

Abstract
Ionizing radiation has long been known to induce heritable mutagenic change in DNA sequence. However, the genome-wide effect of radiation is not well understood. Here we report the molecular properties and frequency of mutations in phenotypically selected mutant lines isolated following exposure of the genetic model flowering plant Arabidopsis thaliana to fast neutrons (FNs). Previous studies suggested that FNs predominantly induce deletions longer than a kilobase in A. thaliana. However, we found a higher frequency of single base substitution than deletion mutations. While the overall frequency and molecular spectrum of fast-neutron (FN)–induced single base substitutions differed substantially from those of “background” mutations arising spontaneously in laboratory-grown plants, G:C>A:T transitions were favored in both. We found that FN-induced G:C>A:T transitions were concentrated at pyrimidine dinucleotide sites, suggesting that FNs promote the formation of mutational covalent linkages between adjacent pyrimidine residues. In addition, we found that FNs induced more single base than large deletions, and that these single base deletions were possibly caused by replication slippage. Our observations provide an initial picture of the genome-wide molecular profile of mutations induced in A. thaliana by FN irradiation and are particularly informative of the nature and extent of genome-wide mutation in lines selected on the basis of mutant phenotypes from FN-mutagenized A. thaliana populations.
Ionizing radiation is pervasive in the environment and acts as a natural mutagen via its DNA-damaging properties (Friedberg et al. 2006). In addition, the mutagenic property of artificial ionizing radiation has been a mainstay of genetic research since the pioneering experiments of Müller (1928). However, while the effects of ionizing radiation on individual genes are now relatively well understood, its genome-wide effects are not. We therefore undertook an analysis of the genome-wide consequences of fast neutron (FN) irradiation in Arabidopsis thaliana, comparing our findings with those of recent studies documenting the frequency and molecular spectrum of spontaneous “background” mutations in the genomes of laboratory-grown “mutation accumulation” (MA) line Arabidopsis plants (Ossowski et al. 2010) and of Arabidopsis plants regenerated in vitro from root tissue explants (Jiang et al. 2011). We found that exposure of Arabidopsis to FNs induces a broader range of mutational lesions than previously suspected, and that both the incidence and spectrum of FN-induced mutations are distinct from those of “spontaneous” mutations. These discoveries have important consequences for the use of FNs in experimental plant mutagenesis and provide an indication of the likely mutagenic effects of environmental ionizing radiation on organisms living in the wild (Hinton et al. 2007).
Results
FN irradiation, mutant generation, and isolation
Our analyses began with a multiply mutant Arabidopsis line [predominantly Landsberg erecta (Ler) background carrying the mutations gai-t6, rga-t2, rgl1-1, rgl2-1, and rgl3-4; henceforth called progenitor] (see Methods) that lacks the growth-regulating DELLA proteins. Progenitor (M1) seeds were exposed to FN irradiation (see Methods), with the long-term aim (although not the focus of this study) of isolating and characterizing novel growth-regulation mutants in a DELLA-deficient genetic background. Accordingly, M2 seedlings were screened for elongated hypocotyls, a mutant phenotype in which exaggerated growth of the hypocotyl (embryonic stem structure) results in a longer hypocotyl than that of progenitor controls. This class of mutant commonly contains loss-of-function mutations in genes that mediate the light inhibition of hypocotyl growth (Chen et al. 2004). We identified more than 300 independent putative elongated hypocotyl mutants and selected six of these (E71, E99, E125, E128, E138, and E216) to confirm heritability and for analysis in subsequent generations.
Creation of a progenitor reference sequence and alignment of progenitor reads to Col-0 reference
To identify mutations in the genomes of the six selected elongated hypocotyl mutants, we first needed to obtain a whole-genome reference sequence of the progenitor from which they were derived. We therefore obtained Illumina Genome Analyzer DNA sequence data from a single progenitor line plant and used these data to create a progenitor reference genome (see Methods). Initial mapping of the progenitor paired-end sequencing reads to the Arabidopsis Information Resource (TAIR 9) (http://www.arabidopsis.org) Col-0 reference genome (using MAQ v0.7.1) (Li et al. 2008) revealed single nucleotide polymorphisms (SNPs) and insertions and deletions (INDELs). This observation is consistent with the fact that only 3.2 Mb of the ∼120-Mb progenitor genome is derived from Col-0 (Fig. 1), while the remaining ∼97% of the genome is of Ler origin. To eliminate the progenitor versus TAIR 9 variants observed and to aid the identification of the genetic lesions caused by FN mutagenesis, we created our own progenitor reference genome sequence (see Methods).
Figure 1.

Density of single nucleotide polymorphisms (SNPs) detected following alignment of (A) A. thaliana Landsberg erecta (Gan et al. 2011) and (B) progenitor reads to the Col-0 (TAIR 9) reference genome sequence. Histograms of SNPs along the length of chromosome 5 are shown. The Col-0 segment of progenitor chromosome 5 (containing the rgl3-4 T-DNA insertion) is clearly visible in (B), between 3 Mb and 6.25 Mb, where the number of variants (versus TAIR 9) falls close to baseline. The site of the RGL3 locus is shown in the Col-0 DNA segment (in gray; see inset).
