

Abstract
Chimeric RNAs comprise exons from two or more different genes and have the potential to encode novel proteins that alter cellular phenotypes. To date, numerous putative chimeric transcripts have been identified among the ESTs isolated from several organisms and using high throughput RNA sequencing. The few corresponding protein products that have been characterized mostly result from chromosomal translocations and are associated with cancer. Here, we systematically establish that some of the putative chimeric transcripts are genuinely expressed in human cells. Using high throughput RNA sequencing, mass spectrometry experimental data, and functional annotation, we studied 7424 putative human chimeric RNAs. We confirmed the expression of 175 chimeric RNAs in 16 human tissues, with an abundance varying from 0.06 to 17 RPKM (Reads Per Kilobase per Million mapped reads). We show that these chimeric RNAs are significantly more tissue-specific than non-chimeric transcripts. Moreover, we present evidence that chimeras tend to incorporate highly expressed genes. Despite the low expression level of most chimeric RNAs, we show that 12 novel chimeras are translated into proteins detectable in multiple shotgun mass spectrometry experiments. Furthermore, we confirm the expression of three novel chimeric proteins using targeted mass spectrometry. Finally, based on our functional annotation of exon organization and preserved domains, we discuss the potential features of chimeric proteins with illustrative examples and suggest that chimeras significantly exploit signal peptides and transmembrane domains, which can alter the cellular localization of cognate proteins. Taken together, these findings establish that some chimeric RNAs are translated into potentially functional proteins in humans.
Chimeric mRNAs are distinct from conventionally spliced mRNA isoforms as they are produced by joining exons from two or more different gene loci (Pirrotta 2002; Horiuchi and Aigaki 2006; Robertson et al. 2007; Li et al. 2008; Gingeras 2009; Douris et al. 2010; Herai and Yamagishi 2010; McManus et al. 2010a,b; Pettitt et al. 2010; Allen et al. 2011). In humans, chimeric transcripts are generated in several ways: trans-splicing of pre-mRNAs (Gingeras 2009; Li et al. 2009c), RNA transcription runoff (Akiva et al. 2006; Parra et al. 2006), from other errors in RNA transcription processing (Gingeras 2009), or represent artifacts of RNA sequencing. Alternatively, chimeric transcripts can be the products of gene fusion following inter-chromosomal translocations or intra-chromosomal rearrangements (Gingeras 2009; Maher et al. 2009b; Herai and Yamagishi 2010). Specific cellular phenotypes are characterized by expression of chimeric transcripts, for example, the fused BCR/ABL, FUS/ERG, MLL/AF6, and MOZ/CBP genes are expressed in acute myeloid leukemia (AML) (Panagopoulos et al. 2003; Nambiar et al. 2008), and the TMPRSS2/ETS chimera is associated with overexpression of the oncogene in prostate cancer (Nambiar et al. 2008). In principle, chimeric transcripts can augment the number of gene products available in a given genome and are suspected to function not only in cancer (Thomson et al. 2000; The ENCODE Project Consortium 2007; Gingeras 2009) but also in normal cells (Akiva et al. 2006; Parra et al. 2006).
A systematic analysis of the location of the 5′ termini of coding genes expressed in various cell lines was initiated as part of the ENCODE pilot project (Denoeud et al. 2007; The ENCODE Project Consortium 2007; Tress et al. 2007; Djebali et al. 2008). This project discovered that gene boundaries extend well beyond the annotated termini in 65% of cases, often extending into neighboring genes, leading to the production of chimeric RNAs (Gingeras 2009). A more recent revision of this analysis focusing on chromosomes 21 and 22 revealed additional cases of chimeric transcripts not only connecting neighboring genes but, rather, encompassing distal genes (Djebali et al. 2008, 2012). Characterization of these chimeric transcripts has highlighted that the information stored in the genome and expressed in the transcriptome is not as linear as previously believed (Guigó et al. 2006; Gingeras 2009).
Although some tissue-specific chimeric transcripts as well as inter-chromosomal and intra-chromosomal chimeras have been identified by paired-end transcriptome sequencing (Maher et al. 2009a,b), only a limited number of chimeric transcripts and their associated protein products have been characterized to date, the majority resulting from chromosomal translocations and associated with cancer (Mitani 2004; Miura et al. 2004; Eguchi et al. 2006; Candel et al. 2009; Maher et al. 2009b; Silberg et al. 2010). For instance, gene fusion in chronic myelogenous leukemia (CML) leads to an mRNA transcript that encompasses the 5′ end of the BCR gene and the 3′ end of the ABL gene. Notably, translation of this transcript produces a chimeric BCR–ABL protein that possesses increased tyrosine kinase activity (Rabbitts 1994; Nambiar et al. 2008).
Various studies have used expressed sequence tag (EST) coverage to search for chimeric transcripts (Akiva et al. 2006; Parra et al. 2006); Li et al. (2009c) performed EST screen in humans, mice, fruit flies, and budding yeast. Of the 25 chimeric transcript candidates identified in fly and five in yeast, 30% have been confirmed by RT-PCR (Li et al. 2009c). An even higher RT-PCR confirmation rate has been reported for human chimeric transcript candidates, ranging from 45% (Akiva et al. 2006) to 34% (Parra et al. 2006). As mentioned, the availability and function of cognate chimeric proteins has been examined in only a few cases. One notable example is a chimera in normal human cells generated by trans-splicing of the 5′ exons of the JAZF1 gene on chromosome 7p15 and the 3′ exons of JJAZ1 (SUZ12) on chromosome 17q1 (Li et al. 2009b). This chimeric RNA is translated in endometrial stroma cells and encodes an anti-apoptotic protein (Gingeras 2009; Li et al. 2009b).
The apparently large discrepancy between the number of putative chimeric transcripts and chimeric proteins reported to date (100:1) could indicate that most chimeric transcripts are not translated and perhaps serve to regulate processes at the RNA level. However, the discrepancy could reflect the problem that current protocols tend to overestimate the true number of chimeric transcripts. Indeed, most protocols used to identify chimeric transcripts rely on a reverse transcription step and the reverse transcriptase is known to switch templates, thus creating chimeric artifacts in vitro (Houseley and Tollervey 2010). Therefore, it remains unclear what proportion of putative chimeric transcripts are genuine, and of these how many are translated.
Here we report screening of 7424 human chimeric transcript candidates from GenBank (Benson et al. 2005), which were previously collected in the data sets of chimeric RNAs (Li et al. 2009c; Kim et al. 2010). We employed functional annotation, high throughput RNA sequencing and mass spectrometry experiments. In this way, we confirmed the expression of 175 chimeric RNAs and we identified 12 novel chimeric proteins in humans. We also assessed the tissue specificity of the chimeric RNAs and we compared the expression of chimeric proteins with that of the parental wild-type proteins. Based on our analysis of the chimeric transcripts, the largest collection identified to date, we define two features of chimeric proteins. First, chimeras exploit signal peptides and transmembrane domains to alter the cellular localization of the associated activities. Second, though chimeras themselves are tissue-specific transcripts expressed at low levels, chimeras incorporate parental genes that are expressed at a high level. Such chimeras could be produced in cancer cells and those associated with other diseases, as well as in response to stress in normal cells. To illustrate the proposed characteristic features of chimeras, we focused on the chimeric proteins validated by RNA-seq at the RNA level, as well as those validated by shotgun and targeted mass spectrometry at the protein level.
