

Abstract
Double-stranded DNA is able to form triple-helical structures by accommodating a third nucleotide strand in its major groove. This sequence-specific process offers a potent mechanism for targeting genomic loci of interest that is of great value for biotechnological and gene-therapeutic applications. It is likely that nature has leveraged this addressing system for gene regulation, because computational studies have uncovered an abundance of putative triplex target sites in various genomes, with enrichment particularly in gene promoters. However, to draw a more complete picture of the in vivo role of triplexes, not only the putative targets but also the sequences acting as the third strand and their capability to pair with the predicted target sites need to be studied. Here we present Triplexator, the first computational framework that integrates all aspects of triplex formation, and showcase its potential by discussing research examples for which the different aspects of triplex formation are important. We find that chromatin-associated RNAs have a significantly higher fraction of sequence features able to form triplexes than expected at random, suggesting their involvement in gene regulation. We furthermore identify hundreds of human genes that contain sequence features in their promoter predicted to be able to form a triplex with a target within the same promoter, suggesting the involvement of triplexes in feedback-based gene regulation. With focus on biotechnological applications, we screen mammalian genomes for high-affinity triplex target sites that can be used to target genomic loci specifically and find that triplex formation offers a resolution of ∼1300 nt.
Nucleic acid triple helices, also called triplexes, are oligonucleotide complexes made of three strands (DNA and/or RNA) (Felsenfeld et al. 1957) that have been implicated in a variety of cellular mechanisms including transcriptional regulation, chromatin organization, DNA repair, and RNA processing. Evidence for their existence and function in vivo is, however, predominately based on in vitro experiments and computational analysis (for review, see Buske et al. 2011). Their unique ability to target DNA in a sequence-specific manner without requiring unwinding of the double helix holds great potential in biotechnological and biomedical applications such as site-directed recombination, mutagen delivery, and, ultimately, gene therapy (Schleifman et al. 2008; Simon et al. 2008).
Triplex formation is governed by sequence-specific binding rules that are conceptually similar to the familiar Watson-Crick base-pairing rules. The third nucleotide strand binds in the major groove of the duplex by forming Hoogsteen or reverse Hoogsteen hydrogen bonds with the purine-rich strand of the duplex (see Fig. 1; Hoogsteen 1959; Sun and Hélène 1993). Throughout this study, we use the term “triplex-forming oligonucleotide” (TFO) for the part of a single-stranded nucleotide strand that is able to form such hydrogen bonds with the duplex. The term “triplex target site” (TTS) refers to the polypurine·polypyrimidine tract of a duplex able to accommodate a TFO.
Figure 1.

Six-nucleotide triads form the basis of canonical triplex formation. The third nucleotide forms Hoogsteen or reverse Hoogsteen hydrogen bonds with the purine of the duplex, resulting in parallel and anti-parallel orientation of the third strand to the purine tract of the duplex, respectively.
The stability of a triplex is constrained by steric features as well as by the availability of hydrogen donor and acceptor groups to establish the Hoogsteen hydrogen bonds (Sun and Hélène 1993). This effectively limits triplex formation to three basic “motifs,” all of which permit two stabilizing hydrogen bonds between the nucleotide of the third strand and the purine of the duplex. These motifs are referred to by the bases of the third strand that participate in the formation: [T,C] (pyrimidine motif), [G,A] (purine motif), and [G,T] (purine–pyrimidine motif) (Morgan and Wells 1968; Cooney et al. 1988; Beal and Dervan 1991). In the pyrimidine motif, T:AT and C+:GC triads are formed in Hoogsteen configuration, and the cytosine of the third strand needs to be protonated (as indicated by our notation) in order to form the second hydrogen bond, thus requiring a slightly acidic pH (illustrated in Fig. 1). In the purine motif, reverse Hoogsteen bonds are formed in the triads G:GC and A:AT, while the [G,T] motif allows a mixed purine–pyrimidine TFO and forms G:GC and T:AT triads in either Hoogsteen or reverse Hoogsteen configuration. (T refers to uracil in case a strand is made of RNA.)
Over the past decades, these rules have been scrutinized with respect to various determinants of triplex formation such as the chemistry of the nucleotides present in each strand (e.g., nucleotide backbone [Nielsen et al. 1991; Roberts and Crothers 1992; Escude et al. 1993;], sugars [Alam et al. 2007], bases [Hogeland and Weller 1993], and modifications [Lee et al. 1984]), the impact of pH (Sugimoto et al. 2001), ionic environment (Wu et al. 2002), sequence composition (Völker and Klump 1994), and base mismatches (Mergny et al. 1991) using a multitude of complementary experimental setups (for more information, see the review Duca et al. 2008). While each of the different determinants affects the triplex stability, these studies demonstrate that the rule set can be used to model triple-helix formation.
Despite these canonical rules being known for some time, no computational effort has been undertaken to systematically identify all potential TFOs and their targets in genomes. Previous computational approaches limited themselves to predicting the genomic targets (TTSs) only (Ussery et al. 2002; Goñi et al. 2004) and left it up to the user to manually assess the compatibility of any specific TFO–TTS pair (Gaddis et al. 2006; Jenjaroenpun and Kuznetsov 2009). Such a manual annotation approach obviously does not scale well. Recently, Lexa et al. (2011) proposed a dynamic-programming approach to predict intramolecular triple helices, leveraging their approximate palindrome characteristic. The success of this approach is, however, reliant on the scoring matrix and cannot be applied to intermolecular triple-helix formation in a straightforward manner.
Here, we present Triplexator, short for triple-helix locator, a computational toolkit for querying all three pillars of nucleic acid triplex formation, i.e., (1) identifying potential TFOs in single-stranded oligonucleotides; (2) identifying suitable target sites (TTS) in double-stranded nucleotide sequences; and, most important, (3) assessing the compatibility of potential partners according to the canonical triplex formation rules described above. Our algorithm reports all possible maximal matches satisfying a set of user-defined constraints. Moreover, it supports a flexible error model in which the tolerated number of noncanonical triads is allowed to grow with the length of the predicted triplex. Such an error rate is motivated by the observation that a longer triplex is stabilized by more hydrogen bonds and should thus be stable despite the additional noncanonical triads.
The scope and efficiency of Triplexator allows us, for the first time, to address the fundamental biological question of the existence and function of triplex formation in vivo by mining next-generation sequencing data for the footprints of triplex formation. Here, we use the TFO-prediction functionality of Triplexator to investigate whether nuclear RNAs have an enriched potential to bind genomic DNA by means of triplex formation.