Detection of mutations in FN-induced mutant lineages
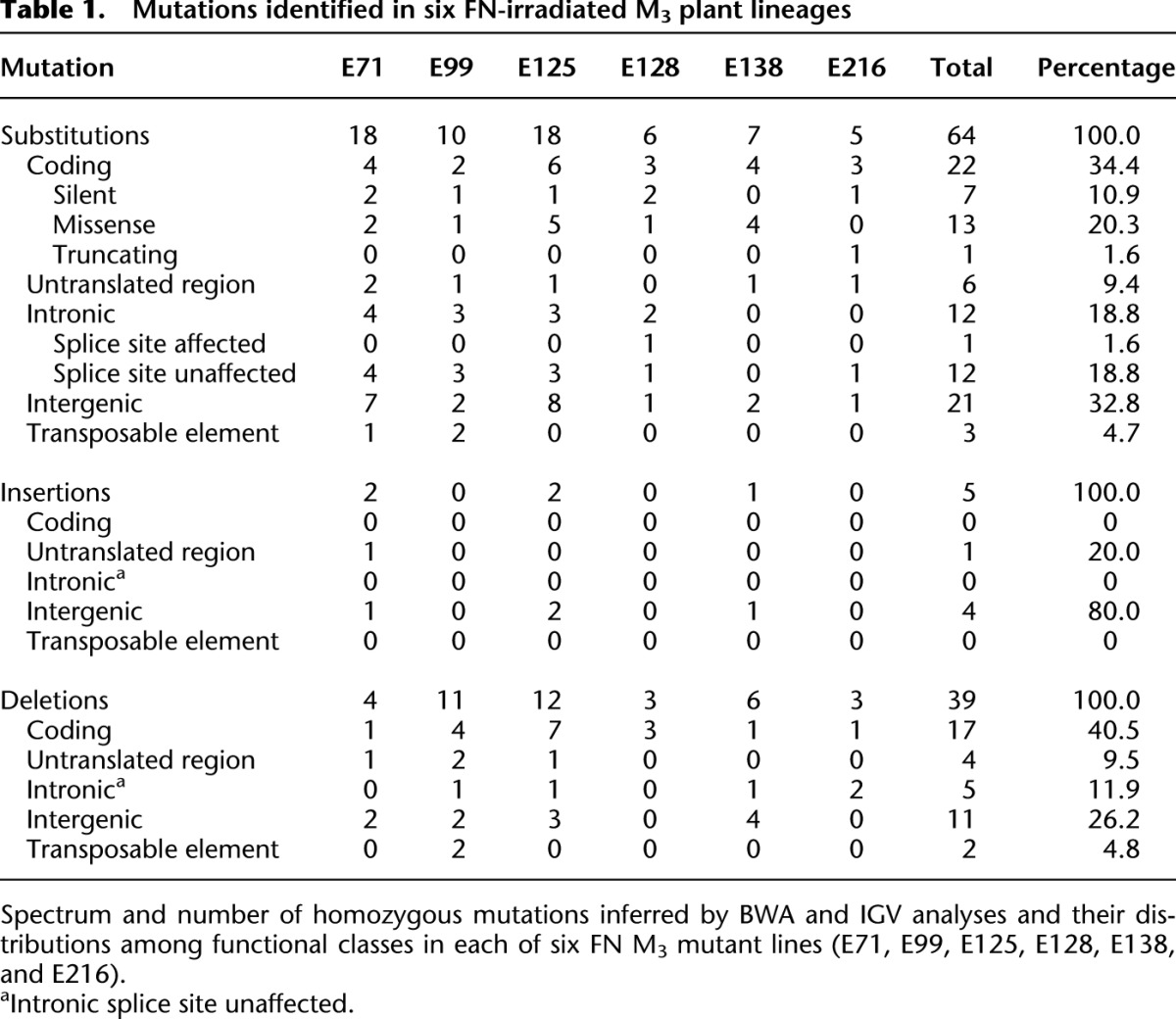
We next generated Illumina Genome Analyzer DNA sequence data from single M3 plants homozygous for the phenotype-conferring mutations in the six selected mutant lineages. We were able to identify a near-complete set of homozygous DNA sequence variants (versus the progenitor reference) in the nonrepetitive fractions of these mutant genomes. A mutation (base substitution or INDEL) was called if 95%–100% of aligned reads in a sample differed from the progenitor reference sequence (see Methods). We detected a total of 108 homozygous mutations (see Fig. 2; Table 1; Supplemental Table 1). Of the 56 variants detected in lines E99 and E125, all but one was independently confirmed by direct PCR amplification and Sanger sequencing, indicating that our false-positive discovery rate was negligible. The unconfirmed variant in E125 was a G:C>A:T substitution that was difficult to confirm because it lies in a region of sequence that is represented repetitively in the Arabidopsis genome.
Figure 2.

Distribution across chromosomes of 108 mutations detected in genomes of six FN-exposed M3 Arabidopsis lineages. The labels indicate the type of mutation, and color their functional class or predicted consequence. Small INDELs are indicated by base-designating letters with a preceding plus or minus sign. Large deletions are indicated by a minus sign (with the number of deleted base pairs). Individual colors define intergenic region (red); intron (yellow); nonsynonymous substitution, shift of reading frame for short INDELs, or gene deletion for large deletions (blue); synonymous substitution (green); untranslated region (purple); transposable element (white). (A) E71; (B) E99; (C) E125; (D) E128; (E) E138; (F) E216. The gene symbols represent likely phenotype causal mutations conferring increased hypocotyl length: hy5 (Ang et al. 1998), hy1 (Davis et al. 1999), frs2 (Lin and Wang 2004), col2 (Ledger et al. 2001), hy2 (Kohchi et al. 2001), cry1 (Ahmad et al. 1998), and phyB (Somers et al. 1991).
Table 1.
Mutations identified in six FN-irradiated M3 plant lineages
Estimation of original numbers of mutations in FN-irradiated plants
While the above 108 homozygous mutations were detected in M3 plant genomes, they had originally been induced by FN irradiation as heterozygous mutations in M1 plant genomes (Fig. 3). We therefore calculated the original mutation numbers in the following way. Assuming recessivity of the initial elongated hypocotyl mutation, each original M2 mutant seedling would have been homozygous for the mutation causing that phenotype (and any neighboring mutations) (see Fig. 3). In addition, there was an approximately one-fourth chance of the original M2 mutant seedling being homozygous for induced mutations (of unknown phenotypic consequence) in other regions of the genome. These regions of “fixed” homozygosity would have been uniformly transmitted to the subsequent M3 generation. Further mutations, heterozygous in the original M2 seedling, will have been segregating in M3 seedlings (the genome of one of which was sequenced). Thus, of the total 108 detected homozygous M3 mutations (Supplemental Table 1), 72 were likely homozygous in the M2, with the remaining quarter (36) resulting from an estimated 144 variants that had been heterozygous in the M2 and had then become homozygous in the M3 (see Fig. 3). Mutations heterozygous in the M3 generation were overwhelmingly likely to have been discarded from variant lists (and thus not detected) because of failure to reach the 95%–100% read difference criterion described above. In summary, we estimate that the 108 homozygous mutations detected in M3 plants (Fig. 2) imply an original number of ∼288 heterozygous FN-induced mutations in M1 plants (Fig. 3).
Figure 3.

Estimation of original numbers of mutations in M1 plants. M1 seeds were irradiated with FNs, germinated, and resultant plants self-pollinated to yield M2 seed. Elongated hypocotyl mutants were identified in the M2 and self-pollinated to yield M3 seed. The genomes of six mutant M3 plants were sequenced. Mendelian segregation laws were used to estimate the number of variants, both homozygous (Homo) and heterozygous (Het), in the generations preceding the six genome-sequenced FN M3 mutant lines. As shown, all homozygous mutations “fixed” in the M2 (in red) will have remained homozygous in the M3. In addition, a quarter of M2 heterozygous mutations (144; blue text) will have segregated in a Mendelian 1:2:1 (homozygous mutation:heterozygous mutation:homozygous nonmutant) ratio, with approximately one-quarter becoming homozygous mutations (36; blue text) in the M3. Thus, a total of 108 homozygous variants (detected by our methods) along with a further predicted 72 segregating heterozygous variants (not detected by our methods) are estimated for the M3. To summarize, in the original FN-exposed M1 plants, a total of 288 (black text) heterozygous mutations are estimated to have arisen. One-quarter of these mutations will have become homozygous in the M2 (72; red text), half becoming heterozygous (144; blue text), with the remaining quarter being homozygous nonmutant. These considerations permit calculation (see Supplemental Table 5) of FN-induced mutation rates.
Detection of spontaneous mutations in the progenitor line
The progenitor line lacks the five growth-regulating DELLA proteins and is substantially derived from the Ler genetic background. To be sure that the mutations detected in E71, E99, E125, E128, E138, and E216 were genuinely FN-induced and were not simply of spontaneous origin, we determined the frequency of spontaneous mutation in the progenitor line. We sequenced the genomes of single plants from three independent control progenitor lineages. Each of these three plants (F2-1 to -3) was an F2 generation plant (the product of two successive self-pollination generations) derived from a control, nonirradiated progenitor plant (and hence the control equivalent of the M2) (Fig. 3). Using our above-described mutation detection methods, we identified only one single mutation in these three genomes (an A:T>C:G substitution in the F2-2 line) (Supplemental Table 2).