Results
Expression of chimeric transcripts in normal cells
We collected 7424 sequences of candidate human chimeric RNAs from GenBank (Benson et al. 2005), previously collected in the chimeric RNA data sets (Li et al. 2009c) and the ChimerDB database (Kim et al. 2010). To determine if these chimeric sequences are indeed expressed as transcripts and to assess their level of expression, we screened RNA sequencing data sets (Human Body Map 2.0: see Methods) in 16 tissues (Supplemental Table S1). Briefly, we identified the junction sites for each chimeric sequence and then searched for matching “chimeric reads,” which did not map linearly to annotated transcripts or novel exons in the human genome (see Methods). To define if a chimeric read validates a junction, we required it to map with at least six nucleotides (nt) on each side of the junction (and we allowed for a maximum of three mismatches). Our screening procedure inherently excludes reads mapping to multiple locations in the genome (repetitive regions), as the chimeric reads by definition do not map to any location in the genome or to the annotated transcriptome.
Among the 7224 ESTs and mRNAs in ChimerDB (Kim et al. 2010), we found that 333 (4.5%) had at least two matching reads from the Human Body Map data set, 212 (3%) had matching reads in two tissues, and 156 (2.2%) matched at least two nonidentical chimeric reads, i.e., mapped to distinct nucleotide positions in the chimera junction site (Table 1; http://chimera.bioinfo.cnio.es/). We focused on the cases validated by at least two distinct reads in order to rule out synthetic duplicates created during the RNA-sequencing protocols. In such cases, the number of reads confirming the junction may be high but they would all align to the same position. Demanding at least two distinct mapping positions is a useful strategy to avoid this type of bias, and in practice, this reduces the number of confirmed chimeras from 333 to 156 (see Table 1). Furthermore, half of the remaining 156 cases are validated by two to 12 reads, while the other half are validated by 12 to 2694 reads. Since the chimeric ESTs were primarily identified in cancer cells, it is noteworthy that some are expressed also in normal tissues (Supplemental Material). These findings corroborate those of other studies showing that some fusion transcripts originate from normal tissues (Akiva et al. 2006; Parra et al. 2006). Of note, as a negative control we used the data set of 300 fusion proteins found in cancers, generated by translocations and listed in the dbCRID database (Kong et al. 2011). Remarkably, we did not find any reads in normal tissues of the Human Body Map 2.0 that matched the junction sites of these cancer-associated chimeras. This latter finding confirms that chimeras generated by these chromosomal translocations are not expressed in the considered normal tissues, or at least not at a detectable level.
Table 1.
The expression of chimeric transcripts was confirmed using paired-end RNA-seq reads from various tissues
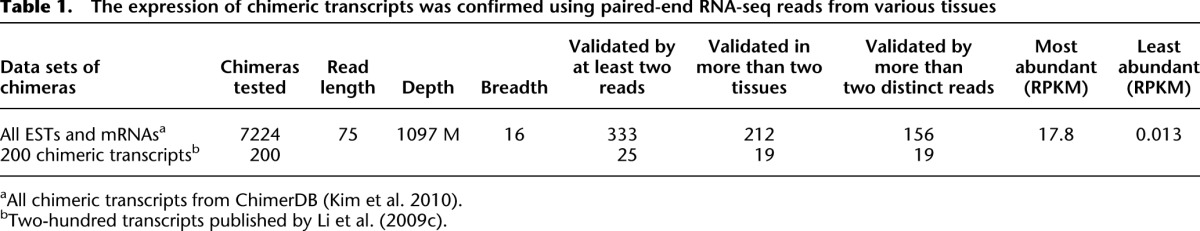
To estimate the expression level of chimeras, we used the measure introduced by Mortazavi and colleagues in 2008 (Mortazavi et al. 2008), namely RPKM (Reads Per Kilobase per Million mapped reads; see Methods), which takes into account the depth of sequencing and the length of the considered “junction” region. Calculations for the human chimeras were performed with the total number of reads set at 1097 million and the “junction” size of 138 nt (= 2 × [75 nt {a read size} − 6 nt]). The most weakly expressed, yet detectable, chimera had two matching reads, corresponding to 0.013 RPKM (Table 1). In general, we observed that most chimeras are lowly expressed transcripts (Fig. 1). Noticeably, most of the parental genes participating in the formation of chimeras (whether expressed or not) are moderately to highly expressed, with expression ranging from 0 to 2495 RPKM and a median expression level of 12.6 (Fig. 1). Though some genes, like tumor necrosis factor (TNF), are not expressed at all in normal tissues, the expression levels of most parental genes fall into the third quartile of the gene expression distribution. Moreover, our data show that genes participating in translated chimeras, i.e., chimeras for which we have evidence of translation (as explained below), are even highly expressed (Wilcoxon test, P-value < 5 × 10−6). In light of these observations, we concluded that, in general, the formation of chimeras is associated with parental genes that exhibit high expression levels. Furthermore, there is an association between detectable expression of the chimera at the protein level and the level of expression of its parental genes (Fig. 1; Table 2; Supplemental Material).
Figure 1.

Expression levels of genes and chimeric transcripts in humans. The expression of genes in human tissues ranges from 0.001 to 15,700 RPKM, with a median of 0.588, whereas the expression of chimeras ranges from 0.006 to 17.8 RPKM with a median of 0.02. Most of the genes involved in the formation of chimeras are moderately to highly expressed, as their expression ranges from 0 to 2495 RPKM, with a median of 12.6. The trend is also observed for the translated chimeras and their parental genes (Wilcoxon test, P-value < 5 × 10−6). The whiskers of the boxplot extend to the data extremes (see also Supplemental Fig. S4).
Table 2.
The expression of chimeric proteins from ChimerDB was confirmed using mass spec peptides matching the junction site

Chimeras are lowly expressed transcripts
To study the expression levels of chimeric transcripts relative to other human transcripts we produced density plots of all transcript expression levels as described recently (Hebenstreit et al. 2011). We found that the distribution of the expression of all genes is clearly bimodal (Fig. 2). The interpretation is that the first peak corresponds to lowly expressed and putatively nonfunctional mRNAs, while the second peak encompasses highly expressed mRNAs (Hebenstreit et al. 2011). Our chimeric transcripts clearly fall within the first peak, with the exception of two chimeras that fall within the second peak. The first one (ESTid = “CD109591.1”) contains regions from genes RAB21 and RN45S, where gene RAB21 is on chromosome 22 and RNA45S corresponds to an rRNA gene located within an unplaced contig. We suspect that this chimera results from read-through transcription of the two genes; and the unplaced contig, or another copy of the rRNA is actually on chromosome 22, next to gene RAB21. The second highly expressed chimera (ESTid = “AB042558.1”) comprises exons from PDE4DIP and NBPF11, the two genes located on chromosome 1. However, this chimera cannot be due to read-through transcription as the two genes are located on different strands and separated by >100 kb. Noticeably, PDE4DIP overlaps NBPF9, raising the possibility that some kind of recombination event has occurred involving NBPF9 and NBPF11 genes either at the genomic or transcriptome level, favoring the formation of this chimera.
Figure 2.

A density plot of RPKM expression levels for all genes versus chimeras. The total number of chimeras is much lower than the total number of genes. Hence, the densities of the distributions are plotted and not the counts. The height of the bars does not correspond to number of transcripts, but to the proportion of transcripts in a given expression category. The distribution for all genes is bimodal, with chimeras falling in the low expressed genes distribution.
