

Abstract
Prions containing misfolded prion protein (PrPSc) can be formed with cofactor molecules using the technique of serial protein misfolding cyclic amplification. However, it remains unknown whether cofactors materially participate in maintaining prion conformation and infectious properties. Here we show that withdrawal of cofactor molecules during serial propagation of purified recombinant prions caused adaptation of PrPSc structure accompanied by a reduction in specific infectivity of >105-fold, to undetectable levels, despite the ability of adapted “protein-only” PrPSc molecules to self-propagate in vitro. We also report that changing only the cofactor component of a minimal reaction substrate mixture during serial propagation induced major changes in the strain properties of an infectious recombinant prion. Moreover, propagation with only one functional cofactor (phosphatidylethanolamine) induced the conversion of three distinct strains into a single strain with unique infectious properties and PrPSc structure. Taken together, these results indicate that cofactor molecules can regulate the defining features of mammalian prions: PrPSc conformation, infectivity, and strain properties. These findings suggest that cofactor molecules likely are integral components of infectious prions.
Keywords: phospholipid, bioassay, repertoire, convergence, diversity
Transmissible spongiform encephalopathies such as Creutzfeldt–Jakob disease, bovine spongiform encephalopathy, chronic wasting disease, and scrapie are caused by unconventional infectious agents termed “prions” that lack informational nucleic acids (1). The most fundamental event in the formation of infectious prions is the conformational change of a host-encoded glycoprotein termed “PrPC” into a misfolded conformer termed “PrPSc” (2, 3); purified PrPSc molecules can induce the conversion of additional PrPC molecules into the PrPSc conformer in a self-propagating manner (4, 5).
Much effort has been made to determine the chemical nature of infectious mammalian prions. According to the “protein-only” hypothesis, infectious prions potentially are composed solely of PrPSc (6–9). However, several groups have shown that relatively low levels of specific infectivity using only pure PrP molecules as a substrate (10–12). In contrast, chemical reactions containing purified PrP molecules plus nucleic acid and lipid molecules spontaneously generate and propagate PrPSc molecules associated with moderate levels of specific infectivity (5). Additional evidence that cellular factors other than PrP influence the efficiency of prion propagation is provided by studies showing that various clonal lines of cultured neuroblastoma cells have different levels of susceptibility to prion infection (13, 14). It is unknown currently whether cofactor molecules are simply catalysts for prion formation or whether they also play an essential role maintaining a specific infectious conformation of PrPSc. Studies with purified prions containing a photolabile oligonucleotide cofactor showed that polyanionic cofactors are not required to maintain infectivity (15), but the prions used in those studies contained copurified lipids, whose role in maintaining infectivity remains unknown.
Interestingly, prions can exist as different “strains” characterized by distinctive clinical and neuropathological features that are recapitulated faithfully upon serial passage within the same animal species (16, 17). Recent studies suggest that individual strains of mammalian prions may be composed of a mixture of PrPSc conformers and that the relative distribution of those conformers may be subject to selective pressure during the process of strain adaptation, e. g., by transmission between different animal species (18) or passage in cloned cell lines (19). However, the molecular mechanism by which a variety of PrPSc conformers can be produced and selected during the process of strain adaptation has not yet been elucidated. One possible mechanism is that each PrPSc conformer might require a unique set of cofactors to propagate efficiently, and the distribution of these putative cofactor molecules may vary in different animal species and cell types. Consistent with this concept, reconstitution studies have revealed the existence of multiple classes of cofactors for prion propagation in vitro (20).
We recently identified the endogenous activity responsible for facilitating mouse prion propagation in vitro (20) as phosphatidylethanolamine (PE) (21). PE robustly facilitates the formation of infectious recombinant mouse prions as a solitary cofactor without RNA, providing a unique tool to test whether cofactor molecules can regulate PrPSc conformation, prion strain properties, and infectivity in a minimal in vitro prion propagation system.
Results
Withdrawal of Cofactor During Serial Propagation Produces an Adapted Self-Propagating “Protein-only” PrPSc Conformer.