In a separate determination of the frequency of spontaneous mutation in the progenitor, we sequenced the genomes of two individual M4 plants (progeny of E99 and E125 genome-sequenced M3 lines) and compared the mutations detected in the M3 with those detected in the M4 (Supplemental Fig. 1). While all mutations previously detected in the M3 were also detected in the M4, a total of seven previously undetected mutations were additionally detected in the E99 and E125 M4 data sets (Supplemental Table 3). Because these additionally detected mutations may have become detectable (homozygous) in the M4 having been undetectable (heterozygous) in the M3, we used Mendelian segregation laws (as in Fig. 3) to predict that there should have been 8.8 such newly detected homozygous variants, a figure slightly higher than the number (seven) actually observed (Supplemental Fig. 1; Supplemental Tables 3, 4). Thus spontaneous mutations do not significantly inflate the number of detected mutations in M4 plants beyond that predicted on the basis of Mendelian segregation of FN-induced heterozygous mutations in M3 plants.
These two separate control experiments indicate that the frequency of spontaneous mutations in the multiple DELLA knockout progenitor line is comparable to that previously observed in the Col-0 wild-type (WT) line (Ossowski et al. 2010). Because the frequency of mutations in FN-irradiated lineages (Figs. 2, 3) is one to two orders of magnitude higher than in the control lineages, we conclude that the mutations detected in our FN-irradiated lineages are indeed substantially caused by FN irradiation.
Chromosomal distribution and frequencies of FN-induced mutations
The 108 detected FN-induced mutations (Fig. 2) ranged in size from single base substitutions (SBSs) and insertions to deletions of one or two bases, to larger deletions (3 bases to 1 kb or greater) (Table 1; Supplemental Table 1). The distribution of mutations across chromosomes in each of the six lineages is shown in Figure 2. The number of mutations per line ranged from eight to 32 (Fig. 2; Table 1). These differences are significant (G-test, P = 1.7 × 10−4) and may reflect heterogeneity in radiation dosage or in water content of irradiated seeds (seed radio-sensitivity is dependent on water content) (Kamra et al. 1960).
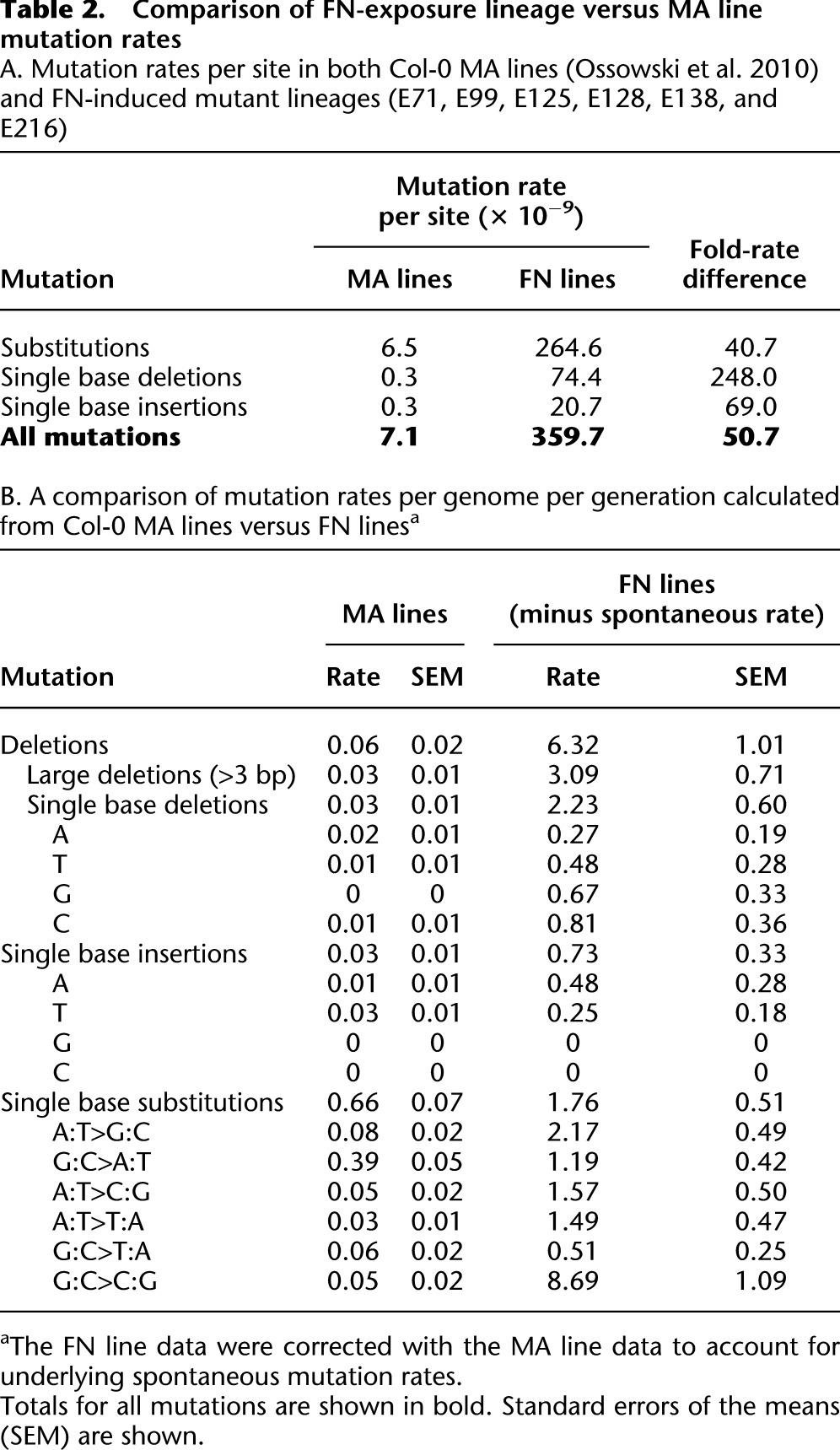
FN exposure increases both overall mutation frequency and the frequency of specific molecular mutation classes. Taking all detected single base mutations together (SBSs and INDELs), we estimated the mutation rate to be 359.7 × 10−9 per site in the irradiated generation following correction for underlying spontaneous mutations (Table 2A; Supplemental Table 5). In essence, a 60-Gy FN dose caused a transient single-generational ∼50-fold increase in mutation rate above the spontaneous rate (7.1 × 10−9 per site per generation) (Ossowski et al. 2010).
Table 2.
Comparison of FN-exposure lineage versus MA line mutation rates A. Mutation rates per site in both Col-0 MA lines (Ossowski et al. 2010) and FN-induced mutant lineages (E71, E99, E125, E128, E138, and E216)
On a genome-wide scale, 35% of the 108 detected FN mutations were in genic regions (see Supplemental Fig. 2), which is higher than the expected 33% but not significant (G-test, P = 0.6). This observation suggests that mutations are not preferentially located in gene-coding, gene regulatory, or intergenic regions (Fig. 2; Supplemental Table 1), despite previous indications of mutational bias associated with chromatin structural difference (Prendergast et al. 2007) and the possibility that local differences in chromatin architecture might differentially affect DNA repair mechanisms, and hence localized mutation rate.