Taken together, these observations indicate that most chimeras are expressed at one or two copies per cell on average, and notably this expression involves highly expressed genes. Although these findings are compatible with a potentially unregulated production of chimeric transcripts, we show below that some chimeras likely exert biological roles as they are expressed at the protein level.
Tissue specificity of chimeric transcripts
We evaluated the tissue specificity of chimeras relative to all other genes. Tissue specificity was measured using Shannon entropy (see Methods). Low entropy values correspond to high tissue specificities (Fig. 3). We found that, overall, chimeras were more tissue specific than other genes (Wilcoxon test, P-value < 2 × 10−16). However, we noticed that in general tissue specificity correlated with expression level, such that highly expressed genes tend to be broadly expressed, and thus acknowledged that the low expression level of most chimeras could be a confounding factor. We therefore performed a new test, now controlling for the expression level as a potential confounding factor and we still found that chimeras were more tissue specific than other genes (ANCOVA, P-value < 7.7 × 10−13). We conclude that, irrespective of expression level, chimeras are significantly more tissue specific than non-chimeric transcripts (Fig. 3).
Figure 3.

Tissue specificity of all genes versus chimeras. All genes are presented in red and chimeras in blue. The expression of chimeras is more tissue specific across the different expression levels (ANCOVA, P-value < 7.7 × 10−13). The bins are chosen so as to cover the expression range of all chimeras and have an equal number of chimeras per bin.
Chimeric transcripts are detected at the protein level
To determine if chimeras are expressed at the protein level and rule out the possibility that they are artifacts of reverse transcription, we used both computational and experimental approaches. First, we produced a comprehensive search for unique peptides–spectra matches in mass spectrometry databases using the chimeric sequences translated in six frames. We considered only unique peptides spanning the gene–gene junctions of the chimeras (three amino acids at each side of the junction) with a maximum false discovery rate (FDR) of 1% (see Methods). Second, we conducted in-house experiments to detect chimeric proteins using both shotgun proteomics and targeted analysis of the identified unique peptides spanning the chimeric junctions. Thus, using these approaches we identified 16 unique peptides that span the junction sites of human chimeras (FDR < 1%) (Methods; Supplemental Fig. S1; Supplemental Material), confirming translation of the 12 cognate chimeric transcripts (Table 2). Notably, chimeric reads spanning the junction sites of three of these chimeras were identified in different tissues of the Human Body Map (the chimera CN306050.1, with only one read; BG978110.1, 10 reads; and BM838228.1, three reads). Finally, we confirmed two putative chimeric proteins by the targeted mass spectrometry analysis, termed selective reaction monitoring (SRM) using specifically synthesized heavy-labeled standards (Supplemental Figs. S2, S3).
Remarkably, one chimera identified initially in the EST collection of ChimerDB (Kim et al. 2010), ESTid = “BM838228.1,” was evident in 18 different mass spectrometry experiments in PeptideAtlas (FDR < 1%, placental tissue and embryonic stem cells) (Supplemental Material). This is a chimera of the ribosomal RPL13A protein and actin ACTG1 for which two unique overlapping peptides that match the chimera junction site were identified (LWTVSRCLTASHTVPIYEGYALPHAILR, E-value < 5.1 × 10−5; ASHTVPIYEGYALPHAILR, E-value < 1.3 × 10−5) (Table 2). Specifically, the former peptide had 10 supporting peptide–spectra matches in the PeptideAtlas experiments and it contained an overlap of 12 residues spanning the junction site. Targeted mass spectrometry (SRM) was employed to validate and measure the levels of this human chimeric protein using unique peptide matching at its gene–gene junction site (ASHTVPIYEGYALPHAILR) (Supplemental Fig. S2). In addition, we detected three RNA-seq reads in two different normal tissues (ovary and adipose, Human Body Map) (Table 2). The unusually large number of mass spectrometry experiments, in which matching peptides were identified, probably reflects that this chimeric protein is more abundant than other such proteins. Interestingly, we were able to verify this chimera by RT-PCR using TaqMan probes in different RNA samples (Supplemental Material). Notably, this chimera incorporates both highly expressed cytosolic proteins (RPL13A, RPKM = 343.5; ACTG1, RPKM = 851.6). Particularly, Phyre2 structural prediction analysis (Kelley and Sternberg 2009) of this chimera suggests it can fold into a 3D structure with 100% confidence and 85% identity to the Ribonuclease H-like motif fold (Actin-like ATPase domain) (Fig. 4A). Accordingly, we identified a preserved beta strand that appears close to the junction site of the chimera, which corresponds with high confidence (RMSD < 2.1) to the secondary structure of wild-type actin (Fig. 4B). However, an ATP-binding site is missing in the chimeric sequence (Fig. 4B), perhaps indicating an inability to produce the polymerized form of F-actin.
Figure 4.

A chimera with confirmed RNA and protein expression. We detected two overlapping unique peptides that matched the junction site in 18 mass spectrometry experiments and by the targeted mass spectrometry (SRM) analysis, confirming that this transcript (ESTid = “BM838228.1”) from ChimerDB (Kim et al. 2010) is expressed at the protein level. (A) The 3D structure of the chimeric protein is modeled by Phyre2 (Kelley and Sternberg 2009). (Green) The chimeric protein part derived from actin, ACTG1, predicted using homology modeling; (red) the part of the ribosomal protein, RPL13A, predicted using ab initio methods. The structure is modeled using the Ribonuclease H-like motif fold (actin-like ATPase domain) with 100% confidence and 85% identity. (B) The secondary structure modeling by Phyre2 (Kelley and Sternberg 2009) predicts that a highly preserved beta strand appearing in the wild-type actin protein should also feature in the chimera (blue rectangle). The motif “GDGV” (red rectangle) is the ATP-binding site, which is missing in the chimera sequence.
For the chimera ESTid = “CN310211.1,” we found a unique peptide (GRLGQPAMAK, FDR < 1%) (Table 2; Supplemental Material) spanning its gene–gene junction site using the shotgun mass spectrometry analysis of the total proteome of the human cell lines (see Methods). This chimera incorporates full domains: coiled coil domain from COPS7B (RPKM = 3.4) and RAB domain from RAB13 protein (RPKM = 35.4). The unique peptide was confirmed by the targeted mass spectrometry (SRM) analysis using a synthesized standard peptide (Supplemental Fig. S3).
We performed a second round of shotgun proteomic analyses, identifying eight of the 11 chimeras found in the first round: BE837730.1, BM827779.1, BM842093.1, BY796539.2, CN310211.1, CN430188.1, DB154094.1, and DW419036.1 (see Supplemental Material). Moreover, we verified the unique peptide (VISSIEQKTMAAPSVK) of the ESTid = “BF969911.1” chimera using targeted proteomics and fractionation analysis (Fig. 5). Based on these proteomics analyses, we surmise that >70% of the peptides representing chimeric junctions can be verified in multiple rounds of proteomic analysis. In summary, we provide the first unbiased genome-wide evidence that chimeras are indeed expressed at both the transcriptional and protein levels in humans. These chimeric proteins are less abundant than regular proteins and they seem to be highly tissue specific.
Figure 5.

Selective reaction monitoring (SRM) mass spectrometry analysis. The peptide VISSIEQKTMAAPSVK at the junction site of the BF969911.1 chimera was confirmed by SRM analysis using a stable isotope labeled standard. Briefly, a peptide of the same amino acid sequence was synthesized with a heavy lysine residue, which was then spiked into the digested human prostate cancer lysate. The mixture was fractionated by high pH reversed phase liquid chromatography and the fractions analyzed by SRM mass spectrometry. On the basis of the concentration of the labeled standard, the chimera was estimated to be present at a concentration of ∼30 fmol/mL. A signal-to-noise ratio was calculated as root-mean-square (RMS).
