay design compatibility. Differences in the observed absolute levels of DNA methylation might be expected due to the more stringent nature of the MethyLight assay, which requires multiple fully methylated CpG dinucleotides in the region covered by the primers and probe. In spite of this, all genes tested except one (SOCS2) that were found to be hypermethylated in the Infinium study were confirmed to be significantly hypermethylated in the verification set tumors. In addition, both hypomethylated genes tested (Supplemental Fig. 5C, right panels, FAM83A and SFN) were confirmed to be statistically significantly hypomethylated in tumors.
Identification of potentially functionally relevant DNA methylation changes in lung adenocarcinoma
To identify those DNA methylation changes with concomitant changes in gene expression, we integrated the gene expression profiles and DNA methylation profiles of the EDRN tumor and NTL tissue samples. We were able to examine gene expression levels for 709 out of 766 of the differentially methylated genes. An exploratory hierarchical clustering of the expression levels of just these 709 genes completely separated out tumors vs. NTL (Fig. 2A). Using a BH-adjusted P-value cutoff of 0.05, 349 genes were differentially expressed. Of these, 164 genes were statistically significantly hypermethylated and down-regulated (23%), while 57 genes (8%) were significantly hypomethylated and up-regulated (Fig. 2B; Supplemental Table 3), suggesting that abnormal DNA methylation might have functional consequences in approximately one-third of the genes showing differential DNA methylation between tumor and NTL. We used Ingenuity Pathways Analysis to investigate which gene networks might be affected by the aberrant DNA methylation of these 221 genes. The top two gene networks identified involved cell differentiation, on the one hand, and MAPK signaling/cell cycle control, on the other (Fig. 2C). Prominent in the first network were phosphoinositide-3-kinase (PIK3) complex members, JUN transcription factors of the AP1 family, transforming growth factor β, and WNT signaling pathway members. Epigenetic interactions with histones H3 and H4 were also seen in the first network. In the second network, genes of the MAPK and FGF families, and CCNA1 and 2 (cyclin A1 and 2) were central. Analysis of the top functional categories of deregulated genes pointed to cellular movement and development, tissue development, and cellular growth and proliferation (Fig. 2D). We then used the NextBio database (http://www.nextbio.com) (Kupershmidt et al. 2010) to identify biosets (uploaded data sets) that were significantly associated with our list of 221 genes for which DNA methylation changes were significantly inversely correlated with changes in expression. We found highly statistically significant overlaps (all p < 1.0 × 10−20) with several previously published gene expression studies comparing lung adenocarcinoma to non-tumor lung (Fig. 2E; Supplemental Fig. 6A–C; Wrage et al. 2009; Hou et al. 2010; Lu et al. 2010). One hundred and seven of the 221 genes were found in all of the top three most correlated biosets (48%), while 184 of the 221 genes (83%) were found in at least one of the top three most correlated biosets. Additionally, our 221 genes were highly correlated with two colorectal cancer DNA methylation studies (Supplemental Fig. 6D,E; Hinoue et al. 2012; YH Kim and YS Kim, unpubl.).
Figure 2.

Identification of genes showing coordinately changed DNA methylation and gene expression. (A) Two-dimensional hierarchical clustering with 1061 probes corresponding to 709 genes across all tumors (n = 58) and NTL (n = 58). Rows represent probes; columns are samples. (B) Starburst plot integrating differential DNA methylation and gene expression analyses. (X-axis) DNA methylation Q-values (−1 × log10 scale); (y-axis) BH adjusted P-values (−1 × log10 scale). Indicated are genes that are hypermethylated and down-regulated in tumors (red); hypomethylated and up-regulated in tumors (green); hypermethylated and up-regulated in tumors (blue); or hypomethylated and down-regulated in tumors (orange). (C) Top gene networks identified through integrative pathways analysis of significant DNA methylation changes associated with significant inverse gene expression changes. Indicated are genes that are hypomethylated and up-regulated in tumors (green) or hypermethylated and down-regulated in tumors (red). (Solid lines) Direct interaction; (dashed lines) indirect interaction. (D) The most significantly enriched biological process categories within genes showing significant DNA methylation changes associated with significant inverse gene expression changes. (E) Venn diagram of NextBio analysis showing the overlap of our bioset (genes showing significant DNA methylation changes in conjunction with significant inverse gene expression changes) with the three most highly correlated NextBio biosets (see also Supplemental Fig. 6).
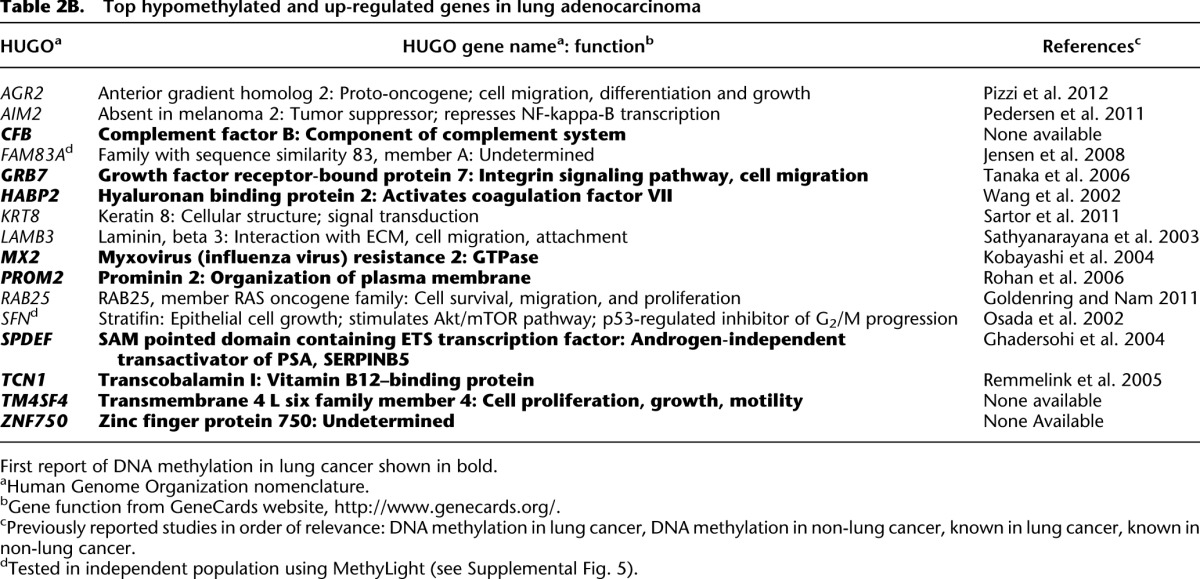
To identify the top changing genes, we applied a twofold cutoff to the average change in gene expression (Fig. 3A), finding 45 genes that were coordinately hypermethylated and down-regulated in tumors, and 16 genes that were coordinately hypomethylated and up-regulated (Tables 2A, 2B). Thus, ∼9% of the genes identified as hypermethylated in lung adenocarcinoma are also down-regulated on average more than twofold in the same tissues, a percentage similar to that found in colorectal cancer (Hinoue et al. 2012). In a more global integration analysis using all genes in common between the DNA methylation and gene expression platforms, we identified these same genes as our top candidates for showing DNA methylation-based deregulation (data not shown). For many of the genes we identified, little to nothing is known about DNA methylation-based deregulation in cancer. In addition to those genes showing inverse relationships between DNA methylation and gene expression changes, five genes were found to be hypermethylated but up-regulated in tumors, while 10 genes were found to be hypomethylated and down-regulated (Supplemental Table 4). We attempted to characterize the different groups of genes by examining whether or not they were associated with CpG islands and/or promoter regions. We found a statistically significant association between group membership and CpG island status (Fisher p < 0.001) (Fig. 3B); genes for which DNA methylation increased were significantly associated with CpG islands, regardless of the direction of the gene expression difference. We found no statistically significant difference between group membership and whether or not probes were located in the promoter region (Fisher p < 0.44) (Fig. 3B). Scatterplots illustrate the negative correlation between DNA methylation and gene expression for select genes as well as the distinct distribution of tumor and NTL sample values (Fig. 3C).
Figure 3.

Genes showing the most significant changes in DNA methylation and gene expression. (A) Three-dimensional starburst plot of 709 genes, integrating significant changes in DNA methylation (x-axis) and gene expression (z-axis), with a mean twofold or greater change in gene expression (y-axis). Colors are as in Figure 2B. (B) Presence of CpG islands and probe locations for genes exhibiting hyper- or hypomethylation and up- or down-regulation. (C) Correlation plots of DNA methylation vs. gene expression in tumors and normal tissues for select genes.
Table 2A.
Top hypermethylated and down-regulated genes in lung adenocarcinoma

Table 2B.
Top hypomethylated and up-regulated genes in lung adenocarcinoma
Examination of the clinical characteristics of the tumors (Table 1) yielded the expected statistically significant correlations of smoking with KRAS mutations and never-smoking with EGFR mutations (Sun et al. 2007). To examine the overall difference in DNA methylation between tissues from smokers and never smokers, we performed a correlation analysis, which showed very similar DNA methylation profiles in both groups, although the correlation for NTL was highest (Fig. 4A,D). We then performed a locus-by-locus differential DNA methylation analysis of smoker vs. never-smoker tissue (tumors as well as NTL) to identify differentially methylated probes. While NTL showed no significant differential DNA methylation between smokers and never-smokers, six genes were statistically significantly different between the tumor tissues of smokers and never-smokers (Fig. 4B,E). IRF8, IHH, LGALS4, IL18BP, and VTN were hypermethylated in current smoker tumors, while KLF11 was hypomethylated (Supplemental Table 5). Only LGALS4 showed a statistically significant corresponding down-regulation in gene expression in current smoker tumors (BH p < 0.0069). A scatterplot of DNA methylation vs. gene expression for LGALS4, which encodes a galactoside-binding protein involved in cell–cell and cell–matrix interactions, demonstrates the dramatic hypermethylation and down-regulation of the gene in current smoker tumors (Fig. 4C).