FN-induced INDEL mutations
Although FN exposure was previously thought to induce a predominance of deletion mutations in plants (Bruggemann et al. 1996; Li et al. 2001), we, in fact, detected substantial impact on the relative frequencies of a range of specific mutational classes. Among INDELs, we detected 19 large deletions (>3 bp), ranging in size from 4 bp to 7.2 kb, and six smaller deletions (2–3 bp) (Fig. 4A) in our FN-exposed lineages. In addition, we detected 14 single base deletions, making this the most prominent class of deletion mutation detected (Fig. 4A), unlike previous suggestions that 2- to 4-kb deletions are the most frequent class of FN-induced deletion mutation (Bruggemann et al. 1996; Li et al. 2001). We also detected five single base insertions. The latter were all A or T insertions, suggesting a possible bias against G or C insertions and consistent with an apparent bias in favor of A:T composition (Table 2B) as previously reported in Col-0 MA lines (Ossowski et al. 2010).
Figure 4.

Molecular spectrum of FN-induced mutations. (A) Frequencies and sizes of deletion mutations identified in the six FN-exposed mutant lineages. (B,C) SBS rates per A:T or G:C site and transition/transversion (Ti/Tv) ratios in (B) Col-0 MA lines (Lynch 2010; Ossowski et al. 2010) and (C) FN-exposed lineages. Complementary mutations, e.g., A>C and T>G, are pooled. The Ti/Tv ratio for each profile is shown. The mutation rate is the mutation rate weighted by the incidence of the base in the Arabidopsis Col-0 genome.
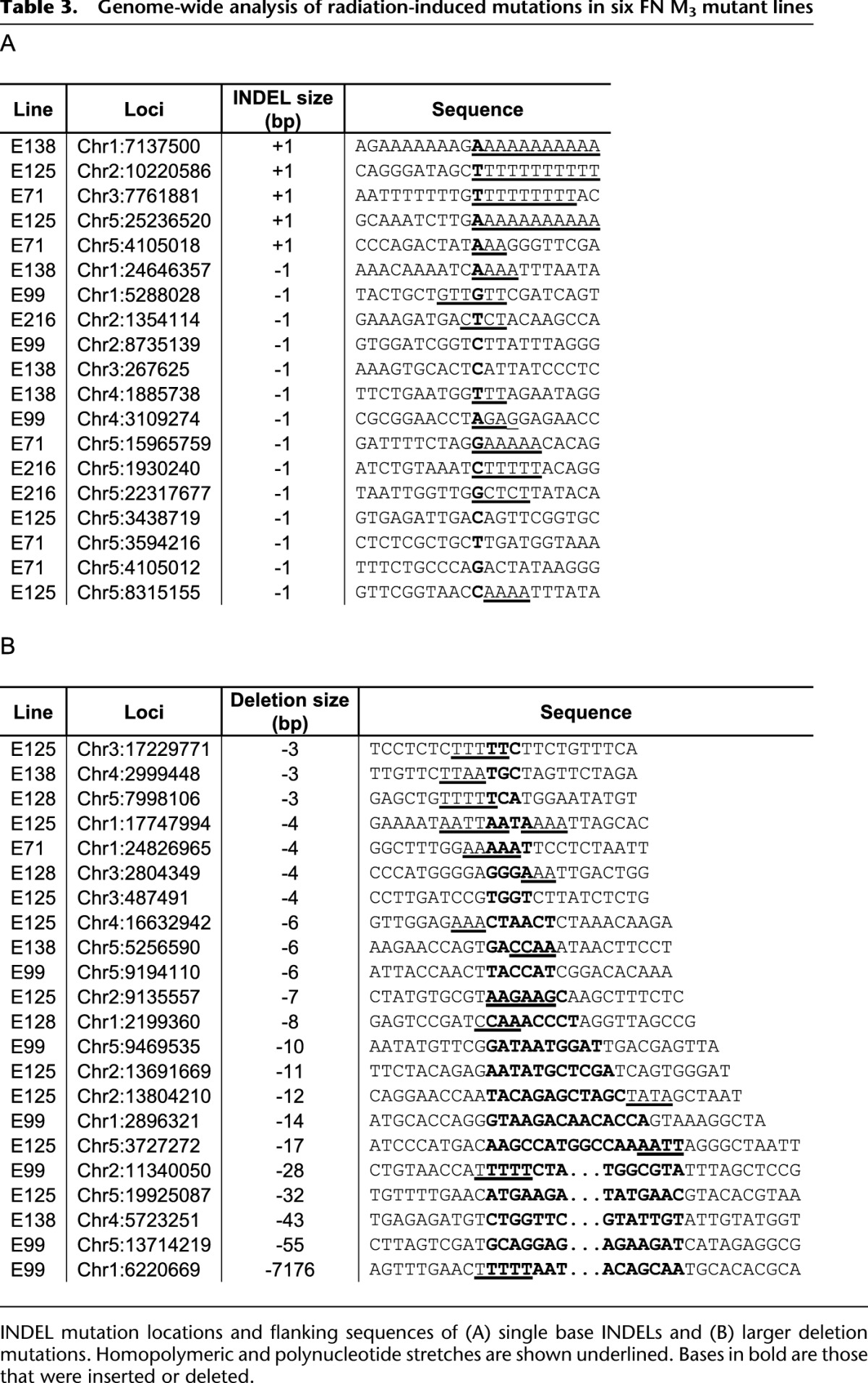
Single base deletion and insertion mutations are often caused by replication slippage at homopolymer or polynucleotide repeat regions (Viguera et al. 2001). Consistent with this, of the 19 FN-induced single base INDELs, all five insertions and nine of the 14 (64%) deletions either occurred within or were adjacent to homopolymeric or polynucleotide repeats (Table 3A). In addition, six of the seven 3-bp or 4-bp deletions were located within adenine or thymine homopolymeric or polynucleotide repeats (although larger FN-induced deletions appear less likely to be associated with mononucleotide or dinucleotide microsatellites; seven of 15; 47%) (Table 3B). These observations suggest the possibility that ionizing radiation may promote the incidence of DNA replication slippage.
Table 3.
Genome-wide analysis of radiation-induced mutations in six FN M3 mutant lines
Ionizing radiation frequently generates gross chromosomal aberration in plants (interstitial inversions, translocations, etc.) (Shikazono et al. 2005). To identify such events, we searched for “distant-pair” signatures (as previously described) (Korbel et al. 2007; Gan et al. 2011; Jiang et al. 2011), where a pair of reads from either end of a genome sequencing fragment that straddles a “breakpoint” (inversion or translocation chromosomal aberration) with respect to the progenitor reference genome align at an unexpected distance apart on that reference. Despite exhaustive searches, we did not detect any distant pairs, indicating the absence of gross chromosomal aberration in our six post-irradiation lineages. However, analysis of a further FN-induced M3 mutant line, E88 (full mutation profile not included in this study), detected a chromosomal rearrangement mutation (an interstitial inversion of ∼215 kb), thus indicating that our methods are robust (Supplemental Fig. 3A). In addition, using our standard methods, an ∼21-kb deletion (larger than observed in the original six FN M3 lines) was identified in the E88 M3 line (Supplemental Fig. 3B).