The flexibility of Triplexator also enables an application-driven investigation of the genome with biotechnological or gene-therapeutic focus. To demonstrate this aspect, we use the TTS-prediction functionality to query the genomes of five model organisms for loci that can be uniquely targeted by triplex-forming molecules.
Finally, the novel ability of Triplexator to predict matching TFOs and TTSs in genome-scale data sets enables us to investigate whether triplex formation is a mechanism by which RNA might be involved in gene regulation (Mattick et al. 2010). Specifically, we study how many human genes could be regulated by triplex formation involving an RNA transcript originating from a second, upstream promoter, as suggested previously for the DHFR gene (Martianov et al. 2007). Triplexator is available at http://bioinformatics.org.au/triplexator.
Results
TFO prediction: Assessing the triplex-forming potential of chromatin-associated RNA
Functional triplex formation in vivo involving a third strand made of RNA implies that (1) RNA molecules residing in the nucleus contain TFOs and (2) these TFOs are able to form triplexes with the DNA under physiological conditions. To test this hypothesis, we use the TFO-prediction feature of Triplexator to assess the number of (predicted) TFOs in small RNAs purified from chromatin, and compare this with what would be expected by chance. We use chromatin-associated RNA transfrags from K562 cells generated by Cold Spring Harbor Laboratory as a part of the ENCODE Consortium as our positive sample (Fejes-Toth et al. 2009). Our negative control consists of matched-length RNA fragments corresponding to randomly selected genomic positions, which is motivated by the observation that the majority of the genome is transcribed at some stage (The ENCODE Project Consortium 2007).
In both data sets, we predict TFOs comprising at least 19 nt that contain fewer than 10% errors (mismatches to one of the canonical triplex motifs). We use these TFOs to score the “triplex-forming potential” (tpot) of each RNA fragment, normalized with respect to sequence length, by calculating the fraction of nucleotides in the sequence that are able to participate in triple-helical formation (see Methods). Furthermore, since actively transcribed mRNA are likely abundant among chromatin-associated RNA, we use Triplexator's low-complexity region filter to avoid any bias originating from poly(A) tails.
We find that small, chromatin-associated RNAs are enriched in TFOs, with both the purine, and, in particular, the purine–pyrimidine motif contributing to this trend (Fig. 2, “GA” and “GT”). The fourfold enrichment of the purine–pyrimidine motif is particularly interesting because this motif has been suggested to be more relevant for physiological conditions (Ayel and Escudé 2010). Conversely, the pyrimidine motif (Fig. 2, “TC”), which is not expected to be functional due to the nonphysiological pH required for the protonation of the cytosines (Sugimoto et al. 2001), is substantially depleted. This may be an artifact of the particular sequencing protocol used to generate the data, which involves addition of cytosines to the 3′ end of the RNA and poly(G) guided amplification. Poly(G) oligonucleotides are likely to also bind cytosine-rich tracts within the targeted RNA, therefore truncating the amplified RNA sequence. Misguided poly(G) amplification will deprive the data set of pyrimidine motif TFOs, which contain cytosines as an integral part, while the other motifs are not affected. Overall, these results suggest that small, chromatin-associated RNAs can function as the third strand in triplex formation with genomic DNA, but they also highlight the importance of sequencing protocols that are designed with triplex formation in mind.
Figure 2.

Potential of small RNAs to participate in triplex formation. Each bar shows the mean length-adjusted triplex potential (Eq. 1) for chromatin-associated RNA (gray bars) or a set of synthetic RNAs sampled from the whole genome (white bars). The potential is shown over all motifs as well as for each motif individually (bars labeled “GA,” “GT,” and “TC”). Lines indicate the 90% confidence intervals based on bootstrapping.
TTS prediction: Identifying unique genomic loci able to form triplexes
Targeting specific genomic loci reliably is still an unresolved issue in biotechnological as well as gene-therapeutic applications. Triplex-forming molecules may represent a solution to this issue by providing an unambiguous addressing system, e.g., to target defective genomic regions for repair.
To assess the potential of triplex-based targeting approaches and showcase the TTS search functionality of Triplexator, we screen the genome of five model organisms to identify how many genes annotated in RefSeq have unique putative TTSs, and to determine the general resolution possible using a triplex-based addressing system. To ensure that these targets can form stable triple helices, we require a minimum guanine rate of 50% in the purine tract of the target (Mills et al. 2002; Vekhoff et al. 2008). In contrast to a previous study (Wu et al. 2007), however, we refrain from filtering out low-complexity regions since it could remove potential targets. Unique TTSs are detected using Triplexator's duplicate filter, which discards TTSs that occur more than once in the genome (either as copy or subsequence of another TTS).
We find that the human genome contains on average one unique TTS every 1366 bases (Table 1, column 5, row 1), which suggests that genomic loci could be targeted with a very high resolution. The mouse genome contains a unique TTS every 1217 bases on average, while in zebrafish the resolution decreases to around one every 4194 bases.
Table 1.
Distribution of unique, putative triplex target sites (uTTS) across various genomes
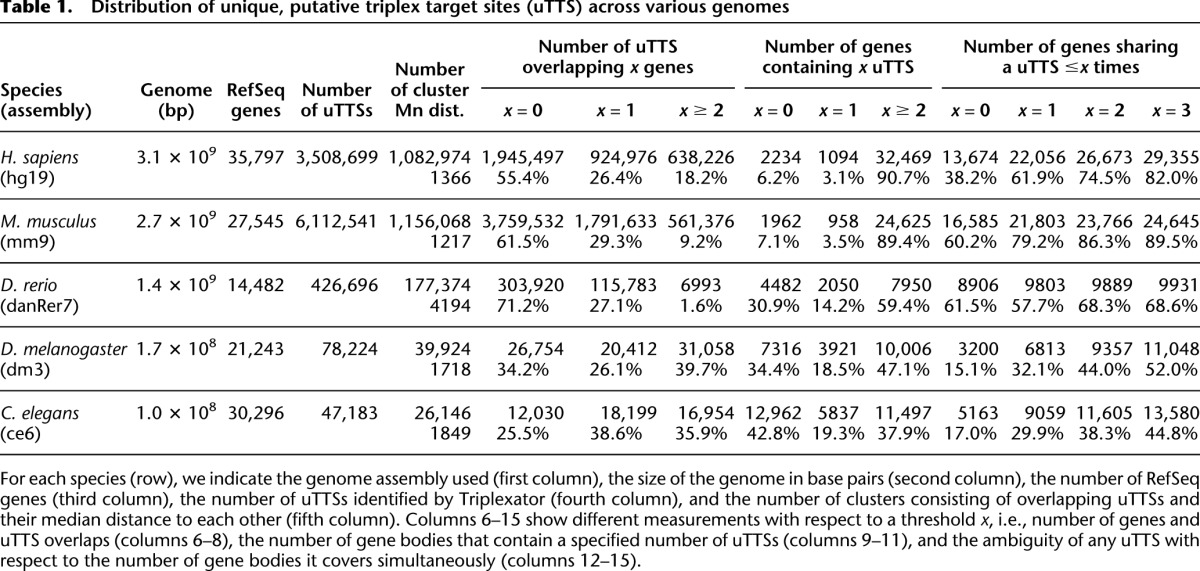
While the density of unique TTSs across the different genomes is similar, their distribution differs between vertebrates and invertebrates. The majority of unique TTSs in vertebrates fall into intergenic regions (Table 1, column 6, x = 0), while at the same time >93% of the combined 2-kb proximal promoter and transcribed genic regions in mammalian genomes contain at least one unique TTS (Table 1, sum of percentages in columns 10 [x = 1] and 11 [x ≥ 2]). Of these, however, only 38%–62% do not share the target site with another gene or alternative transcript that spans the same locus, and could therefore be used to target the gene without immediate off-targets (Table 1, column 12, x = 0).