Figure 4.

Identification of DNA methylation differences between lung adenocarcinoma tumors and NTL with regard to smoking. (A) Correlation matrix of median β-values of tumors from current smokers vs. never-smokers, with the Spearman rho correlation given in the top left corner. (B) Volcano plot of the differential DNA methylation analysis between tumors from smokers and never-smokers. (Vertical dotted lines) 20% change in β-values; (horizontal dotted line) the significance cutoff. (C) Correlation plot of DNA methylation vs. expression for LGALS4. The Spearman rho correlation coefficient is provided on top. (D) Correlation matrix of median β-values of NTL from current smokers vs. never-smokers, with the Spearman rho correlation given in the top left corner. (E) Volcano plot of the differential DNA methylation analysis between NTL from smokers and never-smokers. (Vertical dotted lines) 20% change in β-values; (horizontal dotted line) the significance cutoff. No significant DNA methylation differences are seen between NTL from smokers and never-smokers.
One problem with the studied sample collection was a bias in the ethnic composition (Table 1); only one of the 29 smokers was Asian, whereas 21/30 never-smokers were Asian. To address this bias, we performed a multiple linear regression analysis and found that even with adjustment for race, all five genes that were significantly hypermethylated in smokers remained significant. KLF11 hypomethylation, however, did not. We examined LGALS4, the only gene showing significantly altered gene expression, more closely. When we performed a linear regression analysis with adjustment for race as well as KRAS and EGFR status, the correlation between LGALS4 and smoking remained statistically significant. In addition, a comparison of all Asian vs. all Caucasian tumors did not identify LGALS4 as significantly differentially methylated between the two populations. Lastly, in an analysis limited to only Caucasian tumors (nine never-smokers vs. 28 current-smokers), LGALS4 was found to be differentially methylated (Wilcoxon p < 0.009, median β-value difference = 0.22). These observations support an association between LGALS4 hypermethylation in lung adenocarcinoma and smoking.
The limited number of significant differences between current and never-smoker lung adenocarcinomas and their highly similar global DNA methylation profiles (Fig. 4A) prompted further investigation. We examined 30 genes that had previously been reported to show differences between smokers and never-smokers. Of these, 14 genes showed statistically significant differences in DNA methylation, but only one gene, CDKN2A, showed a median β-value difference of >20% between current smokers and never-smokers (Supplemental Table 6). Our results therefore suggest that smoking status did not greatly influence the DNA methylation profiles of the tumors in our collection.
Class discovery: Identification of DNA methylation subgroups in lung adenocarcinoma
We performed an unsupervised analysis of the entire 59-tumor set to identify any intrinsic DNA methylation-based subclasses that could then be investigated in relation to known clinical features. We used either the top 5000 most variable probes within the tumors (Fig. 5A) or the 766 differentially methylated genes (Supplemental Fig. 7A,C). Both hierarchical clustering analyses identified two distinct clusters, with only two tumors changing memberships between the two clustering approaches. Comparison of DNA methylation of the 5000 most variable probes among the tumor tissues showed considerable hypermethylation: 962 probes (753 genes) were significantly hypermethylated in Cluster 1 vs. Cluster 2, and only one gene was significantly more methylated in Cluster 2 vs. Cluster 1 (RUNX1) (Fig. 5B; Supplemental Table 7). We investigated whether this hypermethylation was tumor-specific and how it related to the probes previously identified as differentially methylated between all tumors vs. all NTL (Fig. 5C,D). Two hundred and sixty-two probes overlapped between the two groups, representing CpGs that were differentially methylated between all tumors and NTL, and that showed significant additional hypermethylation in the tumors of Cluster 1. Notably, 694 probes that had been identified as differentially methylated between all tumors and NTL did not overlap with probes differentially methylated between the two clusters. These probes comprised ones hypomethylated in the tumors as well as probes that were hypermethylated in tumors, but not additionally hypermethylated in Cluster 1 (Fig. 5D, blue dots). This suggests that the hypermethylation seen in Cluster 1 is not a global phenomenon but appears to occur in a particular group of genes. Lastly, 701 probes (red dots) were differentially methylated in Cluster 1 vs. 2 but were not identified as differentially methylated between all tumors vs. NTL. These probes were hypermethylated in tumor vs. NTL samples from Cluster 1, but not from Cluster 2 (Fig. 5E). To determine whether the differential DNA methylation of these latter probes might be of functional relevance, we examined the expression levels of these probes in Cluster 1 tumors vs. Cluster 1 NTL, identifying 13 genes that were concordantly down-regulated and hypermethylated, and three genes that were hypermethylated and up-regulated (Supplemental Table 8). Of interest was the hypermethylation and down-regulation of two genes encoding cell adhesion molecules, junctional adhesion molecule 3 (JAM3) and claudin 11 (CLDN11). Hypermethylation-based silencing of CLDN11 has been implicated in gastric cancer metastasis (Agarwal et al. 2009). Also notable was the hypermethylated and down-regulated gene GFRA1, encoding a membrane protein that interacts with the RET tyrosine kinase (Leppanen et al. 2004). Interestingly, chromosomal translocations that activate RET were recently described in lung adenocarcinoma (Ju et al. 2012; Lipson et al. 2012). The significance of GFRA1 deregulation and other Cluster 1–specific gene deregulatory events is worthy of further investigation, because it might point to patient subgroup-specific targets for epigenetic therapy.
Figure 5.

Hierarchical clustering of tumors identifies two distinct DNA-methylation based clusters. (A) Two-dimensional hierarchical clustering of the top 5000 most variant probes among 59 tumors. Rows are probes; columns are samples. Fisher P-values for different sample parameters are shown on the left; parameters are indicated at right (the listed characteristic is marked as a black tick mark, except as indicated in the key beside the heatmap). The two main clusters are marked in color at the top of the heatmap. (B) Volcano plot showing statistically significant DNA methylation alterations between the two clusters. (Vertical dotted lines) 20% change in β-values; (horizontal dotted line) the significance cutoff. (C) Overlap of probes hypermethylated in all tumors vs. NTL and probes hypermethylated in Cluster 1 vs. Cluster 2. (D) Correlation matrix of median β-values of tumors vs. NTL for the three color groups indicated in C. Note that probes that were differentially methylated in Cluster 1 but had not been identified as differentially methylated in the all tumor vs. all NTL comparison (red dots) fell near the diagonal. (E) Correlation matrix of median β-values of tumors vs. NTL for probes differentially methylated between clusters but not between all tumors vs. NTL (red dots in C). The distribution seen when these probes are examined in either Cluster 1 (green dots) or Cluster 2 (purple dots) samples. Note that within Cluster 1 samples, these probes are hypermethylated in tumors. (F) Associations of cluster membership with KRAS mutation status and (G) smoking status. (H) Volcano plot showing statistically significant gene expression differences (unrelated to DNA methylation) between the two clusters. (Vertical dotted lines) A twofold change in expression; (horizontal dotted line) the significance cutoff.
Investigating the relationship of the two clusters to their clinical and genetic properties, we found no significant differences in clinical stage, age, gender, race, STK11 or EGFR mutation status, or survival between the two clusters (all p > 0.05) (Fig. 5A; Supplemental Fig. 7B). However, we did observe statistically significant positive associations between Cluster 1 membership and KRAS mutation and smoking status (Fig. 5F,G). These results held true with both hierarchical clustering approaches. We determined that within KRAS mutants, there was no association between cluster membership and smoking (Fisher p = 0.21), whereas within current smokers, there was a significant association between KRAS mutation and Cluster 1 (Fisher p = 0.02), indicating that KRAS rather than smoking is associated with the more heavily methylated cluster. We found no significant association between the types of KRAS mutations and cluster membership (Fisher's p < 0.49), although this analysis was limited by the modest number of samples in this study. To further investigate the relationship between KRAS mutational status and cluster membership, we subdivided Cluster 1 and 2 tumors by KRAS mutational status and compared the median DNA methylation β-values among these groups. The median β-values for the KRAS mutant vs. wild-type tumors was well-correlated in both clusters (Supplemental Fig. 8A,B). In addition, Cluster 1 tumors showed higher median β-values in numerous CpGs irrespective of the KRAS mutational status (Supplemental Fig. 8C,D). These observations suggest that while KRAS mutational status is positively associated with Cluster 1 membership, it is not the driver behind Cluster 1–specific DNA methylation.
We next investigated whether the differential DNA methylation between the two clusters could be due to differential expression of proteins known to influence DNA methylation levels. We found no significant differences in expression levels of DNMT1, DNMT3A, DNMT3B, DNMT3L, TET1, TET2, and TET3 between the two clusters (Supplemental Fig. 9). However, when we examined global gene expression differences between the two clusters, we identified 36 genes that were statistically significantly differentially expressed (unrelated to DNA methylation), with seven genes meeting a twofold cutoff (Fig. 5H; Supplemental Table 9). Of these, three were cytokines (CXCL9, CXCL10, and CXCL14), while another gene, PHLDA1, encodes a regulator of IGF1-mediated apoptosis (Toyoshima et al. 2004).