We serially propagated a previously described recombinant prion strain (22) (termed the “OSU strain”) for more than 30 rounds in seeded serial protein misfolding cyclic amplification (sPMCA) reactions using a substrate mixture containing pure α-helical recombinant prion protein (recPrP) molecules and a purified cofactor preparation containing a mixture of mouse brain phospholipids, of which the only active component is PE. Under these conditions, the propagation of an ∼18-kDa PrPSc conformer (which we term “OSU cofactor PrPSc”) is maintained indefinitely (Fig. 1A, first blot). To test whether cofactor molecules are required to maintain a self-propagating PrPSc conformation, we used OSU cofactor PrPSc molecules to seed sPMCA reactions with recPrP as the sole substrate (i.e., without the cofactor preparation). These experiments produced two different sets of outcomes. In ∼40% of these experiments, PrPSc propagation could not be sustained following cofactor withdrawal (Fig. 1A, second blot), but in ∼60% of the experiments we unexpectedly observed step-wise adaptation of the ∼18-kDa OSU cofactor PrPSc seed into a self-propagating ∼16-kDa protease-resistant recPrPSc band (which we term “OSU protein-only PrPSc”) (Fig. 1A, third and fourth blots). The resistance of the OSU protein-only PrPSc conformer to digestion with a 25:1 mass ratio of proteinase K to recPrP, as well as its formation in the absence of 0.1% SDS, distinguishes it from a previously reported recPrP sPMCA product (23). Once formed, the OSU protein-only PrPSc conformer could be propagated indefinitely in sPMCA reactions (Fig. 1B) in the same manner as the cofactor PrPSc conformer. Interestingly, the OSU protein-only PrPSc conformer did not trigger formation of ∼18-kDa recPrPSc when either the purified cofactor preparation or synthetic PE was restored to the substrate mixture (Fig. S1), indicating that the switch from the cofactor PrPSc conformation to the protein-only PrPSc conformation is unidirectional.
Fig. 1.
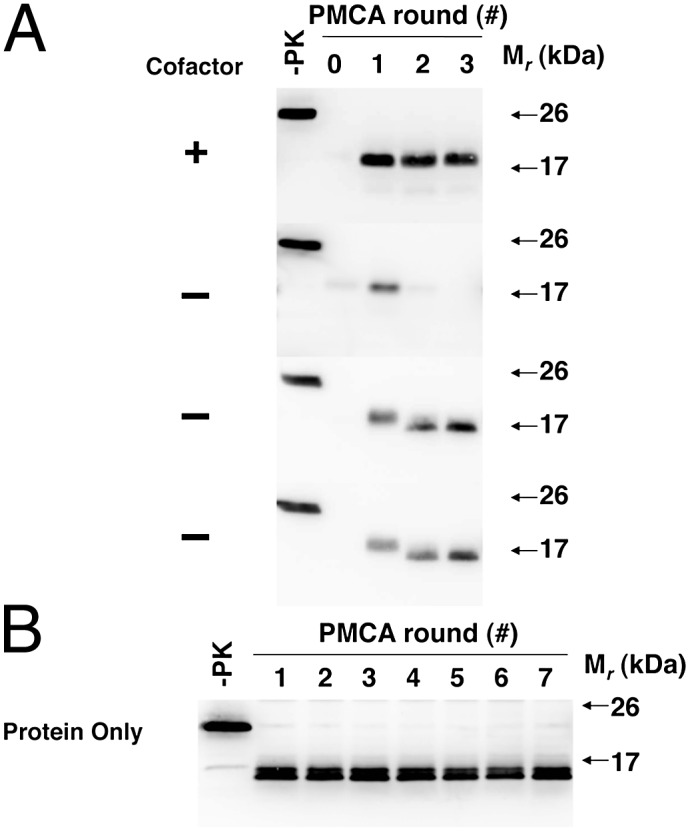
Adaptation of autocatalytic PrPSc molecules. Western blots of reconstituted sPMCA reactions. −PK, samples not subjected to proteinase K digestion; all other samples were proteolyzed. (A) All reactions initially were seeded with OSU cofactor PrPSc molecules and subsequently were propagated in substrate mixtures with or without cofactor, as indicated. (B) Ongoing propagation of OSU protein-only PrPSc molecules in substrate mixture lacking cofactor.
Cofactor PrPSc and Protein-only PrPSc Molecules Have Similar Ultrastructural Features.