In less complex species, the number of genes lacking any unique TTSs rises above 30% (Table 1, column 9, x = 0), which could be due to the more compact gene encoding in these species, the smaller quantity of annotated RefSeq genes (14,482 in zebrafish compared with 35,797 in human), or due to the absence of endogenous triplex formation in these species. Taken together, these results indicate the potential that triplex formation offers to target genes as well as intergenic regions such as cis-regulatory regions in a specific manner. We can conclude that the natural occurrence of unique, high-affinity TTSs provides an ideal prerequisite to leverage triplex-based targeting.
Triplex prediction: Proximity feedback signals in human promoters
It is well-documented that non-protein-coding RNAs participate in the regulation of many genetic and epigenetic processes (Mattick et al. 2010), but not all of the underlying biological mechanisms for their interaction with the genome are known. Triplex formation between RNA molecules and promoter or enhancer regions is one possible mechanism whereby RNA could directly regulate gene expression. Martianov et al. (2007) describe a model of triplex-mediated self-regulation of a gene. In this model, a TFO in the transcript from a secondary promoter of the DHFR gene forms a triplex with a TTS in the primary promoter of the same gene, repressing transcription from the primary promoter (illustrated in Fig. 3).
Figure 3.

Diagram of a triplex-mediated RNA–DNA feedback mechanism. Diagram depicting how a transcript from an upstream promoter p1 could interfere with the transcription mediated by a downstream promoter p2 via formation of a triple helix, as suggested by Martianov et al. (2007).
In this experiment, we use the TFO–TTS matching feature of Triplexator to search the human genome for additional genes that could potentially be regulated in the same way as suggested for the DHFR gene. Our approach is to use Triplexator to determine how many human promoters contain a (predicted) matching TFO–TTS pair, and we compare this with the number expected by chance.
We examine a set of 27,876 promoter sequences defined as the region from 1000 bp upstream of to 200 bp downstream from the gene's transcription start site as annotated in RefSeq. We predict TFO–TTS pairs with a minimum triplex length of 19 nt, 10% error rate, and guanine rate of at least 25%. The latter is to avoid long A-tract duplex DNA, which has been found to handicap triplex formation (Sandström et al. 2002). We refrain from filtering out low-complexity regions in this scenario, since such regions are potentially capable to participate in triplex formation. Although nonspecific binding of such sequences may be possible with respect to the whole genome, the proximity of the TFO source and its target, as well as the local conditions during and after transcription, could promote triplex formation in this local microenvironment.
To model the chance frequency of TFO–TTS pairs, we generate two null models. First, we test the ability of a transcript to form a triplex with any promoter by randomly pairing promoters and searching for a TFO in one promoter that matches a TTS in the other. To ensure the applicability of this background model, we do not allow the source of a TFO to overlap its target when we search for TFO–TTS pairs within a single promoter, which guarantees the independence of the two features. Certain palindromic sequences can encode both the source of a TFO and its matching TTS (Buske et al. 2011), and this might be used by nature. We consequently relax the nonoverlapping criterion and assess significance using a second null model comprising 10,000 sets of 27,876 randomly selected, promoter-length human genomic sequences.
The numbers of promoters containing matching TFO–TTS pairs with and without overlap allowed are both much larger than would be expected by chance. Allowing overlap, we find that 2596 human promoters contain a predicted matching TFO–TTS pair, whereas the second null model predicts at most 1707. In 438 human promoters there is no overlap between the predicted TFO and TTS, nearly four times more than the maximum number (117) predicted by the first null model. This enrichment in TFO–TTS pairs over chance is statistically significant (p ≤ 10−4).
Interestingly, the DHFR promoter is not included in the set of 2596 human promoters with predicted matching TFO–TTS pairs. The TFO–TTS pair reported by Martianov et al. (2007) contains >10% errors, causing it to fail our strict search criteria. We therefore rerun the above experiment with the parameters of the Triplexator set so that the DHFR gene is present in the set of promoters predicted to have a matching TFO–TTS pair. This requires increasing the error rate to a maximum of 20% and reducing the minimum triplex length to 15 nt. Since the reported triplex target sites in the DHFR promoter have a very high content of guanines, which may ensure stable triplex formation (Mills et al. 2002) and compensate for any additional noncanonical nucleotide triads, we set a more strict minimum guanine rate of 65% in this new experiment.
With these relaxed criteria, we find 6779 human promoters that contain at least one predicted matching TFO–TTS. This comprises a significant enrichment over the at most 1688 TFO–TTS pairs found in random promoter-length sequences (p ≤ 10−4). With these search criteria, 2379 promoters contain a nonoverlapping predicted TFO–TTS pair, which cannot be considered significant with respect to the first null model (maximum of 2561 over 10,000 trials, p ≤ 0.7).
These results suggest that feedback-based gene regulation involving triplex formation could be leveraged by hundreds of human genes. Furthermore, genomic loci encoding both a TFO source and its target are particularly suited for such a mechanism due to the immediate proximity of the transcript to its target as well as the redundant encoding of TFO and target, which minimizes the likelihood of a TFO–TTS pair to be disrupted due to evolutionary forces.
Circular dichroism studies
CD spectroscopy leverages the differential absorbance of left and right circularly polarized light. It has been shown to be informative for determining the secondary structure assumed by nucleic acids (Kypr et al. 2009).