Discussion
Using genome-level interrogation of DNA methylation, we identified 766 genes showing significant differential DNA methylation in cancer tissues. These genes may be useful to develop biomarkers for diagnostic or prognostic purposes. For example, the comparison of lung adenocarcinoma DNA methylation profiles with those of white blood cells and other types of cancers (e.g., through The Cancer Genome Atlas; http://cancergenome.nih.gov/), could lead to the development of improved DNA methylation-based blood biomarkers specific for lung adenocarcinoma (Esteller et al. 1999; Usadel et al. 2002). To distinguish DNA methylation events of potential functional significance (“driver events”) from those that do not biologically contribute to tumorigenesis (“passenger events”), we integrated the DNA methylation data with gene expression profiles of the same tumors. One hundred and sixty-four genes were concordantly hypermethylated and down-regulated, and 57 genes were concordantly hypomethylated and up-regulated. Ingenuity Pathways Analysis identified two top deregulated networks: one involved in epithelial-to-mesenchymal transition, the other centered on growth factor signal transduction and cell cycle control. More stringent filtering of genes requiring a minimal mean twofold change in expression yielded 45 hypermethylated and down-regulated genes, including novel methylated genes such as ABCA3, an ATP-binding cassette transporter protein with a critical role in lung development and surfactant metabolism in humans (Shulenin et al. 2004) and mice (Cheong et al. 2007). Also of interest are SOX17, a canonical WNT antagonist previously shown to be functionally hypermethylated in breast and colorectal cancers (Zhang et al. 2008; Fu et al. 2010), and TMEM204, a transmembrane protein that plays a role in cell adhesion and is hypermethylated and down-regulated in pancreatic cancer (Shimizu et al. 2011). To our knowledge, this is the first report of the epigenetic deregulation of these and numerous other genes in lung cancer.
The top hypomethylated and up-regulated gene, FAM83A, has been demonstrated previously to be specifically up-regulated in lung cancer, especially in lung adenocarcinomas (Li et al. 2005). Expression of FAM83A has been used to detect circulating cancer cells in the peripheral blood of lung cancer patients (Liu et al. 2008). FAM83A was also shown to be epigenetically regulated in an in vitro model of arsenic-mediated malignant transformation (Jensen et al. 2008). Also of interest are AGR2, KRT8, and SFN. AGR2 is a known proto-oncogene recently confirmed to be overexpressed in lung adenocarcinoma (Pizzi et al. 2012), and has been shown to promote cell proliferation and migration in several different cancers (Ramachandran et al. 2008; Vanderlaag et al. 2010; Park et al. 2011). KRT8, encoding keratin 8, was previously reported to be up-regulated in lung adenocarcinoma (Wikman et al. 2002); and SFN, encoding stratifin, has been reported to be overexpressed in early invasive lung adenocarcinoma (Shiba-Ishii et al. 2011). Investigating the link between loss of methylation and increased expression of these genes will be important, given the increasing use of epigenetic therapies in cancer treatment.
In addition to genes showing an inverse correlation between DNA methylation and expression, we also identified several genes that were coordinately hypermethylated and up-regulated, or hypomethylated and down-regulated. While these two groups of genes do not fit into the classical paradigm of DNA methylation regulation, increasing evidence from recent deep sequencing studies show that DNA methylation regulation may be more complex. The Illumina Infinium HumanMethylation27 platform is based on probe hybridization and single nucleotide extension, and therefore levels of DNA methylation observed with a particular probe are highly dependent on whether the probe is located within or outside of a CpG island and its proximity to the transcription start site of a gene (Brenet et al. 2011; van Vlodrop et al. 2011). Additionally, the identification of long-range DNA methylation, or spreading of DNA methylation (Clark 2007), as well as roles for DNA methylation on the border or just outside CpG islands, termed “shores” and “shelves” (Irizarry et al. 2009), DNA methylation-regulated alternate transcripts (Maunakea et al. 2010), the role of DNA methylation in chromatin arrangement and organization of the genome (Berman et al. 2012), miRNA regulation (Lopez-Serra and Esteller 2012), and even gene silencing by non-CpG island DNA methylation (Han et al. 2011) add increasing intricacy to the relationship between DNA methylation and gene regulation. The new Illumina Infinium HumanMethylation450 bead array, whole-genome bisulfite-sequencing, and RNA-seq will be able to shed more light onto these possibilities (Berman et al. 2012).
Distinct mutational and gene expression differences between lung adenocarcinomas of smokers and never-smokers have been frequently noted (e.g., Belinsky et al. 2002; Toyooka et al. 2003; Pao et al. 2004, 2005b; Sun et al. 2007; Landi et al. 2008). Previous candidate-gene studies (Belinsky et al. 2002; Pulling et al. 2003) were recapitulated in our study albeit with smaller differences. It should be considered that the second most common form of lung cancer, squamous cell carcinoma, occurs predominantly in smokers, while adenocarcinoma is the most common lung cancer histology in never-smokers. Our study focuses exclusively on lung adenocarcinoma, and the modest differences we detect may therefore be due to different histological compositions as well as to differences in methodology. Two recent studies suggest no simple relationship between tobacco smoke carcinogens and DNA methylation. In a study comparing peripheral blood DNA methylation profiles of smokers and nonsmokers, only one differentially methylated locus was found (Breitling et al. 2011). An in vitro study of human lung cells chronically exposed to a tobacco carcinogen also showed little to no effect on DNA methylation profiles in treated vs. untreated cells (Tommasi et al. 2010). In our genome-scale supervised approach, we noted only six significantly differentially methylated genes between smokers and never-smokers, and of these only LGALS4 showed a corresponding down-regulation in gene expression in current smoker tumors. LGALS4 has been implicated in several cancers, including gastric, colon, and sinusoidal adenocarcinomas (Sakakura et al. 2005; Tripodi et al. 2009; Watanabe et al. 2011). Recently, a mechanism for the involvement of LGALS4 as a tumor suppressor in colorectal cancer was proposed involving the WNT signaling pathway (Satelli et al. 2011). This is especially interesting given the well-established role that WNT signaling plays in the development of lung cancer (Mazieres et al. 2005; Nguyen et al. 2009). Importantly, previous studies have suggested that the WNT pathway is involved in cigarette smoke–induced tumorigenesis (Lemjabbar-Alaoui et al. 2006; Hussain et al. 2009). To our knowledge, this is the first report of differential expression of LGALS4 between smoker and never-smoker lung tumors and of DNA methylation as a potential regulator of LGALS4 expression. Functional validation of LGALS4 regulation in lung adenocarcinoma is a highly interesting avenue of future investigation.
Genome-wide DNA methylation profiling has led to increased knowledge of epigenetic subtypes of colorectal cancer, glioblastoma, and multiple myeloma, among others (Noushmehr et al. 2010; Walker et al. 2011; Hinoue et al. 2012). The best-established DNA methylation-based subgroup is that of CpG island methylator phenotype (CIMP), first identified in colorectal cancer (Toyota et al. 1999). CIMP tumors possess high frequency and levels of cancer-specific DNA methylation at loci that show little or no methylation in non-CIMP tumors. CIMP sometimes shows differences in patient survival and is closely associated with BRAF activating mutations, but this does not appear to drive CIMP (Teodoridis et al. 2008). The existence of CIMP has been suggested in NSCLC (Marsit et al. 2006; Suzuki et al. 2006) using a very limited number of genes. However, another study did not support this conclusion (Vaissiere et al. 2009). Our use of 27,578 probes enabled a more thorough examination of DNA methylation, and we find no evidence for classic CIMP in lung cancer. An additional epigenotype, termed CIMP-low, has been reported and confirmed in several independent populations of colorectal tumors using different methodologies (Ogino et al. 2006; Shen et al. 2007; Yagi et al. 2010; Hinoue et al. 2012). CIMP-low exhibits moderately high levels of DNA hypermethylation at a subset of CIMP-associated loci, and in each study was found to be associated with KRAS mutation. Although the current lung adenocarcinoma study is limited by a modest sample size, we too observe an epigenetic subtype of lung adenocarcinoma with higher DNA methylation levels that is associated with KRAS mutation. In 2006, a higher methylation index in KRAS mutant tumors in comparison to KRAS wild-type tumors was described (Toyooka et al. 2006). However, like CIMP-low in colorectal cancer, KRAS mutations appear not to drive this epigenetic subgroup, and a more complex molecular mechanism may cause the observed epigenetic heterogeneity (Hinoue et al. 2012). The hypermethylated cluster was also associated (albeit more weakly) with smoking status, which is not surprising given the fact that KRAS mutations are most common in smokers (Ahrendt et al. 2001).