FN-induced single base substitutions
Exposure to FNs also induced a relatively high frequency of single base substitutions (SBSs). SBSs can be transitions (purine > purine; pyrimidine > pyrimidine) or transversions (purine > pyrimidine; pyrimidine > purine), and FNs induced both types of substitution (Fig. 4B,C; Supplemental Tables 1, 6). C>T transitions were the most common substitution in MA lines (Fig. 4B; Ossowski et al. 2010), consistent with C being the most spontaneously mutable base, and were also prominent in FN-exposed lineages (Fig. 4C). Another form of radiation, UV, is known to induce the formation of covalent linkages between adjacent pyrimidine residues (where a C or T is adjacent to another C or T) in DNA sequence, resulting in a predominance of UV-induced C>T mutations at dipyrimidine sequences (Daya-Grosjean and Sarasin 2005). We identified 64 FN-induced SBSs of which 51 (80%) were at pyrimidine dinucleotide sites, which is more than expected by chance (Fisher’s exact test, P = 1.9 × 10−6) (Supplemental Tables 1, 6). Of these SBSs, 20 were C>T mutations and 14 of these (70%) were at pyrimidine dinucleotide sites (Supplemental Table 6); this was not found to be significant (Fisher’s exact test, P = 0.1). We conclude that FN exposure induces C>T transitions (and other SBSs) via sequence context-dependent mechanisms preferentially targeting pyrimidine dinucleotides. While previously associated particularly with UV exposure, our results suggest that pyrimidine dinucleotide-associated C>T transitions are actually a feature of exposure to a wider range of radiation classes.
Another prominent class of FN-induced substitution was the G>T transversion (see Supplemental Table 1). Radiation-induced DNA damage is often attributed to the effects of reactive oxygen species (ROS) and hydroxyl radicals generated when water molecules absorb high-energy particles. In particular, ROS are thought to damage guanine residues, thus precipitating G>T mutations (Kawanishi et al. 2001). Our observation of a relatively high frequency of these transversions is thus consistent with these arising through FN-induced ROS-mediated DNA damage.
Overall, the FN-induced SBS mutational spectrum is very different from that of spontaneous SBSs seen in MA lines (Fig. 4B,C; Ossowski et al. 2010). Transversions are relatively more frequent in FN-induced than in spontaneous SBSs, resulting in a much reduced transition/transversion (Ti/Tv) ratio (∼1 for FN-induced mutations versus ∼3 for spontaneous mutations) (Fig. 4B,C). Interestingly, a relatively low Ti/Tv ratio is also characteristic of mutations observed in in vitro regenerant plant lineages (Jiang et al. 2011).
Mutations in the six FN M3 lines likely to confer the elongated hypocotyl phenotype
In each of the six selected M3 lines, we were able to identify the mutation that likely confers the mutant elongated hypocotyl phenotype (mutations in genes HY1, HY2, HY5, FRS2, COL2, CRY1, and PHYB; see below) (Fig. 2). In each line a within-gene deletion or nonsynonymous mutation (unique to that line) presumably confers mutant phenotype, with all other mutations likely having no significant effect on the phenotype. Bioinformatic analyses of the six FN elongated hypocotyl mutant lineages indicated that the likely phenotypic causal mutations were as follows: E71: a 1-bp deletion in an exon of HY5 (a gene encoding a bZIP transcription factor that is a positive regulator of photomorphogenesis) (Ang et al. 1998); E99: a 28-bp deletion that spans part of the promoter, start codon, and first exon of HY1 (a gene that encodes a plastid heme oxygenase necessary for phytochrome chromophore biosynthesis) (Davis et al. 1999); E125: an 11-bp deletion in an exon of FRS (a gene that encodes a protein essential for phytochrome A–controlled far-red light responses) (Lin and Wang 2004) and a 4-bp deletion in an exon of COL2 (a gene that encodes a zinc finger protein with ∼67% amino acid identity to the protein encoded by the flowering-time gene CONSTANS) (Ledger et al. 2001); E128: a 4-bp deletion in HY2 (which like HY1, is necessary for phytochrome chromophore biosynthesis) (Kohchi et al. 2001); E138: a 43-bp deletion in CRY1 (a gene that encodes a cryptochrome blue light photoreceptor) (Ahmad et al. 1998); and E216: a 1-bp nucleotide substitution in an exon of PHYB (a gene encoding a phytochrome red light photoreceptor) (Somers et al. 1991).
Discussion
This study reveals for the first time the genome-wide consequences of irradiation on plant genomes, and the spectrum and rate of mutations that researchers should expect to find in irradiation-mutagenized populations. In contrast to previous reports, we find that FN-induced single base substitutions (SBSs) are more prevalent than FN-induced INDELs (Table 1), and that short deletions <3 bp are more prevalent than larger-scale deletions (Fig. 4A). Among FN-induced SBSs, we detected a bias toward G:C>A:T transitions at dipyrimidine sites and a higher overall frequency of FN-induced transversions versus transitions (with respect to spontaneous SBSs found in MA lines) (Fig. 4B,C).
Overall, the molecular mutational spectrum of FN-induced SBSs is distinct from that of the spontaneous SBSs found in MA lines (Fig. 4B,C). Since MA lines are grown in conditions of relative shelter from environmental mutagenic agents, it is likely that any differences in mutational spectra reflect exposure to a mutagenic environment (in this case, a FN bombardment beam), and imply that the increased frequency of mutations due to FN exposure is not simply due to an accelerated occurrence of the same classes of spontaneous mutations as found in MA lines. There are similarities between the characteristics of FN-induced SBSs and those observed in in vitro regenerant Arabidopsis plants (Jiang et al. 2011), although why this should be the case is not clear.
FN irradiation of plants is traditionally considered to induce deletion mutations of 2–4 kb in size (Bruggemann et al. 1996; Li et al. 2001) and has been used for many years in forward genetics experiments to determine the function of unknown genes (Hoffmann et al. 2007; Hofer et al. 2009). An alternative mutagen is the alkylating agent ethylmethane sulphonate (EMS). While EMS mutagenesis produces both complete and partial loss-of-function mutant alleles (Bowman et al. 1991), almost exclusively via G:C>A:T point mutations (Greene et al. 2003; Martin et al. 2009) at frequencies of ∼1000 mutations per Arabidopsis genome (Jander et al. 2003; Martin et al. 2009), FN mutagenesis was previously thought to produce full loss-of-function alleles by inducing a range of deletions (300 bp to 8 kb) (Bruggemann et al. 1996) and insertions (∼3.4 kb) (Sun et al. 1992). However, our identification of 108 FN-induced genomic molecular variants indicate that these previous studies may have favored the detection of large-scale mutations. Of the 39 deletions that we detected, 38 (97%) were <56 bp in length, with single base deletions being the most frequently observed (36%, 14 of 39) (Table 3) and with the majority of single base deletions (64%, nine of 14) occurring at homopolymeric or dinucleotide sites (Table 3A). Thus the view that FNs predominantly induce relatively large-scale gene-inactivating deletion mutations seems less likely, although we cannot exclude the possibility that some larger deletions (up to megabases), which often have a reduced male transmission rate (Naito et al. 2005) or may cause lethality when homozygous, may have been under-represented in our genome-sequenced M3 lines. Indeed, analysis of an additional FN-induced mutant line, E88, did confirm that FNs can induce larger deletions (than those observed in the original six FN M3 lines) of >20 kb (see Supplemental Fig. 3B), and in addition, a chromosomal rearrangement mutation (an interstitial inversion of ∼215 kb) was identified (see Supplemental Fig. 3A). Taken together, our findings suggest that the six FN lineages analyzed in this study provide a good overall picture of the kinds of variants caused by FNs, although the picture may be biased in favor of mutations less likely to have deleterious effects.