To investigate the specificity of Triplexator, we randomly picked a predicted, unique TTS from the human genome, with a length of 26 nt, not containing any errors and containing ∼50% guanines in the purine tract. This TTS happens to be located in an intron of the gene annexin A4 (ANXA4). We designed purine-motif TFOs against this site and also designed a similar TTS with errors planted in the sequence (see Fig. 4). Figure 5A shows the CD spectra of the native TFO and the TTS sample, as well as that of a 50:50 mixture of the two molecules. The maximal (absolute value) CD difference between the triplex mix and the average of the individual components (Fig. 5B) is observed at 280 nm, with additional peaks around 218 and 240 nm, all of which are indicative of an interaction between the TFO and the TTS (Gray et al. 1995).
Figure 4.

Sequences and anticipated triplex formation for the predicted triplex target site in the ANXA4 locus. A purine-motif TFO (tfo_26) designed to target the unique TTS in the human annexin A4 gene (tts_26) to form a triple helix (tpx_26) is contrasted to a target site that contains a mismatch in the triplex (tpx_26e) targeted by the same TFO as well as a shorter TFO (tfo_18e) potentially forming the triplex (tpx_18e).
Figure 5.

CD spectra of oligo-nucleotide mixtures. (A) CD spectra of single-stranded oligonucleotide (tfo_26), duplex DNA (tts_26), and a mix of both (tpx_26) in phosphate buffer containing 137 mM Na+, 2.7 mM K+, 10 mM MgCl2 (pH 7), at 25°C. (B) Difference CD spectra for the triplex, where the average spectra of the individual components (tfo_26 and tts_26) are subtracted from the spectra of the triplex mix (tpx_26). Wavelengths of maximal difference peak around 280 nm.
To investigate the stability of the triplex, we performed CD spectroscopy at 280 nm as a function of temperature. Figure 6 shows that the spectra of the duplex (tts_26) exhibits a single transition with Tm (tts) at 72°C, which can be attributed to the melting of the two strands. Upon addition of the 26-nt single-stranded oligonucleotide (tfo_26), a second transition appears around 35°C, indicating strand denaturation involving the third strand. Both transitions are also present in the TFO–TTS mix designed to contain a mismatch between the third strand and the duplex (tpx_26e). However, the shorter, 18-nt TFO mixed with the same TTS duplex (tpx_18e) shows only the transition at 72°C, which is characteristic of the duplex alone. This suggests that there is limited or no triplex formation between the error-containing TTS duplex and the shorter (18 nt) TFO.
Figure 6.

Thermal denaturation of oligonucleotide mixtures. (A) CD spectra at 280 nm as a function of temperature showing melting of triplex mixtures tpx_26 (tfo_26 + tts_26), tpx_26e (tfo_26 + tts_26e), and tpx_18e (tfo_18 + tts_26e) as well as the duplex (tts_26) in the same conditions as Figure 5. (B) First derivative of the temperature indicating melting of complexes around 35°C and 72°C.
Our results are in agreement with previously published results on purine-motif triplexes (Xodo 1995; Alunni-Fabbroni et al. 1996; Arimondo et al. 1998; Raghavan et al. 2005).
Runtime and memory footprint
It is possible to construct a data set such that we have to match any position in the duplex with any position in the single-stranded sequence, and therefore need to verify all subsequent positions until we reach the end of the shorter sequence. Therefore, the worst-case runtime of our algorithm is O(|d| · |s| · min(|d|, |s|)), where |s| and |d| are the lengths of the single-stranded and double-stranded sequences, respectively. For genomic and transcriptomic data, however, the last term converges toward the minimum triplex length n because extension of a matching triplex usually terminates before the length of the shorter sequence is reached. The expected runtime is thus on the order of O(|d| · |s| · n). The nonlinear runtime can nevertheless pose a problem for genome-scale analysis. Leveraging multi-core processor architectures is a common approach in bioinformatics to address this challenge. Triplexator uses OpenMP (Dagum et al. 1998) and offers, beside serial processing, parallel processing of the duplex sequences or the putative TTSs.
The average memory consumption and runtime of Triplexator scale with the overall length of the single-stranded sequences providing the TFOs (Table 2). Detecting triplexes serially results in the smallest memory footprint. Choosing one of the parallel processing options trades runtime for memory. If many target sites are expected per duplex (due to the length of the duplex sequences and/or parameter settings), parallel processing of the targets is recommended (Table 2, “individual target”). In the opposite case, it is beneficial to process the duplex sequences in parallel in order to minimize the overhead associated with thread pooling (Table 2, “duplex”).
Table 2.
Runtime and memory footprint of Triplexator

We contrast a brute-force approach for identifying TFO–TTS pairs (Table 2, upper panel) with the q-gram filtering approach that rapidly discards unfit TFO–TTS pairs (Table 2, lower panel). We find that the q-gram approach is about twice as efficient compared with the brute-force approach. This is, however, accompanied by an increased demand in memory resources. It should be noted that the advantage of the q-gram filtering will vanish with decreasing gram size q. We therefore recommend using the brute-force approach for scenarios that employ a high error rate ɛ, small length threshold n, or that refrain from filtering out low-complexity regions. The brute-force approach is also a valid alternative in case memory resources are limited.
Discussion
Nucleic triple-helix formation offers a neat mechanism to target genomic loci in a sequence-specific manner. This unique ability shows great biotechnological and therapeutic potential. Triplexator will prove valuable for the design of highly specific triplex-forming molecules by providing a framework to find and assess the adequacy of genomic targets.
Triplex formation is likely to be leveraged by nature as well. The obvious candidate in vivo for the third strand is ncRNA. Recent advancements in RNA deep-sequencing technology enable us to see beyond the dominating transcripts of the cell, revealing a vast compendium of RNA classes, whose expression is highly dynamic as well as precisely timed (Mattick et al. 2010). Triplexator will help to functionally annotate this ever-growing pool of transcripts, especially since triplex-based DNA interaction does not necessarily require a high transcript abundance for maximum efficiency.
Methods
Problem definition
Our algorithm is based on the assumption that any triplex feature is sufficiently modeled by the commonly used canonical binding rules (illustrated in Fig. 1). It consists of three major parts: (1) identifying sequence features resembling potential, maximal TFOs in single-stranded sequences; (2) identifying sequence features resembling potential, maximal TTSs in double-stranded sequences; and (3) identifying maximal TFO–TTS pairs (triplexes). These sequence features are subject to a set of user-defined constraints, θ = {n, m, ω, ɛ, g}, where n and m specify the minimum and maximum length, respectively; ω represents the maximum number of consecutive errors; ɛ specifies the maximum proportion of errors that can be tolerated by the sequence feature; while g represents the minimum fraction of guanine bases in the target site. The error parameter ω is motivated by the observation that consecutive noncanonical triads counteract stable triplex formation (Gowers and Fox 1997), while the error rate ɛ reflects the observation that a longer triplex is stabilized by more hydrogen bonds and is therefore stable despite additional noncanonical triads. In addition, the triplex-formation rule set, Λ = {GA, GT, TC}, defines the three motifs, each of which dictates the valid canonical triads and the orientation of the three strands to each other.