To further characterize the CIMP-low cluster, we identified genes that show cluster and tumor-specific DNA methylation-based down-regulation, including GFRA1. These genes, which might constitute new DNA-methylation-based deregulated driver genes characteristic for a subset of lung adenocarcinomas, merit further exploration. In addition, we identified seven genes showing statistically significant expression changes of at least twofold, unrelated to DNA methylation. Three out of the seven genes were cytokines recently shown to play a role in tumorigenesis (Andersson et al. 2011), while PHLDA1, a regulator of IGF1-mediated apoptosis (Toyoshima et al. 2004), has recently been suggested to be a putative epithelial stem cell marker in the human intestine (Sakthianandeswaren et al. 2011). The role of cytokines in tumorigenesis is of increasing interest (Dranoff 2004; Li et al. 2011), and the connection between DNA methylation, cytokines, and cancer is an intriguing avenue of investigation (Yoshikawa et al. 2001; Galm et al. 2003; Niwa et al. 2005; Sunaga et al. 2012). While we do not find a survival difference between the two DNA methylation-based clusters, further characterization of the identified molecular differences may lead to the development of new treatment strategies targeted to this subgroup of tumors.
In summary, our DNA methylation profiling of 59 lung adenocarcinomas and matched adjacent non-tumor lung tissue accomplished three goals: (1) the identification of numerous new cancer-specific DNA methylation changes that can be pursued as potential lung adenocarcinoma biomarkers; (2) the identification of potentially functional DNA methylation changes that may constitute driver alterations in lung adenocarcinoma; and (3) the identification of an epigenetic subgroup of lung adenocarcinoma with higher levels of DNA methylation that is reminiscent of CIMP-low in colorectal cancer, is correlated with KRAS mutation, and shows specific gene expression alterations. Our observations lay the groundwork for further diagnostic and mechanistic studies of lung adenocarcinoma that could lead to improvements in detection, patient classification, and therapy.
Methods
Study samples
The Early Detection Research Network (EDRN)/Canary Foundation tissue collection consisted of 60 lung adenocarcinoma tumors and matched adjacent histologically confirmed non-tumor lung (NTL), collected after surgery. Forty-five adenocarcinoma/NTL pairs were obtained from the Vancouver General Hospital (Vancouver, Canada) and 15 adenocarcinoma/NTL pairs from the British Columbia Cancer Agency Tumor Tissue Repository (Vancouver, Canada, BCCA Research Ethics Board #H09-00008). Thirty subjects were never-smokers (defined as less than 100 lifetime cigarettes), and 30 were current smokers (average 53 pack-years, range 11–120 pack-years). One tumor sample was excluded after pathology review later revealed it to be a large cell carcinoma. Subject characteristics for the remaining 59 subjects are detailed in Table 1. For verification of DNA methylation profiling, an independent sample set of 28 lung adenocarcinomas and 27 NTL was used. Subject characteristics for the validation population are detailed in Supplemental Table 2. Of these, 21 tumor and 20 NTL de-identified samples were purchased from the Ontario Tumor Bank (OTB, Ontario, Canada), while seven tumors and seven NTL were collected at the University Hospital at the University of Southern California (USC IRB protocol #HS-06-00447). For MethyLight verification of selected probes, OTB samples were used (26 tumors and 26 NTL), of which 25 pairs were matched. Ten of the tumors and 13 of the NTL samples examined by MethyLight were the same tissues used for the genome-wide verification. EDRN/Canary samples were assessed by an experienced pathologist (A.F.G.), while the verification samples were assessed by a separate expert lung pathologist (M.N.K.). All sample collections were performed conforming to protocols approved by the appropriate local Institutional Review Boards and were acquired with informed consent. The identities of the subjects were not made available to the laboratory investigators.
DNA methylation data production
DNA was extracted by proteinase K digestion following manual microdissection from slides prepared from fresh frozen tissue blocks. The DNA was then bisulfite-converted using the EZ DNA Methylation kit (Zymo Research) with a modification to the manufacturer's protocol in which samples were cycled 16 times for 30 sec at 90°C and 1 h at 50°C. The Illumina Infinium HumanMethylation27 BeadChip assays were performed by the USC Epigenome Center according to the manufacturer's protocols (Illumina). This assay generates DNA methylation data for 27,578 CpG dinucleotides covering 14,473 unique genes. DNA methylation levels are reported as β-values, calculated from mean methylated (M) and unmethylated (U) signal intensities for each locus for each sample using the formula [β = M/(M + U)]. Probes with detection P-values of >0.05 were deemed not significantly different from background noise and were labeled “NA.” Data for all samples are publicly available at the EDRN Public Portal (http://www.cancer.gov/edrn). MethyLight experiments were performed as previously described (Campan et al. 2009). MethyLight measurements are represented as percentage methylated reference (PMR), defined by the GENE:ALU ratio of a sample, wherein ALU refers to a reference primer/probe combination that lacks CpGs and is designed to bind to a subset of ALU repeat sequences (Weisenberger et al. 2006), divided by the GENE:ALU ratio of M.SssI-treated reference DNA. This results in a PMR range of 0–100, where a PMR of 0 indicates no detectable DNA methylation and a PMR of 100 represents fully methylated molecules. Occasionally, a PMR of >100 can be observed, which may result when the reference DNA was not fully methylated at a particular site. To minimize this, the same batch of reference DNA that has been exhaustively enzymatically methylated is used throughout the experiments (Selamat et al. 2011). Previously published MethyLight primer/probe sets were used for the SFN and MAL locus and the ALU reference (Weisenberger et al. 2006; Noushmehr et al. 2010). Other MethyLight primer/probe sequences are detailed in Supplemental Table 10.
DNA methylation data analysis
Data analyses were performed using R (R Development Core Team 2011) and Bioconductor (Gentleman et al. 2004). The analyses of 120 tissue samples necessitated conducting the experiment with the samples randomized and spread over two bisulfite treatment plates and 16 Infinium BeadChips. Batch effect investigations were performed as recommended (Leek et al. 2010) and are illustrated in Supplemental Figure 1. Three tissue samples were excluded from analyses: one tumor/NTL tissue pair (07L36_T/N) found to be a large cell carcinoma instead of a lung adenocarcinoma and one NTL sample (3023_N) for which correlation analyses suggested this was neither a lung adenocarcinoma nor NTL tissue. Probes targeting the X and Y chromosomes were excluded, as were probes containing a known single-nucleotide polymorphism, probes that contain repeat sequences of ≥10 bp, and probes that were found to be non-unique in the genome (Noushmehr et al. 2010). Hierarchical clustering was performed using Ward linkage with Euclidean distance for samples and Pearson correlation coefficients for probes. For each comparison analysis, the top 5000 most variable probes across all samples included in the comparison as measured by SD/SDMAX were retained (Cancer Genome Atlas Research Network 2011). Locus-by-locus analyses were conducted using the nonparametric Wilcoxon rank-sum test, and multiple comparisons correction was performed using Q-values from the qvalue package in R (Storey and Tibshirani 2003). Probes were considered statistically significantly different between the tested groups with a Q < 0.05. We also included an additional filter requiring the median β-value difference between groups to be ≥0.2, or a minimum 20% difference for all group comparisons. MethyLight data were analyzed using Wilcoxon signed-rank tests on GraphPad Prism version 5.00 (GraphPad Software).
Functional classification/gene network analyses
Differentially methylated genes were analyzed for Gene Ontology enrichments in comparison to all genes available on the Illumina Infinium HumanMethylation27 platform using the DAVID Functional Annotation Tool (Huang da et al. 2009). Genes for which expression levels change in concordance with DNA methylation changes were analyzed for gene network and biological processes enrichment using IPA (Ingenuity Systems; http://www.ingenuity.com). Meta-analyses to identify correlated biosets and overlapping genes in publicly available data sets were performed using the NextBio online database (NextBio, Cupertino, CA; http://www.nextbio.com). Accession numbers are GSE10799, GSE17648, GSE19188, GSE19804, and GSE25062.
Integration of gene expression analysis
Gene expression profiles were generated using RNA obtained by TRIzol extraction (Invitrogen) from microdissected alternate sections of the same 59 EDRN lung AD/NTL frozen tissue pairs used for the DNA methylation analysis. Expression data were obtained using the Illumina Human WG-6 v3.0 Expression BeadChips (Illumina) at the Genomics Core at UT Southwestern. Bead-summarized data were obtained using the Illumina BeadStudio software, expression values were log2-transformed, and Robust Spline Normalization (RSN) was performed with the lumi package in R (Du et al. 2008). The ReMOAT annotation of gene expression data was used to include only “perfect” and “good” annotated probes (Barbosa-Morais et al. 2010). Exploratory quality-control analyses revealed no strong batch effects (data not shown), although one tumor sample was excluded (3035_T) due to quality concerns. Out of 766 differentially methylated genes identified (Q < 0.05, median β-value difference ≥0.2), we were able to examine gene expression levels for 709 genes after the probe quality filtering detailed above. For genes with multiple probes, we selected the probe with the highest variance and analyzed differential expression using t-tests and a Benjamini-Hochberg (BH) multiple comparisons correction. Statistical significance was called at BH-adjusted p < 0.05. An additional stringent filter of mean twofold change was used to identify top changing genes. Correlation between gene expression and DNA methylation for each gene was measured using the Spearman correlation coefficient. For genes with multiple probes measuring DNA methylation, we selected the probe with the highest SD/SDmax value for DNA methylation.
Data access
The DNA methylation and expression data generated for the study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE32867. All genome-scale data generated in the study are also publicly available at the EDRN Public Portal (http://edrn.nci.nih.gov/).