Chimeras may alter cellular localizations of proteins
Chimeras incorporate signal peptides
The 1999 Nobel Prize in Physiology or Medicine was awarded to Gunter Blobel for the discovery that proteins have intrinsic signals that govern their transport and localization within the cell (Emanuelsson et al. 2007; Clérico et al. 2008). Indeed, these signal sequences were shown to serve as zipcodes, specifying the eventual destination of the proteins. Signal peptides targeting proteins to the endoplasmic reticulum (ER) membrane in eukaryotes are 15–30 amino acids long, self-contained and removed after targeting (Emanuelsson et al. 2007; Clérico et al. 2008). In eukaryotes, proteins translocated across the ER membrane are by default transported through the Golgi apparatus and then exported by secretory vesicles (Emanuelsson et al. 2007; Clérico et al. 2008). Some chimeras incorporate signal peptides that could direct proteins to the ER and Golgi apparatus. To be functional, these signal peptides must be present in the gene that forms the 5′ end of the transcript, and thereby transport also the product of the gene at the 3′ end of the chimeric transcript.
For example, we found a human chimeric transcript (ESTid = “AJ420584.1”) (Supplemental Material) in the ChimerDB data set (Kim et al. 2010) that comprises Acetyl-CoA:lyso-PAF acetyltransferase (LPCAT2) and the thioredoxin domain containing protein 5 (TXNDC5) and which is translated starting from the signal peptide of TXNDC5 (localized in the ER). Moreover, this chimera incorporates two transmembrane domains of LPCAT2 (Fig. 6A). The signal peptide is predicted to redistribute the chimeric protein from the plasma membrane to the ER lumen (Fig. 6B).
Figure 6.

Putative chimeric proteins often contain the signal peptides or TM domains of the parental proteins. (A) Schematic view of the two proteins participating in the human chimera: thioredoxin domain containing protein 5 (TXNDC5) and lysophosphatidylcholine acyltransferase 2 (LPCAT2). (B) Schematic view of the hypothetical chimera comprising the signal peptide of TXNDC5 and two TM domains of LPCAT2. We predict that this chimera is localized in the ER lumen.
Notably, of the 7224 chimeric transcripts from ChimerDB (Kim et al. 2010), 32% incorporate signal peptides, and of the 175 chimeras confirmed by more than two RNA-seq reads, 29% have these signal peptides (Table 3; Supplemental Material). Additionally, for the data set of Li et al. (2009c), we observed 34% of the chimeras incorporate signal peptides (Table 3). Given the expected percentage for all human genes (22%) (Table3), we concluded that signal peptides are significantly incorporated in chimeras (Table 3, P-value < 0.001).
Table 3.
Signal peptides and TM domain frequencies in chimeras and all human genes

Chimeras are enriched in transmembrane domains
Transmembrane proteins carry out many key functions in cell signaling and transport (Deutsch et al. 2008; Deutsch 2010). Like signal peptides, transmembrane (TM) segments determine the localization of the proteins in cell membranes. Thus, we anticipated that these segments in chimeric proteins lead to the membrane association of cytosolic proteins, thereby altering their molecular interactions and cellular functions. A chimeric protein has been identified that encompasses parts of the matrilin (MATN) and lysosomal-associated protein transmembrane (LAPTM) genes (Maeda et al. 2005). In accord with our hypothesis, the expression and subcellular localization of the MATN–LAPTM chimera differ from those of the parental wild-type genes participating in the chimera (Maeda et al. 2005). Similarly, the TWE-PRIL chimeric protein that comprises two tumor necrosis factors, TWEAK and APRIL, contains the TWEAK cytoplasmic and TM domains combined with the APRIL C-terminal domain (Pradet-Balade et al. 2002). Accordingly, TWE-PRIL was shown to be a membrane protein, positioning the APRIL receptor-binding domain at the cell surface (Pradet-Balade et al. 2002).
For ChimerDB (Kim et al. 2010), we found that 51% (3701/7224) of chimeric transcripts have predicted TM domains (Table 3). Likewise, 50% (88/175) of chimeras confirmed by more than two RNA-seq reads were found to incorporate at least one TM domain (Table 3). In addition, for the data set of Li et al. (2009c), 55% (110/200) of chimeras integrate TM domains (Table 3). To assess the significance of these proportions we used the GENCODE data set of 22,304 human protein coding sequences (Table 3; Harrow et al. 2006). We looked for predicted TM domains and found 23% of the human proteins in this data set contain at least one TM domain (Table 3). We used this finding to calculate the expected number of chimeras containing at least one TM domain, taking into account the fact that chimeras are generated from two genes but assuming an upper boundary for the appearance of TM helices as chimeras are rarely generated from two whole proteins. Given these assumptions, the expected percentage of chimeras incorporating one or more TM domains is 40.2%, and that TM domains are significantly enriched in putative chimeras (Table 3, P-value < 0.001).
Taken together our observations indicate that chimeric transcripts could at least partially explain the origins of proteins with unexpected cellular localizations. Such proteins are frequently evidenced in high throughput protein studies, for example, in the Dynamic Proteomics study, which aim to monitor the position and amount of endogenous proteins in individual human cells (Cohen et al. 2008; Frenkel-Morgenstern et al. 2010).
Discussion
A number of molecular processes, including trans-splicing, translocations or other chromosomal rearrangements, as well as various aberrations of standard co-linear transcription, can produce chimeric transcripts incorporating information from distinct genomic regions. Here, we describe a new comprehensive approach to validating expression of chimeras at the protein level that involves identification of peptides spanning the junction sites of chimeras. Taking this approach along with shotgun proteomics and targeted mass spectrometry analysis, we establish that some chimeras are indeed translated and detectable. It should be noted that, in typical shotgun proteomic experiments, the standard protein sequence databases, such as the UniProtKB (Magrane and UniProt Consortium 2011), do not contain chimeric proteins. Thus chimeric proteins are not taken into account in most proteomic studies. Given the rapid advancement of mass spectrometry instruments and ever increasing sensitivity it seems likely that more and more chimeric protein will be discovered.
Before our study, various chimeric transcripts had been detected in diverse species by RNA sequencing and verified experimentally, such as the 12 gene fusions in humans (Maher et al. 2009a,b) and the multiple chimeric transcripts identified by the ENCODE pilot project in assorted cell types (Denoeud et al. 2007; The ENCODE Project Consortium 2007; Tress et al. 2007; Djebali et al. 2008). Furthermore, early EST assembly experiments suggested the presence of chimeric transcripts in budding yeast, fruit flies, mice, and humans and estimated that up to 25%–49% of all genes could participate in the formation of chimeric transcripts (Akiva et al. 2006; Parra et al. 2006; Li et al. 2009c). This notwithstanding, the present study is the first to systematically survey public databases for chimeras and validate expression at both the transcriptional and protein levels using an unbiased genome-wide approach.