For convenience, we define functions for determining if a string is a TFO or TTS and whether a pair of equal-length strings can form a triplex. Let s be a string representing a single-stranded nucleotide sequence and d be a string representing a double-stranded nucleotide sequence, and let |x| be the length of string x. Furthermore, any reference to a target feature in duplex d is with respect to the strand that provides the purine tract able to form the hydrogen bonds with the third strand.
We define the function is_TFO(s, μ, θ), which returns true if and only if the string s constitutes a TFO of the motif μ subject to the constraints θ, i.e., the length |s| falls in-between n and m, s contains at most ⌊ɛ · |s|⌋ errors with respect to motif μ ∈ Λ but at most ω consecutive errors, and s would bind a target with at least ⌈g · |s|⌉ guanines. For example, in the pyrimidine motif (μ = TC), an error is anything in s that is neither a T nor a C, and the guanine ratio is the fraction of cytosines in s, since cytosine binds guanine in the C:GC triad. Similarly, the function is_TTS(d, θ) returns true if and only if a substring d qualifies as a triplex target site subject to the constraints θ, i.e., the length |d| ranges in-between n and m, d contains at most ⌊ɛ · |d|⌋ pyrimidines (errors) with a maximum of ω consecutive pyrimidines in the strand holding the purine tract, which in addition contains at least ⌈g · |d|⌉ guanines. To evaluate whether a particular TFO–TTS pair qualifies for triplex formation, we define the function is_match(s, d, μ, θ), which returns true if and only if the tuple (s, d) forms valid nucleotide triads with respect to motif μ ∈ Λ subject to constraints θ. Here, an error consists of any noncanonical nucleotide triad that is caused by either (1) an error in the TFO, (2) an error in the target site, or (3) a nucleotide pairing between the TFO and the TTS that violates the motif μ (mismatch). The helical structure of the duplex as well as the steric constraints imposed by the nucleotide stacking of the triads effectively exclude bulges in the participating strands. Although third-strand bulges have been observed, they have a strongly destabilizing effect compared with perfect triplexes (Roberts and Crothers 1991). We therefore disregard indels or any gaps in TFO–TTS pairs; thus the two strings s and d have the same length, that is, |s| = |d|.
Using the functions defined above, we can now describe the set of TFOs or TTSs that is contained in a set of (longer) sequences, as well as the set of triplexes that could be formed between members of two sets of sequences. Each TFO or TTS is a subsequence of some (longer) sequence contained in the set of sequences. We represent these subsequences as substrings. In what follows, if x is a nucleotide sequence, then xij with i < j means the substring of x from i (inclusive) to j (exclusive) that is of length |xij| = j − i.
The set of TFOs contained in a set of single-stranded sequences S with respect to a particular triplex motif, μ ∈ Λ, and a set of constraints θ is given by
Similarly, the set of TTSs contained in a set of double-stranded sequences D is given by
where the function rc(x) returns the reverse complement of x. The set of triplexes that can be formed between a subsequence of a member of set S and a subsequence from a member of set D is given by
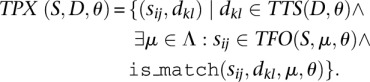 |
The objective of Triplexator is to find maximal TFOs, TTSs, and triplexes—those that cannot be extended on either side without violating either the triplex rules or the given constraints. If substring xij is completely contained in substring xkl and xkl is longer, we write xij ⊂ xkl. Similarly, we say that tuple (sij, dkl) is contained by tuple (smn, dop), and write (sij, dkl) ⊂ (smn, dop), if the respective subsequences are contained in each other: sij ⊂ smn and dkl ⊂ dop. These definitions allow us to define the sets of maximal TFOs, TTSs, and triplexes as
 |
respectively.
Outline of Triplexator
Figure 7 illustrates the workflow of our algorithm, which is implemented in C++. To find all maximal TFOs and TTSs, we use an automaton-based filtering approach to identify initial candidate regions containing at most ω consecutive errors. Subsequently, the set of maximal features contained in these candidate regions are exhaustively identified using a bounded search. The identification of all maximal TFO–TTS pairs, however, is a computational bottleneck. While Triplexator provides a brute-force implementation that exhaustively tests all pairwise combinations of TFOs in the set TFO*(S, μ, θ) and all TTSs in TTS*(D, θ), we also implemented a q-gram-based filtering approach to quickly discard incompatible TFO–TTS pairs.
Figure 7.

Diagram of the implemented pipeline. Triplexator searches for either putative TTSs in a set of duplexes, putative triplex-forming oligonucleotides in a set of single-strand sequences, or the matching pairs from both sets that are able to form nucleic-acid triple helices.
A filter is an algorithm that eliminates a large part of the search space while guaranteeing to preserve any region containing a match. Importantly, this is a necessary but not a sufficient condition, i.e., any region passing the filter does not necessarily contain a match; thus a subsequent verification is required to ensure that the region actually contains a match. Here, we exploit the observation that sequences with Hamming distance ≤k must have a certain number of q -length subsequences (q-grams) in common (Ukkonen 1992). To use such approximate string-matching algorithms, we match the sequence that is targeted by any TFO in S with the set of available targets in D. Any candidate pair passing the filter is extended using the X-drop algorithm (Zhang et al. 2000), and subsequently verified using a bounded search in order to obtain the set of maximal pairs TPX*(S, D, θ). We leverage efficient data structures as well as functionality from the Sequence Analysis template library SeqAn as appropriate (Döring et al. 2008).
Duplicate detection
With respect to biotechnological application, it is important to evaluate how often a particular target occurs in a set of sequences either as an exact copy or as subsequence of a longer feature. We therefore augmented Triplexator with a duplicate detection algorithm that assesses the uniqueness of any feature with respect to the whole set of features spanned by the sequence space of interest. It should be noted that Triplexator performs duplicate detection always with respect to the target site since the primary objective is to assess the uniqueness of putative genomic targets. First, an index of all putative TTSs is generated using an enhanced suffix array data structure. Subsequently, any TTS can be looked up in this index to determine the number of sequences containing this feature as well as their location. Features with numbers of duplicates exceeding a user-defined cutoff are automatically discarded from further processing.