Acknowledgments
This work was supported by the Canary and Labrecque Foundations in partnership with the Early Detection Research Network (EDRN) of the National Cancer Institute/National Institutes of Health (U01 CA086402), with additional support from NIH/NCI grants R01 CA119029 and CA120869 to I.A.L.O., and the Norris Comprehensive Cancer Center core grant NCI CCSG 5P30 CA014089. We thank all of the members of the Canary/EDRN lung team for fruitful discussions; members of the Laird-Offringa laboratory for helpful criticism; Crystal Marconett for comments on the manuscript; Jessica Nishiguchi for manuscript comments and help researching the aberrantly methylated genes; Daniel Weisenberger from the USC Epigenome Center for help with sample processing; and Toshinori Hinoue, Tim Triche Jr., Houtan Noushmehr, and Hui Shen for data analysis advice.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.132662.111.
References
- Acosta D, Suzuki M, Connolly D, Thompson RF, Fazzari MJ, Greally JM, Montagna C 2011. DNA methylation changes in murine breast adenocarcinomas allow the identification of candidate genes for human breast carcinogenesis. Mamm Genome 22: 249–259 [DOI] [PubMed] [Google Scholar]
- Agarwal R, Mori Y, Cheng Y, Jin Z, Olaru AV, Hamilton JP, David S, Selaru FM, Yang J, Abraham JM, et al. 2009. Silencing of claudin-11 is associated with increased invasiveness of gastric cancer cells. PLoS ONE 4: e8002 doi: 10.1371/journal.pone.0008002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahrendt SA, Decker PA, Alawi EA, Zhu YR, Sanchez-Cespedes M, Yang SC, Haasler GB, Kajdacsy-Balla A, Demeure MJ, Sidransky D 2001. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer 92: 1525–1530 [DOI] [PubMed] [Google Scholar]
- Andersson A, Srivastava MK, Harris-White M, Huang M, Zhu L, Elashoff D, Strieter RM, Dubinett SM, Sharma S 2011. Role of CXCR3 ligands in IL-7/IL-7Rα-Fc-mediated antitumor activity in lung cancer. Clin Cancer Res 17: 3660–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Dunning MJ, Samarajiwa SA, Darot JF, Ritchie ME, Lynch AG, Tavare S 2010. A re-annotation pipeline for Illumina BeadArrays: Improving the interpretation of gene expression data. Nucleic Acids Res 38: e17 doi: 10.1093/nar/gkp942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, Lin L, Chen G, Gharib TG, Thomas DG, et al. 2002. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med 8: 816–824 [DOI] [PubMed] [Google Scholar]
- Belinsky SA 2004. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer 4: 707–717 [DOI] [PubMed] [Google Scholar]
- Belinsky SA, Palmisano WA, Gilliland FD, Crooks LA, Divine KK, Winters SA, Grimes MJ, Harms HJ, Tellez CS, Smith TM, et al. 2002. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res 62: 2370–2377 [PubMed] [Google Scholar]
- Bendik I, Schraml P, Lludwig CU 1998. Characterization of MAST9/Hevin, a SPARC-like protein, that is down-regulated in non-small cell lung cancer. Cancer Res 58: 626–629 [PubMed] [Google Scholar]
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. 2012. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet 44: 40–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P, Ladd C, Beheshti J, Bueno R, Gillette M, et al. 2001. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc Natl Acad Sci 98: 13790–13795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D 1996. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 85: 803–815 [DOI] [PubMed] [Google Scholar]
- Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H 2011. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet 88: 450–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenet F, Moh M, Funk P, Feierstein E, Viale AJ, Socci ND, Scandura JM 2011. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 6: e14524 doi: 10.1371/journal.pone.0014524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock MV, Hooker CM, Ota-Machida E, Han Y, Guo M, Ames S, Glockner S, Piantadosi S, Gabrielson E, Pridham G, et al. 2008. DNA methylation markers and early recurrence in stage I lung cancer. N Engl J Med 358: 1118–1128 [DOI] [PubMed] [Google Scholar]
- Campan M, Weisenberger DJ, Trinh B, Laird PW 2009. MethyLight. Methods Mol Biol 507: 325–337 [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong N, Zhang H, Madesh M, Zhao M, Yu K, Dodia C, Fisher AB, Savani RC, Shuman H 2007. ABCA3 is critical for lamellar body biogenesis in vivo. J Biol Chem 282: 23811–23817 [DOI] [PubMed] [Google Scholar]
- Chioni AM, Brackenbury WJ, Calhoun JD, Isom LL, Djamgoz MB 2009. A novel adhesion molecule in human breast cancer cells: Voltage-gated Na+ channel β1 subunit. Int J Biochem Cell Biol 41: 1216–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen BC, Marsit CJ, Houseman EA, Godleski JJ, Longacker JL, Zheng S, Yeh RF, Wrensch MR, Wiemels JL, Karagas MR, et al. 2009. Differentiation of lung adenocarcinoma, pleural mesothelioma, and nonmalignant pulmonary tissues using DNA methylation profiles. Cancer Res 69: 6315–6321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SJ 2007. Action at a distance: Epigenetic silencing of large chromosomal regions in carcinogenesis. Hum Mol Genet 16: R88–R95 [DOI] [PubMed] [Google Scholar]
- Clausen KA, Blish KR, Birse CE, Triplette MA, Kute TE, Russell GB, D'Agostino RB Jr, Miller LD, Torti FM, Torti SV 2011. SOSTDC1 differentially modulates Smad and β-catenin activation and is down-regulated in breast cancer. Breast Cancer Res Treat 129: 737–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich D, Krispin M, Dietrich J, Fassbender A, Lewin J, Harbeck N, Schmitt M, Eppenberger-Castori S, Vuaroqueaux V, Spyratos F, et al. 2010. CDO1 promoter methylation is a biomarker for outcome prediction of anthracycline treated, estrogen receptor-positive, lymph node-positive breast cancer patients. BMC Cancer 10: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divine KK, Pulling LC, Marron-Terada PG, Liechty KC, Kang T, Schwartz AG, Bocklage TJ, Coons TA, Gilliland FD, Belinsky SA 2005. Multiplicity of abnormal promoter methylation in lung adenocarcinomas from smokers and never smokers. Int J Cancer 114: 400–405 [DOI] [PubMed] [Google Scholar]
- Dranoff G 2004. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer 4: 11–22 [DOI] [PubMed] [Google Scholar]
- Du P, Kibbe WA, Lin SM 2008. lumi: A pipeline for processing Illumina microarray. Bioinformatics 24: 1547–1548 [DOI] [PubMed] [Google Scholar]
- Esteller M 2003. Relevance of DNA methylation in the management of cancer. Lancet Oncol 4: 351–358 [DOI] [PubMed] [Google Scholar]
- Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB, Herman JG 1999. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res 59: 67–70 [PubMed] [Google Scholar]
- Fu DY, Wang ZM, Li C, Wang BL, Shen ZZ, Huang W, Shao ZM 2010. Sox17, the canonical Wnt antagonist, is epigenetically inactivated by promoter methylation in human breast cancer. Breast Cancer Res Treat 119: 601–612 [DOI] [PubMed] [Google Scholar]
- Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG 2003. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 101: 2784–2788 [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. 2004. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 5: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghadersohi A, Odunsi K, Lele S, Collins Y, Greco WR, Winston J, Liang P, Sood AK 2004. Prostate derived Ets transcription factor shows better tumor-association than other cancer-associated molecules. Oncol Rep 11: 453–458 [PubMed] [Google Scholar]
- Goldenring JR, Nam KT 2011. Rab25 as a tumour suppressor in colon carcinogenesis. Br J Cancer 104: 33–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Shinjo K, Kondo Y, Shen L, Toyota M, Suzuki H, Gao W, An B, Fujii M, Murakami H, et al. 2009. Epigenetic profiles distinguish malignant pleural mesothelioma from lung adenocarcinoma. Cancer Res 69: 9073–9082 [DOI] [PubMed] [Google Scholar]
- Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G 2011. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet 20: 4299–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D, Malik S, Pan F, Noushmehr H, van Dijk CM, et al. 2012. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 22: 271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka N, Yamazaki-Itoh R, Ino Y, Mizuguchi Y, Yamada T, Hirohashi S, Kanai Y 2011. CXCL17 and ICAM2 are associated with a potential anti-tumor immune response in early intraepithelial stages of human pancreatic carcinogenesis. Gastroenterology 140: 310–321 [DOI] [PubMed] [Google Scholar]