To compare the ultrastructural characteristics of the OSU cofactor PrPSc conformer with those of the OSU protein-only PrPSc conformer, we performed atomic force microscopy on both types of PrPSc molecules. This comparison revealed that the two conformers generally displayed similar ultrastructural features (the predominant species observed in both samples being an ∼2-nm sphere), although the distribution of heights differed slightly between the two samples (Fig. S2 A and B). Rings ∼100 nm in diameter also were seen in ∼1% of the scanned fields in the sample containing protein-only PrPSc molecules (Fig. S2A, Right). Interestingly, these recPrPSc spheres and rings are reminiscent of previously described “dots and rings” formed by yeast prions (24, 25). No fibrils were observed in scans of either OSU cofactor PrPSc or OSU protein-only PrPSc molecules.
Protein-only PrPSc Molecules Are Not Infectious in Vivo and Cannot Trigger Native PrPSc Formation in Vitro.
We next sought to compare the infectivity of OSU cofactor PrPSc and OSU protein-only PrPSc molecules. To do so rigorously, we generated a closely matched set of internally controlled samples. Substrate mixtures were prepared from two aliquots of a single stock solution of recPrP in buffer. Then purified cofactor was added to one of the aliquots to complete the cofactor PrPSc mixture, and an equal volume of water was added to the other aliquot to make the protein-only mixture. We then simultaneously propagated OSU cofactor PrPSc and OSU protein-only PrPSc molecules in their appropriate substrate mixtures using equidistant, concentric locations of a single circular microplate horn. SDS/PAGE of the final round products shows that similar quantities of PrPSc were produced in all the processed samples (Fig. S3). Thus, the availability of these well-matched and simultaneously processed samples provided a unique opportunity to test in isolation the role of cofactor molecules in maintaining prion infectivity.
We performed end-point titration bioassays of these simultaneously processed samples in wild-type C57BL mice. The results of these assays indicate that recPrPSc molecules formed with cofactor caused scrapie at dilutions from 10−1 to 10−5 (Table 1), as confirmed by pathology (Fig. S4) and Western blot (Fig. S5). Based on the end-point titration data and Western blot quantitation of PrPSc in the inoculum, the specific infectivity of OSU cofactor recPrPSc molecules is ∼2.2 × 106 LD50 units/μg PrP. In contrast, OSU protein-only recPrPSc molecules derived from the same original recPrPSc seed failed to cause scrapie in mice even at the highest concentration tested (Table 1). The brains of age-matched, asymptomatic animals inoculated with protein-only recPrPSc molecules were histologically normal (Fig. S4) and lacked PrPSc as judged by Western blot (Fig. S5).
Table 1.
Bioassay of in vitro-generated recombinant PrPSc molecules in normal C57BL mice
| Inoculum | Dilution | n/n0 | IP (days)* |
| Cofactor PrPSc | 10−1 | 7/7 | 356 ± 12 |
| 10−2 | 3/3 | 451 ± 16 | |
| 10−3 | 4/4 | 481 ± 42 | |
| 10−4 | 4/4 | 501 ± 28 | |
| 10−5 | 1/3 | 539 | |
| 10−6 | 0/4 | >570 | |
| Protein only PrPSc Sample A | 10−1 | 0/4 | >570 |
| 10−2 | 0/4 | >570 | |
| 10−3 | 0/4 | >570 | |
| 10−4 | 0/4 | >570 | |
| Protein only PrPSc Sample B | 10−1 | 0/4 | >570 |
| 10−2 | 0/4 | >570 | |
| 10−3 | 0/4 | >570 | |
| 10−4 | 0/4 | >570 |
*Incubation period (IP) of scrapie sick animals, mean ± SE.
To determine whether the >105-fold difference in specific infectivity is caused by differences in the ability of the two PrPSc conformers to trigger native PrPC conversion, we compared the ability to these two conformers to seed sPMCA reactions using crude brain homogenate as substrate. The results show that although the OSU cofactor PrPSc molecules effectively seeded PrPSc formation, the OSU protein-only PrPSc conformer failed to trigger native prion formation in all three rounds of the sPMCA assay (Fig. 2). Moreover, the inability of the OSU protein-only conformer to trigger native PrPC conversion could not be overcome by preliminary propagation for four rounds in a substrate mixture containing PE before sPMCA with brain homogenate (Fig. S6), confirming that the switch to the inactive conformation is most likely irreversible and that phospholipid molecules do not simply protect or enhance delivery of PrPSc. Collectively, the results of the bioassay and sPMCA experiments show that controlled removal of cofactor causes adaptation of self-propagating, protease-resistant recPrPSc molecules into a conformation that is unable to trigger native PrPSc formation in vivo or in vitro.