We suspect that the generation of chimeric transcripts and subsequent translation into chimeric proteins serve to create novel proteins with substantially altered functions compared with the constitutive and alternative isoforms. The altered functions include modified localization (Thomson et al. 2000; Pradet-Balade et al. 2002) and tissue specificity (Akiva et al. 2006; Parra et al. 2006), and could be linked to specific conditions or diseases, such as cancer (Edgren et al. 2011; Kannan et al. 2011). We also provide support for our premise that chimeras are enriched in signal peptides and trans-membrane domains, which alter the cellular localization of proteins participating in the chimeras. Notably, this hypothesis accords with the tissue specificity of chimeras, as Schug et al. (2005) have proposed that most tissue-specific proteins are extracellular and mid-tissue specific proteins are membrane proteins. In addition, we evidence that chimeras connecting more distal genes than neighboring genes tend to incorporate highly expressed genes. This latter observation accords with the “RNA polymerase-induced” mechanism for the chimera production elaborated in Gingeras (2009). Notably, the trend is even more stringent for the translated chimeras, because the genes involved in translated chimeras are even more highly expressed than genes involved in chimeras that are not translated. In light of our data, we hypothesize that the protein products of trans-splicing or genomic alterations generated during evolution serve to control the activity of parental proteins during certain cellular processes or in response to stress. Understanding the principles of functional chimera design and production is an urgent goal in modern Proteomics and Genomics.
To conclude, we establish here that most chimeric RNAs are tissue specific and weakly expressed but can be detected by RNA sequencing techniques. Since the chimeric transcripts analyzed were primarily derived from cancer cells, it is intriguing that matching chimeric reads were found in the Human Body Map data sets for tissues of healthy individuals. Our observations coincide with other recent studies on chimeric transcripts detected in cancers as well as in normal cells (Akiva et al. 2006; Parra et al. 2006; Li et al. 2009b). Having validated the existence of chimeric proteins in eukaryotes, we caution that chimeric proteins should be considered when designing future experimental studies of protein localization in both normal and cancer cells. Finally, we suggest that chimeras should be taken into account when protein–protein interactions are studied, and especially when developing therapeutics.
Methods
Data sets and annotation
To investigate the potential functional role of chimeras, all publicly reported 7424 human chimeric RNA transcripts were analyzed. Specifically, we screened the chimeric ESTs found in human cells by Li et al. (2009c) (200 transcripts), together with all the chimeric ESTs and mRNAs (7224 transcripts) in ChimerDB (Kim et al. 2010). All these chimeric RNAs have well-defined junction sites (at least three nucleotides on either side of the junction). However, only a few of the chimeric sequences exhibit canonical splice-junction sites (Li et al. 2009c).
Initially, sequence similarities between the chimeric RNA transcripts of Li et al. (2009c) and human genomic regions were identified using in-house software and the UCSC BLAT search (Kent 2002; Rosenbloom et al. 2012) to annotate the genes participating in each chimera. NCBI BLAST (Altschul et al. 1997) was applied to delineate the wild-type protein domains corresponding to the genomic regions contained within each chimeric mRNA. All the domain annotation results were manually inspected. WU BLAST (Lopez et al. 2003) was employed to define more precisely short or “strange” genomic regions, as WU BLAST has proven most efficient when transcript composition is unknown (Elizabeth Cha and Rouchka 2005). Finally, FASTA (Pearson and Lipman 1988) was used to find the 100% identical sequence matches for peptides identified by the experimental mass spectrometry analyses for the gene–gene junction of chimeras.
Confirmation of chimeric transcripts by RNA-seq
To assess if a chimeric transcript is present in some RNA sample, we aligned (mapped) the RNA-seq reads to the sequence of each chimera and its junction sites. To ensure that the read could be unambiguously assigned to the chimera, and not to another location in the genome, we performed the following mapping protocol. First, we mapped the RNA-seq reads to the reference genome, in order to identify which reads can be assigned to exons, i.e., exonic reads. In order to identify junction reads, we constructed, for each gene, the combination of all pairs of exons, yielding a collection of all possible intra-gene exon junctions and mapped the RNA-seq reads to these junctions. We then selected the reads, which are not mapped in the previous stages (i.e., reads not mapping to annotated transcripts or novel exons) and mapped them to the chimeric transcripts. Finally, we selected only the reads that mapped precisely to the junction of the chimera, with a minimum of six nucleotides, two codons, mapping on each side of the junction. The number of reads mapping to the junction was taken as an indication of the abundance of the chimera in the RNA sample. Our mapping protocol can be considered stringent as it ensures that, if a read maps both to a known transcript and to a chimeric transcript, it will be assigned to the known transcript. This procedure naturally excludes reads mapping to multiple locations on the genome (repetitive regions), since chimeric reads correspond to reads not mapping to any location on the genome, or the annotated transcriptome. All the mappings were performed using GEM (http://sourceforge.net/apps/mediawiki/gemlibrary) allowing for a maximum of three mismatches (Djebali et al. 2008).
The RNA-seq data sets used for the mapping protocol were the Human Body Map 2.0 data generated on the HiSeq 2000 by Illumina in 2010. The data set comprises 1097 million (M) paired-end reads of 75 nt, resulting from sequencing RNA taken from 16 different tissues.
Quantifying chimeric transcripts with RNA-seq data
Since chimeric transcripts are combinations of annotated transcripts, their identification and quantification is challenging. To quantify a chimeric transcript, we only considered reads unambiguously mapping to its junction. However, this number necessarily depended on the depth of sequencing and on the length of the considered region (in this case the junction). Therefore, we adopted the measure introduced by Mortazavi et al. (2008), namely RPKM. RPKM is defined by the formula (Mortazavi et al. 2008):
| [1] |
where total_reads_identified_junction is the number of reads that have been mapped to a chimeric junction, reads_length_junction(KB) is the size of the region considered to cover the junction, mapped_reads(millions) is the overall number of mapped reads in millions of reads.
In our case, the size of the considered region of reads_length_junction, J, is not the sum of exon length (usually used), but simply the size of the junction calculated as follows:
| [2] |
where L is the read size and M the minimum number of nucleotides required on each side of the junction to assign the read to the junction.
Thus, for the human RNA-seq data set, L = 75 and M = 6, therefore J = 138 nt, or 0.138 kb. The total number of reads sequenced is 1097 M. Hence, for example, chimera BP305895.1, which has 291 reads mapping to its junction, has an expression level of 291/1097/0.138 = 1.92 RPKM.
The correspondence between RPKM and the number of transcripts per cell is still not clearly established. Mortazavi et al. (2008) consider that 3 RPKM corresponds to approximately one transcript per cell in mouse liver, whereas Klisch et al. (2011) suggest that 1 RPKM corresponds to between 0.3 and 1 transcripts per cell.
Tissue specificity of chimeras
The tissue specificity of any transcript was measured using Shannon entropy (Schug et al. 2005). The expression level of a gene in some tissue and the entropy were calculated (Schug et al. 2005). The entropy has units of bits and ranges from zero for genes expressed in a single tissue to log2(N) for genes expressed uniformly in all N = 16 tissues considered.
Identification of chimeric proteins by evidences in PeptideAtlas and GPM
To assess which chimeric transcripts are translated into chimeric proteins, we sought to identify unique mass spec peptides that match the gene–gene junctions of the chimeras. To this end, we searched the mass spectra of two publicly available proteomic databases, the GPM (Craig et al. 2004) and PeptideAtlas (Deutsch et al. 2008; Deutsch 2010), for evidence of such peptides using the default options of the X!Tandem search engine (Muth et al. 2010). We used the GENCODE annotation (version 3C) of the human genome (Harrow et al. 2006, The ENCODE Project Consortium 2007) as the set of known protein sequences and generated randomized decoy sets of the same size and composition as the GENCODE 3C and chimera search sets. We combined the experimental peptide-spectrum matches (PSM) found in each individual experiment in PeptideAtlas and GPM using the P-values generated by X!Tandem.