Low-complexity region filtering
Low-complexity regions such as “GA” and “GAA” repeats fit the triplex motifs and can put a substantial strain on the detection of TFO–TTS pairs. We therefore enhanced Triplexator by providing a repeat-region filter that discards such low-complexity regions. We tap into SeqAn's (Döring et al. 2008) repeat finding functionality, which is based on an index using the advantages of a lazy suffix tree (Giegerich et al. 1999). Low-complexity regions are defined by a minimum repeat length and a maximum period length of the repeat pattern. By default, Triplexator ignores low-complexity regions. It should be kept in mind that such regions can resemble valid triplex features (Zheng et al. 2010; Buske et al. 2011), and the user may consider disabling repeat filtering.
Scoring the triplex-formation potential of sequence sets
To assess the potential of a sequence or sequence pair to engage in triplex formation, one can simply count the number of maximal triplex features—TFO*({s}, μ, θ), TTS*({d}, θ), or TPX*({s}, {d}, θ), respectively. It is obvious that the absolute number of maximal triplex features is expected to increase with the length of the sequence. Similarly, a shorter triplex feature has a higher likelihood to occur than a longer one. These observations suggest that an absolute measure is not adequate when we want to compare the triplex potential of sequences let alone sequence sets with each other because it requires an identical length or length distribution, respectively. Nucleotide sequences obtained from next-generation sequencing are unlikely to satisfy this criterion.
To address this issue, we introduce the triplex potential tpot as a measure that accounts for different length distributions in sets of nucleotide sequences. When considering a single sequence (duplex or third strand), tpot represents the fraction of nucleotide subsequences that are able to participate in triplex formation subject to the constraints θ. Let 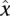 be the complete set of subsequences of x of length l with n ≤ l ≤ m. That is,
be the complete set of subsequences of x of length l with n ≤ l ≤ m. That is, 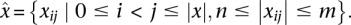 We define tpot for single- and double-stranded features as:
We define tpot for single- and double-stranded features as:
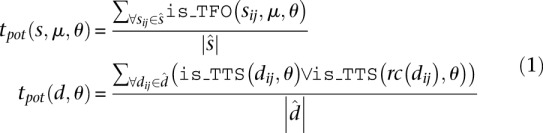 |
This concept is extended to both participating sequences when considering TFO–TTS pairs. We define the triplex potential between sequences s and d as the fraction of equal-length subsequences 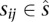 and
and 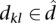 that can form a triplex:
that can form a triplex:
 |
We illustrate the effect of the absolute measure (number of triplex features) and the length-normalized measure (tpot) using an arbitrary genomic sequence of length 107, which is split into sets of sequences mimicking different length distributions. As expected, the average number of triplex features scales with sequence length, assigning a sequence of length 2500 nt, ∼5× the number of features as a sequence of length 500 (Fig. 8, upper panel, “TFO potential” and “TTS potential”). This effect is amplified when the two sets of sequences are matched against each other to detect TFO–TTS pairs (Fig. 8, upper panel, “Triplex potential”). Our length-adjusted tpot, on the other hand, calculates a comparable potential for sequence sets of various length distribution (Fig. 8, lower panel). Importantly, all sequence sets contain the same triplex features because they are generated from the same overall sequence; however, some triplex features are being destroyed by the partitioning. The latter contributes to the slight but insignificant differences we observe for the length-adjusted tpot measure. We conclude that the tpot should be favored over an absolute measure when comparing the triplex potentials of nucleotide sequence sets with each other.
Figure 8.

Effect of sequence length normalization using random data. The plots show the accumulated triplex potential of single-stranded sequence sets (left), double-stranded sequence sets (middle), and between both sets of sequences (right) either counting the number of triplex features (upper panel) or using a length-adjusted scoring scheme (lower panel) with lines indicating the 90% confidence intervals. All sequence sets are derived from the same 107 nucleotide sequence but partitioned into sets of varying length distribution as indicated by the mean sequence length m ∈ {50, 100, 250} for single-stranded and m ∈ {500, 1000, 2500} for double-stranded sequence sets.
Experimental setup
All computational experiments are run with Triplexator v1.2.
TFO prediction: Assessing the triplex-forming potential of chromatin-associated RNA
We obtain transfrags from small RNA sequencing data on K562 cells that were generated by Cold Spring Harbor Laboratory as a part of the ENCODE Consortium (Fejes-Toth et al. 2009). As a control, we create a synthetic set of RNAs that is randomly sampled from the human genome, mimicking the length distribution of the transfrag set. Using Triplexator, we detect putative TFOs of length at least n = 19 nt, allowing ɛ = 10% errors, and remove poly(A) tail contamination by enabling filtering for low-complexity regions. We compute the mean tpot on a set of 10,000 RNAs that are randomly sampled with replacement (bootstrapping) and compute the 90% confidence interval computed based on 1000 repeats.
TTS prediction: Identifying unique genomic loci able to form triplexes
We detect all putative TTSs that cover 15 ≤ n ≤ 30 nt, contain more than g = 50% guanines and at most ɛ = 10% pyrimidine interruptions in the genomes of five species: Homo sapiens (hg19), Mus musculus (mm9), Danio rerio (danRer7), Drosophila melanogaster (dm3), and Caenorhabditis elegans (ce6). Putative TTSs with more than one copy are removed by enabling Triplexator's duplicate filter. In addition, we obtain the RefSeq gene annotations for these species and define a gene body to be the transcribed region of a gene combined with its 2-kb proximal promoter. Custom scripts are used to intersect putative TTSs and gene body regions.
Triplex prediction: Proximity feedback signals in human promoters
We assemble a data set containing the promoters (1000 bp upstream of and 200 bp downstream from the transcription start site) of all human RefSeq genes (hg19) combining genes that share the same promoter region. The resulting 27,876 promoter sequences are input into Triplexator as both the sequences providing the third strand (transcripts from an upstream promoter) and the duplexes providing the targets (gene promoter). We set the parameters to a minimum triplex length of n = 19 nt, an error rate of ɛ = 10%, and a guanine rate of at least g = 25%. To ensure that the DHFR promoter is in the positive set, i.e., contains a TFO that can bind a TTS in the promoter as illustrated in Figure 3, we run the experiment a second time with the parameters adjusted to n = 15, ɛ = 20%, g = 65%, and in addition allow up to two consecutive errors, ω = 2.
In the first null model, we shuffle the relationship between transcripts and promoters 10,000 times to assess the triplex-formation potential between a transcript that is assigned to a promoter at random. We do not allow the TFO source and its target to overlap in order to ensure independence of the features. As a second null model, we sample promoter-length sequences from the human genome at random and test the triplex-formation potential of these regions. Here, bootstrapping is used to calculate empirical P-values using 10,000 repeats.