Fig. 2.

Seeding of brain homogenate sPMCA reactions. Western blot of three-round sPMCA reactions using normal mouse brain homogenate substrate seeded with various samples as indicated. −PK, samples not subjected to proteinase K digestion; all other samples were proteolyzed. Native PrPSc was isolated as PrP27-30 molecules from the brains of Me7-infected mice as previously described (35), and PrP amyloid was generated as previously described (36).
Cofactor Molecules Are Physically Associated with PrPSc Aggregates.
Because the foregoing results indicate that a cofactor preparation containing PE is required to maintain PrPSc infectivity, we sought to determine whether PE becomes physically incorporated into the recombinant prion aggregates as they are formed. To study this question, we used the compound 1-oleoyl-2-{12-[(7-nitro-2–1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-sn-glycero-3-phosphoethanolamine [18:1–12:0 nitrobenzoxadiazole (NBD):PE], in which a fluorescent NBD group is covalently attached as a probe to the C2 fatty acid adduct of synthetic PE. We performed seeded four-round sPMCA reactions with a substrate mixture containing recPrP and NBD-PE to produce NBD-PE PrPSc molecules (Fig. 3A). We then used a microscopic dual-channel fluorescence assay to determine whether NBD-PE became incorporated into complexes with recPrPSc during the sPMCA reactions. The results of this assay show that the fluorescent cofactor is present and colocalizes with PrPSc aggregates detected by antibody staining (Fig. 3B), indicating that the recombinant prions do contain PE in addition to PrPSc molecules. Quantitation of total NBD fluorescence within the washed recPrPSc pellet suggests a protein:lipid molar ratio of ∼1:4. It is unlikely that the fluorescent lipid molecules are weakly bound to the solvent-accessible surface of PrPSc aggregates because the samples were washed extensively with detergent before analysis.
Fig. 3.

NBD-PE PrPSc colocalization assay. (A) Western blot of a four-round sPMCA reaction using recPrP and NBD-PE as substrate. −PK, samples not subjected to proteinase K digestion; all other samples were proteolyzed. (B) Dual-channel fluorescence micrographs showing representative images of the final product of the sPMCA reaction shown in A, after purification with detergent washes as described in Experimental Procedures. PrPSc aggregates immunostained with anti-PrP mAb D13 and Alexa Fluor 568-labeled secondary antibody are shown in red (PrP), and colocalized NBD-PE molecules are shown in green (NBD-PE).
Cofactor-Induced Modulation of Strain-Dependent Neurotropism and Prion Incubation Times.
The observation that cofactor molecules participate in maintaining the infectious conformation of PrPSc raises the possibility that they also may play a role in encoding strain properties. Castilla et al. (26) previously established that several different murine prion strains maintain their specific biochemical and infectious properties when serially propagated in vitro using crude brain homogenate as a substrate. Therefore, we decided to use our chemically defined in vitro prion propagation system to test whether different prion strains are able to maintain distinctive properties when only one active cofactor (PE) is available to form new PrPSc molecules.
For these experiments, we used the original OSU isolate [which was produced de novo in sPMCA reactions from recPrP, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG), and RNA substrates (22)] as well as two easily distinguishable native mouse prion strains (301C and Me7) to seed a uniform substrate mixture containing recPrP and purified cofactor preparation. For each strain, sPMCA produced self-propagating cofactor recPrPSc molecules with a protease-resistant core ∼18 kDa in size (Fig. S7A).
We inoculated wild-type C57BL mice with cofactor PrPSc molecules produced by 18-round sPMCA propagation of each strain along with the original seed material (input samples) for each strain and negative control samples. Mock-propagated samples originally seeded with each strain and processed in parallel with the experimental samples were noninfectious, confirming that 18 rounds of sPMCA were sufficient to eliminate the original infectious seeds by serial dilution (Table 2). In contrast, cofactor PrPSc molecules derived from all three strains caused scrapie in the inoculated animals. Interestingly, the incubation periods for cofactor PrPSc molecules were at least twice as long as the incubation periods caused by the input samples for all three strains (Table 2).