- Hou J, Aerts J, den Hamer B, van Ijcken W, den Bakker M, Riegman P, van der Leest C, van der Spek P, Foekens JA, Hoogsteden HC, et al. 2010. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 5: e10312 doi: 10.1371/journal.pone.0010312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, Wang YP, Xiao HS, Han ZG 2010. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest 120: 223–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu-Lowe DD, Chen E, Zhang L, Watson KD, Mancuso P, Lappin P, Wickman G, Chen JH, Wang J, Jiang X, et al. 2011. Targeting activin receptor-like kinase 1 inhibits angiogenesis and tumorigenesis through a mechanism of action complementary to anti-VEGF therapies. Cancer Res 71: 1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, Caragacianu D, Schrump DS 2009. Tobacco smoke induces Polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res 69: 3570–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibragimova I, Ibanez de Caceres I, Hoffman AM, Potapova A, Dulaimi E, Al-Saleem T, Hudes GR, Ochs MF, Cairns P 2010. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev Res (Phila) 3: 1084–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa JP 2004. CpG island methylator phenotype in cancer. Nat Rev Cancer 4: 988–993 [DOI] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D 2011. Global cancer statistics. CA Cancer J Clin 61: 69–90 [DOI] [PubMed] [Google Scholar]
- Jensen TJ, Novak P, Eblin KE, Gandolfi AJ, Futscher BW 2008. Epigenetic remodeling during arsenical-induced malignant transformation. Carcinogenesis 29: 1500–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA 2002. DNA methylation and cancer. Oncogene 21: 5358–5360 [DOI] [PubMed] [Google Scholar]
- Ju YS, Lee WC, Shin JY, Lee S, Bleazard T, Won JK, Kim YT, Kim JI, Kang JH, Seo JS 2012. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res 22: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Leszczynska K, Abraham S, Scarcia M, Hiltbrunner S, Marshall CJ, Mavria G, Bicknell R, Heath VL 2011. RhoJ/TCL regulates endothelial motility and tube formation and modulates actomyosin contractility and focal adhesion numbers. Arterioscler Thromb Vasc Biol 31: 657–664 [DOI] [PubMed] [Google Scholar]
- Kerr KM, Galler JS, Hagen JA, Laird PW, Laird-Offringa IA 2007. The role of DNA methylation in the development and progression of lung adenocarcinoma. Dis Markers 23: 5–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Yamada D, Fukami T, Masuda M, Sakurai-Yageta M, Williams YN, Maruyama T, Asamura H, Matsuno Y, Onizuka M, et al. 2005. Promoter methylation of DAL-1/4.1B predicts poor prognosis in non-small cell lung cancer. Clin Cancer Res 11: 2954–2961 [DOI] [PubMed] [Google Scholar]
- Kim H, Lapointe J, Kaygusuz G, Ong DE, Li C, van de Rijn M, Brooks JD, Pollack JR 2005. The retinoic acid synthesis gene ALDH1a2 is a candidate tumor suppressor in prostate cancer. Cancer Res 65: 8118–8124 [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Nishioka M, Kohno T, Nakamoto M, Maeshima A, Aoyagi K, Sasaki H, Takenoshita S, Sugimura H, Yokota J 2004. Identification of genes whose expression is upregulated in lung adenocarcinoma cells in comparison with type II alveolar cells and bronchiolar epithelial cells in vivo. Oncogene 23: 3089–3096 [DOI] [PubMed] [Google Scholar]
- Kohda M, Hoshiya H, Katoh M, Tanaka I, Masuda R, Takemura T, Fujiwara M, Oshimura M 2001. Frequent loss of imprinting of IGF2 and MEST in lung adenocarcinoma. Mol Carcinog 31: 184–191 [DOI] [PubMed] [Google Scholar]
- Kubo T, Yamamoto H, Ichimura K, Jida M, Hayashi T, Otani H, Tsukuda K, Sano Y, Kiura K, Toyooka S 2009. DNA methylation in small lung adenocarcinoma with bronchioloalveolar carcinoma components. Lung Cancer 65: 328–332 [DOI] [PubMed] [Google Scholar]
- Kupershmidt I, Su QJ, Grewal A, Sundaresh S, Halperin I, Flynn J, Shekar M, Wang H, Park J, Cui W, et al. 2010. Ontology-based meta-analysis of global collections of high-throughput public data. PLoS ONE 5: e13066 doi: 10.1371/journal.pone.0013066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et al. 2008. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One 3: e1651 doi: 10.1371/journal.pone.0001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JE, Pavey SJ, Passmore LH, Bowman RV, Hayward NK, Fong KM 2007. Gene expression signature predicts recurrence in lung adenocarcinoma. Clin Cancer Res 13: 2946–2954 [DOI] [PubMed] [Google Scholar]
- Leandro-Garcia LJ, Leskela S, Landa I, Montero-Conde C, Lopez-Jimenez E, Leton R, Cascon A, Robledo M, Rodriguez-Antona C 2010. Tumoral and tissue-specific expression of the major human β-tubulin isotypes. Cytoskeleton (Hoboken) 67: 214–223 [DOI] [PubMed] [Google Scholar]
- Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA 2010. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet 11: 733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtonen ST, Svensk AM, Soini Y, Paakko P, Hirvikoski P, Kang SW, Saily M, Kinnula VL 2004. Peroxiredoxins, a novel protein family in lung cancer. Int J Cancer 111: 514–521 [DOI] [PubMed] [Google Scholar]
- Lemjabbar-Alaoui H, Dasari V, Sidhu SS, Mengistab A, Finkbeiner W, Gallup M, Basbaum C 2006. Wnt and Hedgehog are critical mediators of cigarette smoke-induced lung cancer. PLoS ONE 1: e93 doi: 10.1371/journal.pone.0000093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen VM, Bespalov MM, Runeberg-Roos P, Puurand U, Merits A, Saarma M, Goldman A 2004. The structure of GFRα1 domain 3 reveals new insights into GDNF binding and RET activation. EMBO J 23: 1452–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Dong X, Yin Y, Su Y, Xu Q, Zhang Y, Pang X, Chen W 2005. BJ-TSA-9, a novel human tumor-specific gene, has potential as a biomarker of lung cancer. Neoplasia 7: 1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lee KM, Han W, Choi JY, Lee JY, Kang GH, Park SK, Noh DY, Yoo KY, Kang D 2010. Estrogen and progesterone receptor status affect genome-wide DNA methylation profile in breast cancer. Hum Mol Genet 19: 4273–4277 [DOI] [PubMed] [Google Scholar]
- Li N, Grivennikov SI, Karin M 2011. The unholy trinity: Inflammation, cytokines, and STAT3 shape the cancer microenvironment. Cancer Cell 19: 429–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind GE, Ahlquist T, Lothe RA 2007. DNA hypermethylation of MAL: A promising diagnostic biomarker for colorectal tumors. Gastroenterology 132: 1631–1632 [DOI] [PubMed] [Google Scholar]
- Lipson D, Capelletti M, Yelensky R, Otto G, Parker A, Jarosz M, Curran JA, Balasubramanian S, Bloom T, Brennan KW, et al. 2012. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med 18: 382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Liao GQ, He P, Zhu H, Liu PH, Qu YM, Song XM, Xu QW, Gao Q, Zhang Y, et al. 2008. Detection of circulating cancer cells in lung cancer patients with a panel of marker genes. Biochem Biophys Res Commun 372: 756–760 [DOI] [PubMed] [Google Scholar]
- Lopez-Serra P, Esteller M 2012. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene 31: 1609–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu TP, Tsai MH, Lee JM, Hsu CP, Chen PC, Lin CW, Shih JY, Yang PC, Hsiao CK, Lai LC, et al. 2010. Identification of a novel biomarker, SEMA5A, for non-small cell lung carcinoma in nonsmoking women. Cancer Epidemiol Biomarkers Prev 19: 2590–2597 [DOI] [PubMed] [Google Scholar]
- Marsit CJ, Houseman EA, Christensen BC, Eddy K, Bueno R, Sugarbaker DJ, Nelson HH, Karagas MR, Kelsey KT 2006. Examination of a CpG island methylator phenotype and implications of methylation profiles in solid tumors. Cancer Res 66: 10621–10629 [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazieres J, He B, You L, Xu Z, Jablons DM 2005. Wnt signaling in lung cancer. Cancer Lett 222: 1–10 [DOI] [PubMed] [Google Scholar]
- Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, Politi LS, Gentner B, Brown JL, Naldini L, et al. 2011. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 19: 512–526 [DOI] [PubMed] [Google Scholar]
- Mura M, Swain RK, Zhuang X, Vorschmitt H, Reynolds G, Durant S, Beesley JF, Herbert JM, Sheldon H, Andre M, et al. 2012. Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene 31: 293–305 [DOI] [PubMed] [Google Scholar]
- Nguyen DX, Chiang AC, Zhang XH, Kim JY, Kris MG, Ladanyi M, Gerald WL, Massague J 2009. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138: 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara H, Maeda M, Oda A, Tsuda M, Sawa H, Nagashima K, Tanaka S 2002. DOCK2 associates with CrkL and regulates Rac1 in human leukemia cell lines. Blood 100: 3968–3974 [DOI] [PubMed] [Google Scholar]
- Niwa Y, Kanda H, Shikauchi Y, Saiura A, Matsubara K, Kitagawa T, Yamamoto J, Kubo T, Yoshikawa H 2005. Methylation silencing of SOCS-3 promotes cell growth and migration by enhancing JAK/STAT and FAK signalings in human hepatocellular carcinoma. Oncogene 24: 6406–6417 [DOI] [PubMed] [Google Scholar]