The combined P-values were used to rank the PSMs, and the simple FDR (number of decoys divided by number of correct matches) that could be estimated from the peptides in the decoy and GENCODE sets was corrected using a multiple testing method. We assigned q-values (the minimal FDR threshold at which a given peptide is accepted; Käll et al. 2009) to the PSM. Chimeric transcripts with expressed peptides corresponding to their gene–gene junction site confirmed by a PSM below the corrected FDR threshold (1% for all the cases confirmed by RNA-seq, or not) were considered potential true positives.
Shotgun proteomics experiments
To witness chimeric proteins we employed “bottom-up” shotgun proteomics using two-dimensional liquid chromatography coupled with high-resolution tandem mass spectrometry. The platform was operated in data independent mode as described in Levin et al. (2011).
Human cell lines
Three human cancer cell lines were subjected to proteomic analysis: the MCF7 human breast epithelial cell derived from mammary gland adenocarcinoma (HTB-22), the OVCAR-3 human epithelial cell line derived from ovary (HTB-161), and the DU-145 human epithelial carcinoma derived from prostate (HTB-81). The cells were prepared as explained in the Supplemental Methods.
Proteome sample preparation
Proteins in the cell lysates were reduced by addition of dithiolthreitol (Sigma; 5 mM) and incubation for 30 min at 60°C and then alkylated by addition of iodoacetemide (Sigma; 10 mM) and incubation in the dark for 30 min at 21°C. The proteins were then digested by incubation with trypsin (Promega) for 16 h at 37°C, added at a ratio of 1:50 (w/w trypsin/protein). Digestions were stopped by the addition of 1% trifluroacetic acid (TFA). Following digestion, detergents were removed using the Pierce Detergent Removal spin columns according to the manufacturer's procedure. The samples were stored at −80°C in aliquots.
Liquid chromatography–mass spectrometry
Digested protein (15 μg) from each sample was analyzed by nano-Ultra Performance Liquid Chromatography (10 kpsi nanoAcquity; Waters) in high-pH/low-pH reversed phase (RP) 2 dimensional liquid chromatography mode, coupled to high resolution, high mass accuracy mass spectrometry (Synapt G2 HDMS, Waters). The quadrupole ion mobility time-of-flight mass spectrometer was tuned to 20,000 mass resolution (full width at half height). Data were acquired in HDMSE positive ion mode in data independent acquisition (for further details see Supplemental Methods).
Bioinformatics procedure
Raw data processing and database searching was performed using Proteinlynx Global Server (IdentityE) version 2.5. Database searching was carried out using the Ion Accounting algorithm described by Li et al. (2009a). Briefly, the algorithm detects the 250 most abundant peptides and performs an initial pass through the database in order to identify these peptides (with mass tolerance of 7 ppm for precursor ions and 15 ppm for fragment ions). These 250 peptides were used to calibrate 14 predetermined models of specific, physicochemical attributes (such as retention time and fragmentation prediction, fragment to precursor ratios, etc.). These peptides are depleted from the database before the remaining peptides are sought in the database. The cycle continues to the next most abundant peptides, which are identified and then depleted from the database. The tentative peptide identifications are ranked and scored based on how well they conform to the 14 physiochemical models and reported in a final list. Trypsin was set as the protease, two missed cleavages were allowed, and fixed modification was set to carbamidomethylation of cysteines. Variable modification included oxidation of methionine.
The data set of combined human Swiss-Prot and all ESTs (translated in six frames) from ChimerDB (Kim et al. 2010) was employed. The criteria for protein identification were set to minimum of three fragments per peptide, five fragments per protein, and FDR < 1% (Keller et al. 2002; Nesvizhskii et al. 2003). The peptide score of 6.7 was estimated as a threshold for FDR of 1% using all sequences from our data set (“target set”) versus all reversed sequences (“decoy set”) (Supplemental Fig. S1). Finally, search results were imported into Scaffold v3.2 for manual inspection and reporting.
Targeted analysis in selective reaction monitoring mode (SRM)
The liquid chromatography mass spectrometry in SRM mode technique is widely used in proteomics for targeted analysis (Addona et al. 2009; Stergachis et al. 2011). The peptides were synthesized (JPT Peptide Technologies, http://www.jpt.com/) with heavy isotopic labels: C terminus R (15N6, 13C4) or C terminus K (15N6, 13C2) and added to the cell lysates prior to the analysis.
Sample preparation for SRM
An aliquot was taken from the digested samples outlined in the previous section. Samples were diluted to 0.5 μg/μL in 97:3% H2O:ACN+0.1% TFA.
Liquid chromatography
ULC/MS grade solvents were used for all chromatographic steps. Each sample was loaded using split-less nano-Ultra Performance Liquid Chromatography (10 kpsi nanoAcquity; Waters). The mobile phase was: (A) H2O + 0.1% formic acid and (B) ACN + 0.1% formic acid. Desalting of samples was performed online using a reverse-phase C18 trapping column (180 μm i.d., 20 mm length, 5 μm particle size; Waters). The peptides in samples were separated using a C18 T3 HSS nano-column (75 μm i.d., 150 mm length, 1.8 μm particle size; Waters) run at 0.4 μL/min. Peptides were eluted from the column and into the mass spectrometer using the following gradient: 3%–30%B over 40 min, 30%–95%B over 5 min, maintained at 95% for 7 min and then back to initial conditions.
Mass spectrometry analysis
The nanoLC was coupled online through a nanoESI emitter (7 cm length, 10 mm tip; New Objective) to a tandem quadrupole mass spectrometer (Xevo TQ-S, Waters). Data were acquired in SRM using Masslynx 4.1. Data were then imported into Skyline (Maclean et al. 2010a, MacLean et al. 2010b) for final processing and evaluation. Signal-to-noise ratio was calculated by root-mean-square in Masslynx software (Waters) with no extra processing. Minimum criteria were 5:1 signal to noise.
Signal peptides and transmembrane domain analysis
All chimeras translated in six frames were submitted to the SignalP 3.0 and TMHMM 2.0 servers (Emanuelsson et al. 2007). A chimera was considered as transmembrane (TM) if the TMHMM short report predicted one (or more) TM domain in at least one translated frame, and the chimera did not have a predicted signal peptide at the overlapping region of the TM domain. The summary outputs of SignalP 3.0 and TMHMM 2.0 are available in the Supplemental Material (Supplemental Data and http://chimera.bioinfo.cnio.es/).
Data access
The raw spectra identified for the three human cell lines and the Skyline projects that contain the SRM traces have been uploaded to Tranche (www.proteomecommons.org, Access password: chimera), using the hashes listed in the Supplemental Methods; for the cell lines: prostate cancer (HTB-81), breast cancer (HTB-22), ovary cancer (HTB-161). All the sequences of the chimeric RNAs presented in this study are ESTs or mRNAs from GenBank (http://www.ncbi.nlm.nih.gov/genbank) and can be found using ESTid listed for all the chimeras in the manuscript.