We have here described the establishment of six elongated hypocotyl mutant lineages from mutant homozygotes identified in the M2 generation. In each case, we were able to identify the likely phenotype-conferring mutation (see Fig. 2). However, a major previous concern in mutant screens of this nature has been the unknown frequency, chromosomal distribution, and phenotypic consequence of additional mutagen-induced mutations. This concern is the basis of the traditional approach of sequential backcrossing of novel phenotype-causal mutant alleles to the nonmutagenized progenitor, with the aim of segregating out unknown additional mutations (although mutations genetically linked to the phenotype-causal allele will tend to be retained). Because we analyzed the genomes of lineages selected on the basis of mutant phenotype, it is possible that the number of detected additional mutations is somewhat different from what would have been found in unselected M3 plants. However, our results show for the first time that additional mutations, although they clearly occur, are not as frequent as might have been supposed, and that the traditional backcrossing approach is indeed a relatively effective means of “cleaning up” the genetic background of mutant lineages. Most importantly, our results identify the genome-wide spectrum of mutations that researchers can expect to find as additional mutations in mutants selected from FN-irradiation mutagenized material.
Our observations have consequences for the use of FN irradiation as a mutagen in plant genetic studies, including publicly available resources such as those for Arabidopsis (http://www.lehleseeds.com), soybean (http://www.soybase.org), and rice (http://www.ars.usda.gov). They indicate the spectrum of mutations that researchers can expect when using FN irradiation in reverse genetic screens. The high frequency of single base variants suggests that likely causal mutations may not be as readily identifiable using traditional deletion-detection approaches as previously supposed. In addition, our results provide an initial picture of the genome-wide mutational effect of exposure to ionizing radiation, identifying in particular an increase in the relative frequency of transversion mutations. The presence in the environment of cosmic radiation and natural and artificial radionuclides implies a consequential radiation exposure of all organisms. Genome-wide DNA sequence analysis can now be used as a measure of previous exposure to ionizing radiation. Next-generation sequencing of plant genomes could contribute to our understanding of the effect and consequences of environmental ionizing radiation on natural plant populations, thus helping in the implementation of effective environmental protective measures (Copplestone et al. 2010).
Our analyses reveal for the first time the genome-wide consequences of FN irradiation on plant genomes and the spectrum and rate of mutations that researchers should expect to find in FN-mutagenized plant populations. In addition, our study provides a valuable benchmark for future studies of the effects of ionizing radiation on genome integrity.
Methods
Plant material
All experiments used the Landsberg erecta (Ler) laboratory strain of A. thaliana as genetic background. The progenitor “global-DELLA” (gai-t6, rga-t2, rgl1-1, rgl2-1, and rgl3-4) line was isolated from the F3 progeny of a cross between Ler “quadruple-DELLA” (gai-t6, rga-t2, rgl1-1, and rgl2-1) (Achard et al. 2006) and a line carrying a T-DNA insertion in the A. thaliana Columbia-0 RGL3 locus (rgl3-4) obtained from the publicly available SAIL (Syngenta Arabidopsis Insertion Lines) collection. This rgl3-4 line had previously been backcrossed six times sequentially to the Ler background.
Plant mutagenesis, growth conditions, and mutant screening
Progenitor seeds were mutagenized by FN bombardment at a dose of 60 Gy at the KFKI Atomic Energy Research Institute (Budapest, Hungary) and then sown on soil in glasshouses with a 16-h light/8-h dark photoperiod at 22°C–24°C (irradiance 120 μmol of photons/m2 per second). The resultant M1 plants were allowed to self-pollinate in pools of about 1000 plants. M2 and M3 plants were grown in controlled environment rooms (CERs) with a 16-h light/8-h dark photoperiod at 22°C–24°C (irradiance 120 μmol of photons/m2 per second).
Genetic screens of M2 seedlings for elongated hypocotyl mutants were performed by sowing seeds on soil in CERs and allowing them to germinate and grow for 5–7 d. Seedlings were screened visually for elongated hypocotyls. From a total of about 400,000 seeds, more than 300 mutant seedlings displaying elongated hypocotyls were isolated, grown, and self-pollinated. Segregation analysis based on hypocotyl length was performed on 40 M3 plants per line, identifying homozygous lines E71, E88, E99, E125, E128, E138, and E216.
Genome sequencing
Young leaf samples were taken from single nonirradiated progenitor F2, or FN M3 and M4 mutants as well as progenitor plants. DNA was isolated using a Plant DNeasy Mini kit (QIAGEN). Samples representing three independent nonirradiated progenitor derived F2 lines (F2-1 to -3; control plants equivalent to the M2 generation in Fig. 3), seven elongated hypocotyl M3 mutant lines (E71, E88, E99, E125, E128, E138, and E216), an individual E99 M4 and an E125 M4 mutant plant, and the progenitor itself were sequenced using the Illumina Genome Analyzer II platform according to the manufacturer’s instructions at the Wellcome Trust Center for Human Genetics, Oxford, UK, GeneServices, UK or the Beijing Genomics Institute, China. Several lanes of 36-, 51-, 75-, 76-, 90-, and/or 101-bp paired-end runs were produced for each line. A 24–85 (raw sequencing data) and 17–58 (aligned sequencing data) fold depth sequence coverage was obtained for each sample (Supplemental Table 7; sequencing data statistics for the E88 M3 line are not shown).
Creation of progenitor reference sequence
To identify DNA sequence variants in the genomes of the six FN-irradiated M3 mutant lineages (E71, E99, E125, E128, E138, and E216; versus progenitor), it was necessary to create our own reference genome. We developed a software package called Iterative Read-Mapping and Realignment (IMR) v0.3.0 (http://mus.well.ox.ac.uk/19genomes/IMR-DENOM/) (Gan et al. 2011), which was used to create the progenitor genome reference sequence. The algorithm underlying IMR iteratively modifies and corrects the reference, such that in each iteration, sequencing reads are mapped to the current version of the reference using MAQ v0.7.1 (Li et al. 2008) and variants identified with high confidence are used to update and improve the reference sequence. The reads are then remapped and the process repeated until convergence. For our data five iterations were run.
In each iteration base substitution calling is performed using an algorithm based on SAMtools v0.1.5c varFilter (Li et al. 2009). The default behavior of varFilter (which we follow) is to accept isolated base substitutions (with less than two other variants within 10 bp) if:
(1) coverage ≥3 and <100; and
(2) the root mean square (RMS) of the mapping qualities (Phred scores) of the aligned reads >25.