Assessment of triplex potential, runtime, and memory consumption
To study the triplex potential as a function of sequence length, we generate six data sets that mimic different length distributions using an arbitrary 107-nt region from the human genome. For single-stranded data sets Sm, we split the region into sequences mimicking a Gaussian length distribution with mean m ∈ {50, 10, 250} and standard deviation std ∈ {5, 10, 25}, respectively, while for the duplex sets Dm, we model with mean (std) of 500 (50), 1000 (100), and 2500 (250), respectively. Importantly, all six data sets contain exactly the same overall sequence; however, triplex features located at break points will be lost. To evaluate the runtime and memory footprints, we partition the same region into sequence chunks of lengths 103 and 105 to form the duplex sets and pick an overall sequence length of 105, 106, and 107 to serve as the single-stranded sequence providing the TFOs. These experiments are performed with the default settings: n = 19, ɛ = 10%, and low-complexity filtering with minimum repeat length of 7 and maximum repeat period of 3.
Circular dichroism
HPLC-purified oligonucleotides were ordered from IDT (http://www.idtdna.com) with sequences and predicted triplex formation as shown as in Figure 4. CD was performed on a Jasco J-810 spectropolarimeter equipped with a Peltier-type temperature controller. Spectra were recorded between 200 and 320 nm (data pitch 0.5, scan mode: continuous, sensitivity: 10 mdeg, speed: 10 nm/min, response: 1 sec, bandwidth: 1 nm, four accumulations) using a 0.1-cm path length cuvette containing DNA at 11 μM in single strand (or duplex or triplex). The scan of the buffer was subtracted from the average scan of each sample. Denaturation experiments were performed in the temperature range of 10°C–95°C adjusting the temperature at 5°C/min. Spectra were normalized to total species concentration and presented in Δɛ = ɛL − ɛR in units of M−1 cm−1.
Acknowledgments
We thank the SeqAn development team and especially Manuel Holtgrewe and David Weese for their support with the template library. F.A.B. is supported by the UQ Research Scholarship and the UQ International Research Tuition Award; D.C.B. is a research scientist at CSIRO; J.S.M. is funded by the National Health & Medical Research Council Australia Fellowship 631668; T.L.B. is funded by the Australian Research Council Centre of Excellence in Bioinformatics and the National Institutes of Health (RO-1 RR021692-01).
Footnotes
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.130237.111.
References
- Alam MR, Majumdar A, Thazhathveetil AK, Liu S-T, Liu J-L, Puri N, Cuenoud B, Sasaki S, Miller PS, Seidman MM, et al. 2007. Extensive sugar modification improves triple helix forming oligonucleotide activity in vitro but reduces activity in vivo. Biochemistry 46: 10222–10233 [DOI] [PubMed] [Google Scholar]
- Alunni-Fabbroni M, Manzini G, Quadrifoglio F, Xodo LE 1996. Guanine-rich oligonucleotides targeted to a critical R·Y site located in the Ki-ras promoter. The effect of competing self-structures on triplex formation. Eur J Biochem 238: 143–151 [DOI] [PubMed] [Google Scholar]
- Arimondo PB, Barcelo F, Sun JS, Maurizot JC, Garestier T, Hélène C 1998. Triple helix formation by (G,A)-containing oligonucleotides: asymmetric sequence effect. Biochemistry 37: 16627–16635 [DOI] [PubMed] [Google Scholar]
- Ayel E, Escudé C 2010. In vitro selection of oligonucleotides that bind double-stranded DNA in the presence of triplex-stabilizing agents. Nucleic Acids Res 38: e31 doi: 10.1093/nar/gkp1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal PA, Dervan PB 1991. Second structural motif for recognition of DNA by oligonucleotide-directed triple-helix formation. Science 251: 1360–1363 [DOI] [PubMed] [Google Scholar]
- Buske FA, Mattick JS, Bailey TL 2011. Potential in vivo roles of nucleic acid triple-helices. RNA Biol 8: 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney M, Czernuszewicz G, Postel EH, Flint SJ, Hogan ME 1988. Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science 241: 456–459 [DOI] [PubMed] [Google Scholar]
- Dagum L, Menon R, Inc S 1998. OpenMP: An industry standard API for shared-memory programming. IEEE Comput Sci Eng 5: 46–55 [Google Scholar]
- Döring A, Weese D, Rausch T, Reinert K 2008. SeqAn an efficient, generic C++ library for sequence analysis. BMC Bioinformatics 9: 11 doi: 10.1186/1471-2105-9-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duca M, Vekhoff P, Oussedik K, Halby L, Arimondo PB 2008. The triple helix: 50 years later, the outcome. Nucleic Acids Res 36: 5123–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447: 799–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escude C, Francois JC, Sun JS, Ott G, Sprinzl M, Garestier T, Helene C 1993. Stability of triple helices containing RNA and DNA strands: Experimental and molecular modeling studies. Nucleic Acids Res 21: 5547–5553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejes-Toth K, Sotirova V, Sachidanandam R, Assaf G, Hannon GJ, Kapranov P, Foissac S, Willingham AT, Duttagupta R, Dumais E, et al. 2009. Post-transcriptional processing generates a diversity of 5′-modified long and short RNAs. Nature 457: 1028–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenfeld G, Davies DR, Rich A 1957. Formation of a three-stranded polynucleotide molecule. J Am Chem Soc 79: 2023–2024 [Google Scholar]
- Gaddis SS, Wu Q, Thames HD, DiGiovanni J, Walborg EF, MacLeod MC, Vasquez KM 2006. A web-based search engine for triplex-forming oligonucleotide target sequences. Oligonucleotides 16: 196–201 [DOI] [PubMed] [Google Scholar]
- Giegerich R, Kurtz S, Stoye J 1999. Efficient implementation of lazy suffix trees. In Proceedings of the 3rd International Workshop on Algorithm Engineering, WAE '99, pp. 30–42. Springer-Verlag, London [Google Scholar]