Acknowledgments
We thank Begoña Aguado, Alberto Rastrojo, and Eloy D. Hernández for help with the PCR analysis; Ricardo Ramos and Jose Pedro Borges for help in RT-PCR analysis; Sarah Djebali and David Gonzalez Knowles for their help in mapping the RNA-seq data; David Pisano Gonzalez for help with the GEO database; Jose Manuel Rodriguez for the ELM, SignalP, and TargetP scripts; Mark Wass for help with the Phyre tutorial; Susana Velasco and Eva Pilar Lospitao for the WI38 (human fibroblasts), MCF7 (breast cancer), and DU145 (prostate cancer) cell lines; and Federico Abascal, Miguel Vazquez, Edward Trifonov, Juan Cruz Cigudosa, Florian Leitner, and Dan Michaeli for valuable discussions. The authors also thank Professor Sanghyuk Lee and Professor Seokmin Shin for the availability of chimeric transcripts in the ChimerDB databases, and the ENCODE consortiums for the availability of the human genome annotation (GENCODE 3C). The work of M.F-M. is supported by the CNIO (Caja Navarra International Postdoctoral Program) and the Miguel Servet (FIS) grant. The work of V.L. is supported by the French ANR MIRI BLAN08-1335497 Project and the ERC Advanced Grant Sisyphe. This study is supported by the Spanish National Bioinformatics Institute (INB-ISCIII) and by grants from the Spanish Government (CONSOLIDER CSD2007-00050, BIO2007-666855, and BIO2011-26205), European Commission FP7 project ASSET (HEALTH-F4-2010-259348), and the NHGRI-NIH ENCODE grants (U54 HG00455-04 and U54 HG004557).
Author contributions: A.V. designed the study, interpreted the results, and wrote the manuscript. M.F-M. designed the study, collected the data, analyzed the data, interpreted the results, and wrote the manuscript. V.L. and R.G. performed the RNA-seq analysis and revised the manuscript. I.E., A.D.P., and M.T. searched the chimeras vs. PeptideAtlas; I.E. and M.T. revised the manuscript. J.P. analyzed the genomic alignments. R.J. performed the PCR analysis and revised the manuscript. Y.L. and A.G. performed the shotgun and SRM experiments; Y.L. interpreted the results and revised the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.130062.111.
Freely available online through the Genome Research Open Access option.
References
- Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, Spiegelman CH, Zimmerman LJ, Ham AJ, Keshishian H, et al. 2009. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol 27: 633–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiva P, Toporik A, Edelheit S, Peretz Y, Diber A, Shemesh R, Novik A, Sorek R 2006. Transcription-mediated gene fusion in the human genome. Genome Res 16: 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen MA, Hillier LW, Waterston RH, Blumenthal T 2011. A global analysis of C. elegans trans-splicing. Genome Res 21: 255–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S, Madden T, Schaffer A, Zhang J, Zhang Z, Miller W, Lipman D 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL 2005. GenBank. Nucleic Acids Res 33: D34–D38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candel AM, Cobos ES, Conejero-Lara F, Martinez JC 2009. Evaluation of folding co-operativity of a chimeric protein based on the molecular recognition between polyproline ligands and SH3 domains. Protein Eng Des Sel 22: 597–606 [DOI] [PubMed] [Google Scholar]
- Clérico EM, Maki JL, Gierasch LM 2008. Use of synthetic signal sequences to explore the protein export machinery. Biopolymers 90: 307–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AA, Geva-Zatorsky N, Eden E, Frenkel-Morgenstern M, Issaeva I, Sigal A, Milo R, Cohen-Saidon C, Liron Y, Kam Z, et al. 2008. Dynamic proteomics of individual cancer cells in response to a drug. Science 322: 1511–1516 [DOI] [PubMed] [Google Scholar]
- Craig R, Cortens JP, Beavis RC 2004. Open source system for analyzing, validating, and storing protein identification data. J Proteome Res 3: 1234–1242 [DOI] [PubMed] [Google Scholar]
- Denoeud F, Kapranov P, Ucla C, Frankish A, Castelo R, Drenkow J, Lagarde J, Alioto T, Manzano C, Chrast J, et al. 2007. Prominent use of distal 5′ transcription start sites and discovery of a large number of additional exons in ENCODE regions. Genome Res 17: 746–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW 2010. The PeptideAtlas Project. Methods Mol Biol 604: 285–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Lam H, Aebersold R 2008. PeptideAtlas: A resource for target selection for emerging targeted proteomics workflows. EMBO Rep 9: 429–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Kapranov P, Foissac S, Lagarde J, Reymond A, Ucla C, Wyss C, Drenkow J, Dumais E, Murray RR, et al. 2008. Efficient targeted transcript discovery via array-based normalization of RACE libraries. Nat Methods 5: 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Lagarde J, Kapranov P, Lacroix V, Borel C, Mudge J, Howald C, Foissac S, Ucla C, Chrast J, et al. 2012. Evidence for transcript networks composed of chimeric RNAs in human cells. PLoS ONE 7: e28213 doi: 10.1371/journal.pone.0028213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douris V, Telford MJ, Averof M 2010. Evidence for multiple independent origins of trans-splicing in Metazoa. Mol Biol Evol 27: 684–693 [DOI] [PubMed] [Google Scholar]
- Edgren H, Murumagi A, Kangaspeska S, Nicorici D, Hongisto V, Kleivi K, Rye IH, Nyberg S, Wolf M, Borresen-Dale AL, et al. 2011. Identification of fusion genes in breast cancer by paired-end RNA-sequencing. Genome Biol 12: R6 doi: 10.1186/gb-2011-12-1-r6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi M, Eguchi-Ishimae M, Knight D, Kearney L, Slany R, Greaves M 2006. MLL chimeric protein activation renders cells vulnerable to chromosomal damage: An explanation for the very short latency of infant leukemia. Genes Chromosomes Cancer 45: 754–760 [DOI] [PubMed] [Google Scholar]
- Elizabeth Cha I, Rouchka EC 2005. Comparison of current BLAST software on nucleotide sequences. IPDPS 19: 8 doi: 10.1109/IPDPS.2005.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Brunak S, von Heijne G, Nielsen H 2007. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc 2: 953–971 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel-Morgenstern M, Cohen AA, Geva-Zatorsky N, Eden E, Prilusky J, Issaeva I, Sigal A, Cohen-Saidon C, Liron Y, Cohen L, et al. 2010. Dynamic Proteomics: A database for dynamics and localizations of endogenous fluorescently-tagged proteins in living human cells. Nucleic Acids Res 38: D508–D512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingeras TR 2009. Implications of chimaeric non-co-linear transcripts. Nature 461: 206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigó R, Flicek P, Abril JF, Reymond A, Lagarde J, Denoeud F, Antonarakis S, Ashburner M, Bajic VB, Birney E et al. 2006. EGASP: The human ENCODE Genome Annotation Assessment Project. Genome Biol 7 (Suppl 1): S2 doi: 10.1186/gb-2006-7-s1-s2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Denoeud F, Frankish A, Reymond A, Chen CK, Chrast J, Lagarde J, Gilbert JG, Storey R, Swarbreck D, et al. 2006. GENCODE: Producing a reference annotation for ENCODE. Genome Biol 7 (Suppl 1): S4 doi: 10.1186/gb-2006-7-s1-s4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, Teichmann SA 2011. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol 7: 497 doi: 10.1038/msb.2011.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herai RH, Yamagishi ME 2010. Detection of human interchromosomal trans-splicing in sequence databanks. Brief Bioinform 11: 198–209 [DOI] [PubMed] [Google Scholar]
- Horiuchi T, Aigaki T 2006. Alternative trans-splicing: A novel mode of pre-mRNA processing. Biol Cell 98: 135–140 [DOI] [PubMed] [Google Scholar]