- Noordhuis MG, Fehrmann RS, Wisman GB, Nijhuis ER, van Zanden JJ, Moerland PD, Ver Loren van Themaat E, Volders HH, Kok M, ten Hoor KA, et al. 2011. Involvement of the TGF-β and β-catenin pathways in pelvic lymph node metastasis in early-stage cervical cancer. Clin Cancer Res 17: 1317–1330 [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, et al. 2010. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS 2006. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: Possible associations with male sex and KRAS mutations. J Mol Diagn 8: 582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. 2007. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet 39: 237–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka R, Sasagawa T, Ninomiya I, Miwa K, Tanii H, Saijoh K 2001. Reduction in the local expression of complement component 6 (C6) and 7 (C7) mRNAs in oesophageal carcinoma. Eur J Cancer 37: 1158–1165 [DOI] [PubMed] [Google Scholar]
- Osada H, Tatematsu Y, Yatabe Y, Nakagawa T, Konishi H, Harano T, Tezel E, Takada M, Takahashi T 2002. Frequent and histological type-specific inactivation of 14-3-3σ in human lung cancers. Oncogene 21: 2418–2424 [DOI] [PubMed] [Google Scholar]
- Oster B, Thorsen K, Lamy P, Wojdacz TK, Hansen LL, Birkenkamp-Demtroder K, Sorensen KD, Laurberg S, Orntoft TF, Andersen CL 2011. Identification and validation of highly frequent CpG island hypermethylation in colorectal adenomas and carcinomas. Int J Cancer 129: 2855–2866 [DOI] [PubMed] [Google Scholar]
- Pao W, Girard N 2011. New driver mutations in non-small-cell lung cancer. Lancet Oncol 12: 175–180 [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. 2004. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci 101: 13306–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H 2005a. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2: e73 doi: 10.1371/journal.pmed.0020073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, Zakowski MF, Heelan RT, Kris MG, Varmus HE 2005b. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med 2: e17 doi: 10.1371/journal.pmed.0020017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K, Chung YJ, So H, Kim K, Park J, Oh M, Jo M, Choi K, Lee EJ, Choi YL, et al. 2011. AGR2, a mucinous ovarian cancer marker, promotes cell proliferation and migration. Exp Mol Med 43: 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen KS, Bamlet WR, Oberg AL, de Andrade M, Matsumoto ME, Tang H, Thibodeau SN, Petersen GM, Wang L 2011. Leukocyte DNA methylation signature differentiates pancreatic cancer patients from healthy controls. PLoS ONE 6: e18223 doi: 10.1371/journal.pone.0018223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepoli A, Cotugno R, Merla G, Gentile A, Augello B, Quitadamo M, Merla A, Panza A, Carella M, Maglietta R, et al. 2009. Promoter methylation correlates with reduced NDRG2 expression in advanced colon tumour. BMC Med Genomics 2: 11 doi: 10.1186/1755-8794-2-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzi M, Fassan M, Balistreri M, Galligioni A, Rea F, Rugge M 2012. Anterior gradient 2 overexpression in lung adenocarcinoma. Appl Immunohistochem Mol Morphol 20: 31–36 [DOI] [PubMed] [Google Scholar]
- Pulling LC, Divine KK, Klinge DM, Gilliland FD, Kang T, Schwartz AG, Bocklage TJ, Belinsky SA 2003. Promoter hypermethylation of the O6-methylguanine-DNA methyltransferase gene: More common in lung adenocarcinomas from never-smokers than smokers and associated with tumor progression. Cancer Res 63: 4842–4848 [PubMed] [Google Scholar]
- Pyle-Chenault RA, Stolk JA, Molesh DA, Boyle-Harlan D, McNeill PD, Repasky EA, Jiang Z, Fanger GR, Xu J 2005. VSGP/F-spondin: A new ovarian cancer marker. Tumour Biol 26: 245–257 [DOI] [PubMed] [Google Scholar]
- R Development Core Team 2011. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/
- Ramachandran V, Arumugam T, Wang H, Logsdon CD 2008. Anterior gradient 2 is expressed and secreted during the development of pancreatic cancer and promotes cancer cell survival. Cancer Res 68: 7811–7818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP 2007. Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci 104: 5527–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmelink M, Mijatovic T, Gustin A, Mathieu A, Rombaut K, Kiss R, Salmon I, Decaestecker C 2005. Identification by means of cDNA microarray analyses of gene expression modifications in squamous non-small cell lung cancers as compared to normal bronchial epithelial tissue. Int J Oncol 26: 247–258 [PubMed] [Google Scholar]
- Risch A, Plass C 2008. Lung cancer epigenetics and genetics. Int J Cancer 123: 1–7 [DOI] [PubMed] [Google Scholar]
- Rodenhiser DI, Andrews J, Kennette W, Sadikovic B, Mendlowitz A, Tuck AB, Chambers AF 2008. Epigenetic mapping and functional analysis in a breast cancer metastasis model using whole-genome promoter tiling microarrays. Breast Cancer Res 10: R62 doi: 10.1186/bcr2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Paredes M, Esteller M 2011. Cancer epigenetics reaches mainstream oncology. Nat Med 17: 330–339 [DOI] [PubMed] [Google Scholar]
- Rohan S, Tu JJ, Kao J, Mukherjee P, Campagne F, Zhou XK, Hyjek E, Alonso MA, Chen YT 2006. Gene expression profiling separates chromophobe renal cell carcinoma from oncocytoma and identifies vesicular transport and cell junction proteins as differentially expressed genes. Clin Cancer Res 12: 6937–6945 [DOI] [PubMed] [Google Scholar]
- Rui X, Tsao J, Scheys JO, Hammer GD, Schimmer BP 2008. Contributions of specificity protein-1 and steroidogenic factor 1 to Adcy4 expression in Y1 mouse adrenal cells. Endocrinology 149: 3668–3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura C, Hasegawa K, Miyagawa K, Nakashima S, Yoshikawa T, Kin S, Nakase Y, Yazumi S, Yamagishi H, Okanoue T, et al. 2005. Possible involvement of RUNX3 silencing in the peritoneal metastases of gastric cancers. Clin Cancer Res 11: 6479–6488 [DOI] [PubMed] [Google Scholar]
- Sakhinia E, Farahangpour M, Tholouli E, Liu Yin JA, Hoyland JA, Byers RJ 2006. Comparison of gene-expression profiles in parallel bone marrow and peripheral blood samples in acute myeloid leukaemia by real-time polymerase chain reaction. J Clin Pathol 59: 1059–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakthianandeswaren A, Christie M, D'Andreti C, Tsui C, Jorissen RN, Li S, Fleming NI, Gibbs P, Lipton L, Malaterre J, et al. 2011. PHLDA1 expression marks the putative epithelial stem cells and contributes to intestinal tumorigenesis. Cancer Res 71: 3709–3719 [DOI] [PubMed] [Google Scholar]
- Sartor MA, Dolinoy DC, Jones TR, Colacino JA, Prince ME, Carey TE, Rozek LS 2011. Genome-wide methylation and expression differences in HPV+ and HPV− squamous cell carcinoma cell lines are consistent with divergent mechanisms of carcinogenesis. Epigenetics 6: 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satelli A, Rao PS, Thirumala S, Rao US 2011. Galectin-4 functions as a tumor suppressor of human colorectal cancer. Int J Cancer 129: 799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyanarayana UG, Toyooka S, Padar A, Takahashi T, Brambilla E, Minna JD, Gazdar AF 2003. Epigenetic inactivation of laminin-5-encoding genes in lung cancers. Clin Cancer Res 9: 2665–2672 [PubMed] [Google Scholar]
- Sato M, Mori Y, Sakurada A, Fujimura S, Horii A 1998. The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum Genet 103: 96–101 [DOI] [PubMed] [Google Scholar]
- Sato N, Fukushima N, Maitra A, Matsubayashi H, Yeo CJ, Cameron JL, Hruban RH, Goggins M 2003. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res 63: 3735–3742 [PubMed] [Google Scholar]
- Schimanski S, Wild PJ, Treeck O, Horn F, Sigruener A, Rudolph C, Blaszyk H, Klinkhammer-Schalke M, Ortmann O, Hartmann A, et al. 2010. Expression of the lipid transporters ABCA3 and ABCA1 is diminished in human breast cancer tissue. Horm Metab Res 42: 102–109 [DOI] [PubMed] [Google Scholar]
- Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, Eden E, Yakhini Z, Ben-Shushan E, Reubinoff BE, et al. 2007. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet 39: 232–236 [DOI] [PubMed] [Google Scholar]
- Selamat SA, Galler JS, Joshi AD, Fyfe MN, Campan M, Siegmund KD, Kerr KM, Laird-Offringa IA 2011. DNA methylation changes in atypical adenomatous hyperplasia, adenocarcinoma in situ, and lung adenocarcinoma. PLoS ONE 6: e21443 doi: 10.1371/journal.pone.0021443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, Hernandez NS, Chen X, Ahmed S, Konishi K, et al. 2007. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci 104: 18654–18659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Ishii A, Kano J, Morishita Y, Sato Y, Minami Y, Noguchi M 2011. High expression of stratifin is a universal abnormality during the course of malignant progression of early-stage lung adenocarcinoma. Int J Cancer 129: 2445–2453 [DOI] [PubMed] [Google Scholar]
- Shimizu H, Horii A, Sunamura M, Motoi F, Egawa S, Unno M, Fukushige S 2011. Identification of epigenetically silenced genes in human pancreatic cancer by a novel method “microarray coupled with methyl-CpG targeted transcriptional activation” (MeTA-array). Biochem Biophys Res Commun 411: 162–167 [DOI] [PubMed] [Google Scholar]