Short INDELs (up to 30 bp) reported by SAMtools (Li et al. 2009) were accepted. Under certain circumstances, SAMtools reported two different deletions at the same location. In such cases, the more likely deletion, usually the longer, was accepted.
One important issue that the IMR algorithm addresses is the problem of clusters of base substitutions and whether these are genuine or artifacts of undetected INDELs (often the alignments of the ends of sequencing reads generate base substitution calls in preference to INDELs). Since varFilter of SAMtools (Li et al. 2009) automatically filters out variant sites where three or more base substitutions occur within 10 bp, these cases were reconsidered as follows: the base substitutions with the greatest confidence (i.e., highest RMS) were accepted, and the two criteria (1) and (2) above were applied. Sites that passed these criteria were modified. Variants that were close to INDELs (i.e., within 10 bp) were ignored since they would be considered in the next iteration.
We also found a number of sites that could not be fixed with IMR because there were two or more different bases in the progenitor sequencing reads aligning to one position in the Col-0 reference sequence. To overcome this, we developed a filter to call base substitutions at these ambiguous sites in the progenitor whereby a base substitution was called at a position if:
(1) at least 80% of reads had the same base;
(2) the number of sequencing reads was within ±20% of the average genome-wide read coverage.
In total, 169.5 million filtered paired-end progenitor reads were aligned to the original Col-0 reference sequence to create our own progenitor genome reference sequence of 119.6 Mb.
Detection of DNA sequence variants
The bioinformatic methods used in this study are the same as those used in Jiang et al. (2011), which found that neither data type nor method significantly altered observed relative frequency or spectrum of detected single base substitutions (SBSs) and small INDELs. In addition, the systematic comparisons performed in Jiang et al. (2011) showed that differences in types of data and data analysis methods between our methods and those used in Ossowski et al. (2010) had negligible effect on detected SBSs and INDELs, and that false-negative rates would have been at comparable small levels in both cases. These observations are also consistent with a recent publication indicating that different softwares are in general comparable (Lunter and Goodson 2011).
Sequencing reads from the three control nonirradiated plants that were derived from the progenitor and grown in independent lineages over two successive generations (F2-1 to -3), seven FN-induced elongated hypocotyl mutants (E71, E88, E99, E125, E128, E138, and E216; M3 generation), and two M4 generation E99 and E125 plants were next mapped to the progenitor reference genome using the Burrows-Wheeler Aligner (BWA) v 0.5.8a (Li and Durbin 2010). Reads with Phred-scaled mapping quality of 20 or less, reads that were not uniquely mapped, and reads for which the mate (the other read of a pair) did not map onto the reference genome were excluded from further analyses. Between 106.6 and 107.9 million nucleotide sites in the six FN M3 lines (E71, E99, E125, E128, E138, and E216) had sufficient read information (at least one read) for calling initial variants compared with the progenitor reference (an average of 107.5 million sites was used to determine the mutation rates per site calculated in Table 2).
The variant lists were generated for each data set using SAMtools (Li et al. 2009) after alignment with the progenitor reference genome sequence. The variant lists were then filtered to exclude putative false positives called at sites with low sequencing read coverage (that could lead to miscalling errors) or high coverage (that are associated with DNA sequences with a high degree of similarity to other sequences in the genome such as transposable elements). A minimum of seven and a maximum of 75 sequencing reads per site were set as the base substitution threshold limits. For INDELs, at least two reads per site were required. Furthermore, base substitution and INDEL false positives called by SAMtools (Li et al. 2009) after re-alignment of the progenitor sequencing reads to the progenitor reference genome sequence using BWA were subtracted from our elongated hypocotyl mutant variant lists.
Following the filtering processes above, the base substitutions and INDEL lists were then checked manually by visually scanning the alignment files (BAM files) generated by SAMtools (Li et al. 2009) using a locally customized version of Integrated Genome Viewer (IGV) (Robinson et al. 2011), which allowed visual comparison of all aligned reads for each mutant data set with the progenitor reference. The six lines analyzed in this study were simultaneously visualized in IGV (for an example, see Supplemental Fig. 4). Based on comprehensive variant analysis using these data sets, the following filters were identified as those that reduced the number of false positives without excluding real mutations:
(a) Base substitutions: a Phred-scaled mapping quality of >63.
(b) Insertion: a minimum seven reads per site and at least 25% of reads had the extra nucleotides.
(c) Deletion: a minimum six reads per site and at least 30% of reads were missing the nucleotides.
Detection of large INDELs and other chromosomal aberrations
We also ran novel protocols (Jiang et al. 2011) to detect larger-scale variants such as insertions, inversions, and translocations. Novel codes were used to generate lists of “distant-pair” read pairs (where two paired reads [“mates”] align at unexpectedly distant regions of the reference sequence [progenitor]). Reads whose mates mapped >750 bp distant with respect to the reference (progenitor) were called, thus creating new (distant-pair) BAM files. Variant lists of covered regions (coverage >5) were generated and visually inspected with IGV. While no such mutations were detected in the six FN M3 lines (E71, E99, E125, E128, E138, and E216 lines), analysis of a further FN M3 line (E88) confirmed the reliability of these methods.
Further exhaustive searches were performed by systematically scanning each of the six FN M3 line BAM files with IGV. These searches did not reveal any genomic variants (large or small) in FN lines additional to the 108 mutations initially identified by the above-described mutant detection procedures.
Correction of FN-lineage mutation data with respect to spontaneous mutations
A total of 108 homozygous variants (SBSs and INDELs) were identified in the genomes of six FN-irradiated M3 Arabidopsis lines. These variants are derived from a combination of both irradiation mutagenesis and also, to a far lesser extent, spontaneous mutational events. To determine the rate and spectrum of FN-induced mutations, the mutation rate and spectrum of spontaneously derived mutations needed to be deducted from our FN data. To do this, we used the results from the Col-0 MA lines experiment, which included a total of 98 substitutions, eight short deletions (ranging in size from 1 to 3 bp), four long deletions (ranging in size from 11 to 5445 bp), and five insertions (all 1 bp) across five MA lines over 30 generations (Ossowski et al. 2010).
To calculate the FN-specific mutation rate observed for single base variants in the different variant categories (substitutions and INDELs) and in the Ti/Tv profiles, we needed to correct the number of mutations observed in the M3 generation in each category twice; first for the spontaneous mutations that accumulated across six lines (E71, E99, E125, E128, E138, and E216) and second for the three generations of single seed descent from the reference strain (progenitor). To do this, we first calculated the frequencies of these events per MA line per generation, and then multiplied them by 18 (= 6 lines × 3 generations) to get the correction factors for different variant categories. These numbers were then subtracted from the number of homozygous mutations observed in M3 generations (e.g., see Table 2A) to obtain the number of homozygous mutations arising as a result of FN treatment.
Data access
The Illumina DNA sequencing data files analyzed in this manuscript have been submitted to the NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) under accession number SRA051285.
Acknowledgments
This publication is based on work supported by Award No. KUK-I1-002-03, made by King Abdullah University of Science and Technology (KAUST) and Biological and Biotechnological Sciences Research Council (BBSRC) grant no. BB/F020759/1. We thank the IGV group (Broad Institute, Cambridge, MA) for help with implementation of IGV. We thank David Buck and colleagues at the Wellcome Trust Centre for Human Genetics (Oxford, UK) Genomics Core for advice, help, and performing most of the genomic sequencing, supported by the Wellcome Trust Core grant no. 090532/Z/09/Z.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.131474.111.