- Houseley J, Tollervey D 2010. Apparent non-canonical trans-splicing is generated by reverse transcriptase in vitro. PLoS ONE 5: e12271 doi: 10.1371/journal.pone.0012271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Storey JD, Noble WS 2009. QVALITY: Non-parametric estimation of q-values and posterior error probabilities. Bioinformatics 25: 964–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan K, Wang L, Wang J, Ittmann MM, Li W, Yen L 2011. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proc Natl Acad Sci 108: 9172–9177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74: 5383–5392 [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ 2009. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc 4: 363–371 [DOI] [PubMed] [Google Scholar]
- Kent WJ 2002. BLAT- the BLAST-like alignment tool. Genome Res 12: 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim P, Yoon S, Kim N, Lee S, Ko M, Lee H, Kang H, Kim J 2010. ChimerDB 2.0—a knowledgebase for fusion genes updated. Nucleic Acids Res 38: D81–D85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klisch TJ, Xi Y, Flora A, Wang L, Li W, Zoghbi HY 2011. In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proc Natl Acad Sci 108: 3288–3293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Zhu J, Wu J, Peng J, Wang Y, Wang Q, Fu S, Yuan LL, Li T 2011. dbCRID: A database of chromosomal rearrangements in human diseases. Nucleic Acids Res 39: D895–D900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin Y, Hradetzky E, Bahn S 2011. Quantification of proteins using data-independent analysis (MSE) in simple and complex samples: A systematic evaluation. Proteomics 11: 3273–3287 [DOI] [PubMed] [Google Scholar]
- Li H, Wang J, Mor G, Sklar J 2008. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 321: 1357–1361 [DOI] [PubMed] [Google Scholar]
- Li GZ, Vissers JP, Silva JC, Golick D, Gorenstein MV, Geromanos SJ 2009a. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 9: 1696–1719 [DOI] [PubMed] [Google Scholar]
- Li H, Wang J, Ma X, Sklar J 2009b. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle 8: 218–222 [DOI] [PubMed] [Google Scholar]
- Li X, Zhao L, Jiang H, Wang W 2009c. Short homologous sequences are strongly associated with the generation of chimeric RNAs in eukaryotes. J Mol Evol 68: 56–65 [DOI] [PubMed] [Google Scholar]
- Lopez R, Silventoinen V, Robinson S, Kibria A, Gish W 2003. WU-Blast2 server at the European Bioinformatics Institute. Nucleic Acids Res 31: 3795–3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ 2010a. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem 82: 10116–10124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ 2010b. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26: 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Horikoshi T, Nakashima E, Miyamoto Y, Mabuchi A, Ikegawa S 2005. MATN and LAPTM are parts of larger transcription units produced by intergenic splicing: Intergenic splicing may be a common phenomenon. DNA Res 12: 365–372 [DOI] [PubMed] [Google Scholar]
- Magrane M, UniProt Consortium 2011. UniProt Knowledgebase: A hub of integrated protein data. Database (Oxford) 2011: bar009 doi: 10.1093/database/bar009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM 2009a. Transcriptome sequencing to detect gene fusions in cancer. Nature 458: 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S, Khrebtukova I, Barrette TR, Grasso C, Yu J, et al. 2009b. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci 106: 12353–12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Coolon JD, Duff MO, Eipper-Mains J, Graveley BR, Wittkopp PJ 2010a. Regulatory divergence in Drosophila revealed by mRNA-seq. Genome Res 20: 816–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus CJ, Duff MO, Eipper-Mains J, Graveley BR 2010b. Global analysis of trans-splicing in Drosophila. Proc Natl Acad Sci 107: 12975–12979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitani K 2004. Molecular mechanisms of leukemogenesis by AML1/EVI-1. Oncogene 23: 4263–4269 [DOI] [PubMed] [Google Scholar]
- Miura TA, Li H, Morris K, Ryan S, Hembre K, Cook JL, Routes JM 2004. Expression of an E1A/E7 chimeric protein sensitizes tumor cells to killing by activated macrophages but not NK cells. J Virol 78: 4646–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Muth T, Vaudel M, Barsnes H, Martens L, Sickmann A 2010. XTandem Parser: An open-source library to parse and analyse X!Tandem MS/MS search results. Proteomics 10: 1522–1524 [DOI] [PubMed] [Google Scholar]
- Nambiar M, Kari V, Raghavan SC 2008. Chromosomal translocations in cancer. Biochim Biophys Acta 1786: 139–152 [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, Aebersold R 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75: 4646–4658 [DOI] [PubMed] [Google Scholar]
- Panagopoulos I, Isaksson M, Lindvall C, Hagemeijer A, Mitelman F, Johansson B 2003. Genomic characterization of MOZ/CBP and CBP/MOZ chimeras in acute myeloid leukemia suggests the involvement of a damage-repair mechanism in the origin of the t(8;16)(p11;p13). Genes Chromosomes Cancer 36: 90–98 [DOI] [PubMed] [Google Scholar]
- Parra G, Reymond A, Dabbouseh N, Dermitzakis ET, Castelo R, Thomson TM, Antonarakis SE, Guigó R 2006. Tandem chimerism as a means to increase protein complexity in the human genome. Genome Res 16: 37–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson WR, Lipman DJ 1988. Improved tools for biological sequence comparison. Proc Natl Acad Sci 85: 2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt J, Harrison N, Stansfield I, Connolly B, Müller B 2010. The evolution of spliced leader trans-splicing in nematodes. Biochem Soc Trans 38: 1125–1130 [DOI] [PubMed] [Google Scholar]
- Pirrotta V 2002. Trans-splicing in Drosophila. Bioessays 24: 988–991 [DOI] [PubMed] [Google Scholar]
- Pradet-Balade B, Medema JP, López-Fraga M, Lozano JC, Kolfschoten GM, Picard A, Martínez-A C, Garcia-Sanz JA, Hahne M 2002. An endogenous hybrid mRNA encodes TWE-PRIL, a functional cell surface TWEAK-APRIL fusion protein. EMBO J 21: 5711–5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbitts TH 1994. Chromosomal translocations in human cancer. Nature 372: 143–149 [DOI] [PubMed] [Google Scholar]
- Robertson HM, Navik JA, Walden KK, Honegger HW 2007. The bursicon gene in mosquitoes: An unusual example of mRNA trans-splicing. Genetics 176: 1351–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Dreszer TR, Long JC, Malladi VS, Sloan CA, Raney BJ, Cline MS, Karolchik D, Barber GP, Clawson H, et al. 2012. ENCODE whole-genome data in the UCSC Genome Browser: Update 2012. Nucleic Acids Res 40: D912–D917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ 2005. Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol 6: R33 doi: 10.1186/gb-2005-6-4-r33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberg JJ, Nguyen PQ, Stevenson T 2010. Computational design of chimeric protein libraries for directed evolution. Methods Mol Biol 673: 175–188 [DOI] [PubMed] [Google Scholar]
- Stergachis AB, Maclean B, Lee K, Stamatoyannopoulos JA, Maccoss MJ 2011. Rapid empirical discovery of optimal peptides for targeted proteomics. Nat Methods 8: 1041–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson TM, Lozano JJ, Loukili N, Carrió R, Serras F, Cormand B, Valeri M, Díaz VM, Abril J, Burset M, et al. 2000. Fusion of the human gene for the polyubiquitination coeffector UEV1 with Kua, a newly identified gene. Genome Res 10: 1743–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress ML, Martelli PL, Frankish A, Reeves GA, Wesselink JJ, Yeats C, Olason PI, Albrecht M, Hegyi H, Giorgetti A, et al. 2007. The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci 104: 5495–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]