- Shiraishi M, Sekiguchi A, Oates AJ, Terry MJ, Miyamoto Y 2002. HOX gene clusters are hotspots of de novo methylation in CpG islands of human lung adenocarcinomas. Oncogene 21: 3659–3662 [DOI] [PubMed] [Google Scholar]
- Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M 2004. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med 350: 1296–1303 [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci 100: 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Schiller JH, Gazdar AF 2007. Lung cancer in never smokers—a different disease. Nat Rev Cancer 7: 778–790 [DOI] [PubMed] [Google Scholar]
- Sunaga N, Imai H, Shimizu K, Shames DS, Kakegawa S, Girard L, Sato M, Kaira K, Ishizuka T, Gazdar AF, et al. 2012. Oncogenic KRAS-induced interleukin-8 overexpression promotes cell growth and migration and contributes to aggressive phenotypes of non-small cell lung cancer. Int J Cancer 130: 1733–1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Shigematsu H, Iizasa T, Hiroshima K, Nakatani Y, Minna JD, Gazdar AF, Fujisawa T 2006. Exclusive mutation in epidermal growth factor receptor gene, HER-2, and KRAS, and synchronous methylation of nonsmall cell lung cancer. Cancer 106: 2200–2207 [DOI] [PubMed] [Google Scholar]
- Suzuki-Ishigaki S, Numayama-Tsuruta K, Kuramasu A, Sakurai E, Makabe Y, Shimura S, Shirato K, Igarashi K, Watanabe T, Ohtsu H 2000. The mouse L-histidine decarboxylase gene: Structure and transcriptional regulation by CpG methylation in the promoter region. Nucleic Acids Res 28: 2627–2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Pero SC, Taguchi K, Shimada M, Mori M, Krag DN, Arii S 2006. Specific peptide ligand for Grb7 signal transduction protein and pancreatic cancer metastasis. J Natl Cancer Inst 98: 491–498 [DOI] [PubMed] [Google Scholar]
- Tao R, Li J, Xin J, Wu J, Guo J, Zhang L, Jiang L, Zhang W, Yang Z, Li L 2011. Methylation profile of single hepatocytes derived from hepatitis B virus-related hepatocellular carcinoma. PLoS ONE 6: e19862 doi: 10.1371/journal.pone.0019862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teodoridis JM, Hardie C, Brown R 2008. CpG island methylator phenotype (CIMP) in cancer: Causes and implications. Cancer Lett 268: 177–186 [DOI] [PubMed] [Google Scholar]
- Tessema M, Belinsky SA 2008. Mining the epigenome for methylated genes in lung cancer. Proc Am Thorac Soc 5: 806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh CK, Gao F, Lim WT, Leong SS, Fong KW, Yap SP, Hsu AA, Eng P, Koong HN, Thirugnanam A, et al. 2006. Never-smokers with lung cancer: Epidemiologic evidence of a distinct disease entity. J Clin Oncol 24: 2245–2251 [DOI] [PubMed] [Google Scholar]
- Tommasi S, Kim SI, Zhong X, Wu X, Pfeifer GP, Besaratinia A 2010. Investigating the epigenetic effects of a prototype smoke-derived carcinogen in human cells. PLoS ONE 5: e10594 doi: 10.1371/journal.pone.0010594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka S, Maruyama R, Toyooka KO, McLerran D, Feng Z, Fukuyama Y, Virmani AK, Zochbauer-Muller S, Tsukuda K, Sugio K, et al. 2003. Smoke exposure, histologic type and geography-related differences in the methylation profiles of non-small cell lung cancer. Int J Cancer 103: 153–160 [DOI] [PubMed] [Google Scholar]
- Toyooka S, Tokumo M, Shigematsu H, Matsuo K, Asano H, Tomii K, Ichihara S, Suzuki M, Aoe M, Date H, et al. 2006. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res 66: 1371–1375 [DOI] [PubMed] [Google Scholar]
- Toyoshima Y, Karas M, Yakar S, Dupont J, Lee H, LeRoith D 2004. TDAG51 mediates the effects of insulin-like growth factor I (IGF-I) on cell survival. J Biol Chem 279: 25898–25904 [DOI] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP 1999. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci 96: 8681–8686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE, et al. 2011. International Association for the Study of Lung Cancer/American Thoracic Society/European Respiratory Society international multidisciplinary classification of lung adenocarcinoma. J Thorac Oncol 6: 244–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripodi D, Quemener S, Renaudin K, Ferron C, Malard O, Guisle-Marsollier I, Sebille-Rivain V, Verger C, Geraut C, Gratas-Rabbia-Re C 2009. Gene expression profiling in sinonasal adenocarcinoma. BMC Med Genomics 2: 65 doi: 10.1186/1755-8794-2-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou JA, Shen LY, Siegmund KD, Long TI, Laird PW, Seneviratne CK, Koss MN, Pass HI, Hagen JA, Laird-Offringa IA 2005. Distinct DNA methylation profiles in malignant mesothelioma, lung adenocarcinoma, and non-tumor lung. Lung Cancer 47: 193–204 [DOI] [PubMed] [Google Scholar]
- Tsou JA, Galler JS, Siegmund KD, Laird PW, Turla S, Cozen W, Hagen JA, Koss MN, Laird-Offringa IA 2007. Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma. Mol Cancer 6: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usadel H, Brabender J, Danenberg KD, Jeronimo C, Harden S, Engles J, Danenberg PV, Yang S, Sidransky D 2002. Quantitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer Res 62: 371–375 [PubMed] [Google Scholar]
- Vaissiere T, Hung RJ, Zaridze D, Moukeria A, Cuenin C, Fasolo V, Ferro G, Paliwal A, Hainaut P, Brennan P, et al. 2009. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res 69: 243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Vlodrop IJ, Niessen HE, Derks S, Baldewijns M, Van Criekinge W, Herman JG, van Engeland M 2011. Analysis of promoter CpG island hypermethylation in cancer: Location, location, location! Clin Cancer Res 17: 4225–4231 [DOI] [PubMed] [Google Scholar]
- Vanderlaag KE, Hudak S, Bald L, Fayadat-Dilman L, Sathe M, Grein J, Janatpour MJ 2010. Anterior gradient-2 plays a critical role in breast cancer cell growth and survival by modulating cyclin D1, estrogen receptor-α and survivin. Breast Cancer Res 12: R32 doi: 10.1186/bcr2586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale L, Frabetti F, Huntsman SA, Canaider S, Casadei R, Lenzi L, Facchin F, Carinci P, Zannotti M, Coppola D, et al. 2007. Sequence, “subtle” alternative splicing and expression of the CYYR1 (cysteine/tyrosine-rich 1) mRNA in human neuroendocrine tumors. BMC Cancer 7: 66 doi: 10.1186/1471-2407-7-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, Davies FE, Ross FM, Morgan GJ 2011. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 117: 553–562 [DOI] [PubMed] [Google Scholar]
- Wang S, El-Deiry WS 2003. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 22: 8628–8633 [DOI] [PubMed] [Google Scholar]
- Wang KK, Liu N, Radulovich N, Wigle DA, Johnston MR, Shepherd FA, Minden MD, Tsao MS 2002. Novel candidate tumor marker genes for lung adenocarcinoma. Oncogene 21: 7598–7604 [DOI] [PubMed] [Google Scholar]
- Wang L, Aakre JA, Jiang R, Marks RS, Wu Y, Chen J, Thibodeau SN, Pankratz VS, Yang P 2010. Methylation markers for small cell lung cancer in peripheral blood leukocyte DNA. J Thorac Oncol 5: 778–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Takemasa I, Kaneko N, Yokoyama Y, Matsuo E, Iwasa S, Mori M, Matsuura N, Monden M, Nishimura O 2011. Clinical significance of circulating galectins as colorectal cancer markers. Oncol Rep 25: 1217–1226 [DOI] [PubMed] [Google Scholar]
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. 2006. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 38: 787–793 [DOI] [PubMed] [Google Scholar]
- Wikman H, Kettunen E, Seppanen JK, Karjalainen A, Hollmen J, Anttila S, Knuutila S 2002. Identification of differentially expressed genes in pulmonary adenocarcinoma by using cDNA array. Oncogene 21: 5804–5813 [DOI] [PubMed] [Google Scholar]
- Wrage M, Ruosaari S, Eijk PP, Kaifi JT, Hollmen J, Yekebas EF, Izbicki JR, Brakenhoff RH, Streichert T, Riethdorf S, et al. 2009. Genomic profiles associated with early micrometastasis in lung cancer: Relevance of 4q deletion. Clin Cancer Res 15: 1566–1574 [DOI] [PubMed] [Google Scholar]
- Yagi K, Akagi K, Hayashi H, Nagae G, Tsuji S, Isagawa T, Midorikawa Y, Nishimura Y, Sakamoto H, Seto Y, et al. 2010. Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res 16: 21–33 [DOI] [PubMed] [Google Scholar]
- Yoshikawa H, Matsubara K, Qian GS, Jackson P, Groopman JD, Manning JE, Harris CC, Herman JG 2001. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet 28: 29–35 [DOI] [PubMed] [Google Scholar]
- Yue W, Dacic S, Sun Q, Landreneau R, Guo M, Zhou W, Siegfried JM, Yu J, Zhang L 2007. Frequent inactivation of RAMP2, EFEMP1 and Dutt1 in lung cancer by promoter hypermethylation. Clin Cancer Res 13: 4336–4344 [DOI] [PubMed] [Google Scholar]
- Zhang W, Glockner SC, Guo M, Machida EO, Wang DH, Easwaran H, Van Neste L, Herman JG, Schuebel KE, Watkins DN, et al. 2008. Epigenetic inactivation of the canonical Wnt antagonist SRY-box containing gene 17 in colorectal cancer. Cancer Res 68: 2764–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]