- Goñi JR, de la Cruz X, Orozco M 2004. Triplex-forming oligonucleotide target sequences in the human genome. Nucleic Acids Res 32: 354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowers DM, Fox KR 1997. DNA triple helix formation at oligopurine sites containing multiple contiguous pyrimidines. Nucleic Acids Res 25: 3787–3794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DM, Hung SH, Johnson KH 1995. Absorption and circular dichroism spectroscopy of nucleic acid duplexes and triplexes. Methods Enzymol 246: 19–34 [DOI] [PubMed] [Google Scholar]
- Hogeland JS, Weller DD 1993. Investigations of oligodeoxyinosine for triple helix formation. Antisense Res Dev 3: 285–290 [DOI] [PubMed] [Google Scholar]
- Hoogsteen K 1959. The structure of crystals containing a hydrogen-bonded complex of 1-methylthymine and 9-methyladenine. Acta Crystallogr 12: 822–823 [Google Scholar]
- Jenjaroenpun P, Kuznetsov V 2009. TTS Mapping: Integrative WEB tool for analysis of triplex formation target DNA sequences, G-quadruplets and non-protein coding regulatory DNA elements in the human genome. BMC Genomics (Suppl 3) 10: S9 doi: 10.1186/1471-2164-10-S3-S9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kypr J, Kejnovská I, Renciuk D, Vorlícková M 2009. Circular dichroism and conformational polymorphism of DNA. Nucleic Acids Res 37: 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Woodsworth ML, Latimer LJ, Morgan AR 1984. Poly(pyrimidine) poly(purine) synthetic DNAs containing 5-methylcytosine form stable triplexes at neutral pH. Nucleic Acids Res 12: 6603–6614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexa M, Martínek T, Burgetová I, Kopeček D, Brázdová M 2011. A dynamic programming algorithm for identification of triplex-forming sequences. Bioinformatics 27: 2510–2517 [DOI] [PubMed] [Google Scholar]
- Martianov I, Ramadass A, Barros AS, Chow N, Akoulitchev A 2007. Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature 445: 666–670 [DOI] [PubMed] [Google Scholar]
- Mattick JS, Taft RJ, Faulkner GJ 2010. A global view of genomic information—moving beyond the gene and the master regulator. Trends Genet 26: 21–28 [DOI] [PubMed] [Google Scholar]
- Mergny JL, Sun JS, Rougée M, Montenay-Garestier T, Barcelo F, Chomilier J, Hélène C 1991. Sequence specificity in triple-helix formation: Experimental and theoretical studies of the effect of mismatches on triplex stability. Biochemistry 30: 9791–9798 [DOI] [PubMed] [Google Scholar]
- Mills M, Arimondo PB, Lacroix L, Garestier T, Klump H, Mergny J-L 2002. Chemical modification of the third strand: Differential effects on purine and pyrimidine triple helix formation. Biochemistry 41: 357–366 [DOI] [PubMed] [Google Scholar]
- Morgan AR, Wells RD 1968. Specificity of the three-stranded complex formation between double-stranded DNA and single-stranded RNA containing repeating nucleotide sequences. J Mol Biol 37: 63–80 [DOI] [PubMed] [Google Scholar]
- Nielsen PE, Egholm M, Berg RH, Buchardt O 1991. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 254: 1497–1500 [DOI] [PubMed] [Google Scholar]
- Raghavan SC, Chastain P, Lee JS, Hegde BG, Houston S, Langen R, Hsieh C-L, Haworth IS, Lieber MR 2005. Evidence for a triplex DNA conformation at the bcl-2 major breakpoint region of the t(14;18) translocation. J Biol Chem 280: 22749–22760 [DOI] [PubMed] [Google Scholar]
- Roberts RW, Crothers DM 1991. Specificity and stringency in DNA triplex formation. Proc Natl Acad Sci 88: 9397–9401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RW, Crothers DM 1992. Stability and properties of double and triple helices: Dramatic effects of RNA or DNA backbone composition. Science 258: 1463–1466 [DOI] [PubMed] [Google Scholar]
- Sandström K, Wärmländer S, Gräslund A, Leijon M 2002. A-tract DNA disfavours triplex formation. J Mol Biol 315: 737–748 [DOI] [PubMed] [Google Scholar]
- Schleifman EB, Chin JY, Glazer PM 2008. Triplex-mediated gene modification. Methods Mol Biol 435: 175–190 [DOI] [PubMed] [Google Scholar]
- Simon P, Cannata F, Concordet J-P, Giovannangeli C 2008. Targeting DNA with triplex-forming oligonucleotides to modify gene sequence. Biochimie 90: 1109–1116 [DOI] [PubMed] [Google Scholar]
- Sugimoto N, Wu P, Hara H, Kawamoto Y 2001. pH and cation effects on the properties of parallel pyrimidine motif DNA triplexes. Biochemistry 40: 9396–9405 [DOI] [PubMed] [Google Scholar]
- Sun J-S, Hélène C 1993. Oligonucleotide-directed triple-helix formations. Curr Opin Struct Biol 3: 345–356 [DOI] [PubMed] [Google Scholar]
- Ukkonen E 1992. Approximate string-matching with q-grams and maximal matches. Theor Comput Sci 92: 191–211 [Google Scholar]
- Ussery D, Soumpasis DM, Brunak S, Staerfeldt HH, Worning P, Krogh A 2002. Bias of purine stretches in sequenced chromosomes. Comput Chem 26: 531–541 [DOI] [PubMed] [Google Scholar]
- Vekhoff P, Ceccaldi A, Polverari D, Pylouster J, Pisano C, Arimondo PB 2008. Triplex formation on DNA targets: How to choose the oligonucleotide. Biochemistry 47: 12277–12289 [DOI] [PubMed] [Google Scholar]
- Völker J, Klump HH 1994. Electrostatic effects in DNA triple helices. Biochemistry 33: 13502–13508 [DOI] [PubMed] [Google Scholar]
- Wu P, Kawamoto Y, Hara H, Sugimoto N 2002. Effect of divalent cations and cytosine protonation on thermodynamic properties of intermolecular DNA double and triple helices. J Inorg Biochem 91: 277–285 [DOI] [PubMed] [Google Scholar]
- Wu Q, Gaddis SS, MacLeod MC, Walborg EF, Thames HD, DiGiovanni J, Vasquez KM 2007. High-affinity triplex-forming oligonucleotide target sequences in mammalian genomes. Mol Carcinog 46: 15–23 [DOI] [PubMed] [Google Scholar]
- Xodo LE 1995. Characterization of the DNA triplex formed by d(TGGGTGGGTGGTTGGGTGGG) and a critical R·Y sequence located in the promoter of the murine Ki-ras proto-oncogene. FEBS Lett 370: 153–157 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Schwartz S, Wagner L, Miller W 2000. A greedy algorithm for aligning DNA sequences. J Comput Biol 7: 203–214 [DOI] [PubMed] [Google Scholar]
- Zheng R, Shen Z, Tripathi V, Xuan Z, Freier SM, Bennett CF, Prasanth SG, Prasanth KV 2010. Polypurine-repeat-containing RNAs: A novel class of long non-coding RNA in mammalian cells. J Cell Sci 123: 3734–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]