References
- Achard P, Cheng H, De Grauwe L, Decat J, Schoutteten H, Moritz T, Van Der Straeten D, Peng J, Harberd NP 2006. Integration of plant responses to environmentally activated phytohormonal signals. Science 311: 91–94 [DOI] [PubMed] [Google Scholar]
- Ahmad M, Jarillo JA, Smirnova O, Cashmore AR 1998. Cryptochrome blue-light photoreceptors of Arabidopsis implicated in phototropism. Nature 392: 720–723 [DOI] [PubMed] [Google Scholar]
- Ang LH, Chattopadhyay S, Wei N, Oyama T, Okada K, Batschauer A, Deng XW 1998. Molecular interaction between COP1 and HY5 defines a regulatory switch for light control of Arabidopsis development. Mol Cell 1: 213–222 [DOI] [PubMed] [Google Scholar]
- Bowman JL, Smyth DR, Meyerowitz EM 1991. Genetic interactions among floral homeotic genes of Arabidopsis. Development 112: 1–20 [DOI] [PubMed] [Google Scholar]
- Bruggemann E, Handwerger K, Essex C, Storz G 1996. Analysis of fast neutron-generated mutants at the Arabidopsis thaliana HY4 locus. Plant J 10: 755–760 [DOI] [PubMed] [Google Scholar]
- Chen M, Chory J, Fankhauser C 2004. Light signal transduction in higher plants. Annu Rev Genet 38: 87–117 [DOI] [PubMed] [Google Scholar]
- Copplestone D, Beresford N, Howard B 2010. Protection of the environment from ionising radiation: Developing criteria and evaluating approaches for use in regulation. J Radiol Prot 30: 191–194 [DOI] [PubMed] [Google Scholar]
- Davis SJ, Kurepa J, Vierstra RD 1999. The Arabidopsis thaliana HY1 locus, required for phytochrome-chromophore biosynthesis, encodes a protein related to heme oxygenases. Proc Natl Acad Sci 96: 6541–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daya-Grosjean L, Sarasin A 2005. The role of UV induced lesions in skin carcinogenesis: An overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res 571: 43–56 [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenburger T 2006. DNA repair and mutagenesis. ASM Press, Washington, DC [Google Scholar]
- Gan X, Stegle O, Behr J, Steffen JG, Drewe P, Hildebrand KL, Lyngsoe R, Schultheiss SJ, Osborne EJ, Sreedharan VT, et al. 2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature 477: 419–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene EA, Codomo CA, Taylor NE, Henikoff JG, Till BJ, Reynolds SH, Enns LC, Burtner C, Johnson JE, Odden AR, et al. 2003. Spectrum of chemically induced mutations from a large-scale reverse-genetic screen in Arabidopsis. Genetics 164: 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton TG, Alexakhin R, Balonov M, Gentner N, Hendry J, Prister B, Strand P, Woodhead D 2007. Radiation-induced effects on plants and animals: Findings of the United Nations Chernobyl Forum. Health Phys 93: 427–440 [DOI] [PubMed] [Google Scholar]
- Hofer J, Turner L, Moreau C, Ambrose M, Isaac P, Butcher S, Weller J, Dupin A, Dalmais M, Le Signor C, et al. 2009. Tendril-less regulates tendril formation in pea leaves. Plant Cell 21: 420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann D, Jiang Q, Men A, Kinkema M, Gresshoff PM 2007. Nodulation deficiency caused by fast neutron mutagenesis of the model legume Lotus japonicus. J Plant Physiol 164: 460–469 [DOI] [PubMed] [Google Scholar]
- Jander G, Baerson SR, Hudak JA, Gonzalez KA, Gruys KJ, Last RL 2003. Ethylmethanesulfonate saturation mutagenesis in Arabidopsis to determine frequency of herbicide resistance. Plant Physiol 131: 139–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Mithani A, Gan X, Belfield EJ, Klingler JP, Zhu J-K, Ragoussis J, Mott R, Harberd NP 2011. Regenerant Arabidopsis lineages display a distinct genome-wide spectrum of mutations conferring variant phenotypes. Curr Biol 21: 1385–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamra OP, Kamra SK, Nilan RA, Konzak CF 1960. Radiation response of soaked barley seeds. Hereditas 46: 152–170 [Google Scholar]
- Kawanishi S, Hiraku Y, Oikawa S 2001. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutat Res 488: 65–76 [DOI] [PubMed] [Google Scholar]
- Kohchi T, Mukougawa K, Frankenberg N, Masuda M, Yokota A, Lagarias JC 2001. The Arabidopsis HY2 gene encodes phytochromobilin synthase, a ferredoxin-dependent biliverdin reductase. Plant Cell 13: 425–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, Kim PM, Palejev D, Carriero NJ, Du L, et al. 2007. Paired-end mapping reveals extensive structural variation in the human genome. Science 318: 420–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledger S, Strayer C, Ashton F, Kay SA, Putterill J 2001. Analysis of the function of two circadian-regulated CONSTANS-LIKE genes. Plant J 26: 15–22 [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Song Y, Century K, Straight S, Ronald P, Dong X, Lassner M, Zhang Y 2001. A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J 27: 235–242 [DOI] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Wang H 2004. Arabidopsis FHY3/FAR1 gene family and distinct roles of its members in light control of Arabidopsis development. Plant Physiol 136: 4010–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunter G, Goodson M 2011. Stampy: A statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21: 936–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107: 961–968 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Ramiro M, Martinez-Zapater JM, Alonso-Blanco C 2009. A high-density collection of EMS-induced mutations for TILLING in Landsberg erecta genetic background of Arabidopsis. BMC Plant Biol 9: 147 doi: 10.1186/1471-2229-9-147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller HJ 1928. The production of mutations by X-rays. Proc Natl Acad Sci 14: 714–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito K, Kusaba M, Shikazono N, Takano T, Tanaka A, Tanisaka T, Nishimura M 2005. Transmissible and nontransmissible mutations induced by irradiating Arabidopsis thaliana pollen with γ-rays and carbon ions. Genetics 169: 881–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledo JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327: 92–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast JG, Campbell H, Gilbert N, Dunlop MG, Bickmore WA, Semple CA 2007. Chromatin structure and evolution in the human genome. BMC Evol Biol 7: 72 doi: 10.1186/1471-2148-7-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP 2011. Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikazono N, Suzuki C, Kitamura S, Watanabe H, Tano S, Tanaka A 2005. Analysis of mutations induced by carbon ions in Arabidopsis thaliana. J Exp Bot 56: 587–596 [DOI] [PubMed] [Google Scholar]
- Somers DE, Sharrock RA, Tepperman JM, Quail PH 1991. The hy3 long hypocotyl mutant of Arabidopsis is deficient in phytochrome B. Plant Cell 3: 1263–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun TP, Goodman HM, Ausubel FM 1992. Cloning the Arabidopsis Ga1 locus by genomic subtraction. Plant Cell 4: 119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguera E, Canceill D, Ehrlich SD 2001. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J 20: 2587–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]