Table 2.
Bioassay of native and in vitro-generated recombinant PrPSc molecules in normal C57BL mice
| Seed (strain) | Inoculum | Dilution | n/n0 | IP (days)* |
| OSU | Input recPrPSc | 100 | 5/5 | 173 ± 1 |
| OSU | Cofactor recPrPSc | 10−1 | 7/7 | 356 ± 12 |
| OSU | Synthetic PE recPrPSc | 10−1 | 8/8 | 381 ± 11 |
| OSU | Serial Passage- Synthetic PE recPrPSc | 1% wt/vol | 8/8 | 175 ± 4 |
| OSU | Mock sPMCA control | 10−1 | 0/4 | >570 |
| Me7 | Input brain homogenate | 10−1 | 7/7 | 158 ± 7 |
| Me7 | Cofactor recPrPSc | 10−1 | 8/8 | 396 ± 20 |
| Me7 | Synthetic PE recPrPSc | 10−1 | 6/6 | 417 ± 21 |
| Me7 | Serial Passage- Synthetic PE recPrPSc | 1% wt/vol | 8/8 | 166 ± 3 |
| Me7 | Mock sPMCA control | 10−1 | 0/4 | >510 |
| 301C | Input brain homogenate | 10−1 | 7/7 | 182 ± 4 |
| 301C | Cofactor recPrPSc | 10−1 | 8/8 | 401 ± 11 |
| 301C | Synthetic PE recPrPSc | 10−1 | 6/7 | 360 ± 9† |
| 301C | Serial Passage- Synthetic PE recPrPSc | 1% wt/vol | 8/8 | 173 ± 3 |
| 301C | Mock sPMCA control | 10−1 | 0/4 | >510 |
| None | Cofactor mixture | 10−1 | 0/4 | >570 |
| None | Synthetic PE mixture | 10−1 | 0/4 | >570 |
*Incubation period (IP) of scrapie sick animals, mean ± SE.
†Ongoing experiment. n/n0 = number of animals diagnosed with prion disease/number of animals inoculated.
The defining characteristic of mammalian prion strains is selective neurotropism in infected hosts, and therefore we analyzed the neuropathological profiles of mice inoculated with input and cofactor PrPSc molecules by scoring brain regions for spongiform change (vacuolation) and PrP deposition (immunohistochemistry). As expected, the three input strains could be distinguished from each other easily by their vacuolation and PrP deposition profiles (Fig. 4 A and E). However, the vacuolation and PrP deposition profiles for all three cofactor PrPSc samples derived from those strains were similar to each other (Fig. 4 B and F) and were different from their parent strains (i.e., the three input samples) (compare vacuolation profiles in Fig. 4 A and B and PrP deposition profiles in Fig. 4 E and F).
Fig. 4.

Regional neuropathology of infected mice. (A–D) Profiles of vacuolation scores of animals inoculated with samples containing (A) input prions, (B) cofactor PrPSc molecules, (C) PE PrPSc prions, or (D) serial-passage PE PrPSc prions. (E–H) Profiles of PrP deposition scores of animals inoculated with samples containing (E) input prions, (F) cofactor PrPSc molecules, (G) PE PrPSc prions, or (H) serial-passage PE PrPSc prions. Prion strains: OSU, red squares; Me7, blue circles; 301C, green triangles. Brain regions: I–II, cerebral cortical layers 1 and 2; III–IV, cortical layers 3 and 4; V–VI, cortical layers 5 and 6; BS, brainstem; Cb, cerebellum; CC, cerebral cortex (all layers); H, hippocampus; HT, hypothalamus; Mid, midbrain; T, thalamus. Mean values ± SEM are shown; n = 5.
At a microscopic level, the most dramatic examples of cofactor-induced changes in neurotropism that we observed were in (i) the degree of vacuolation caused by OSU input versus OSU cofactor PrPSc molecules in the cerebral cortex and hypothalamus (Fig. S8) and (ii) the patterns of PrP immunodeposition induced by 301C input versus 301C cofactor PrPSc molecules in the cerebral cortex (where 301C input causes deposition selectively in cortical layers III–IV, as indicated by the arrowhead in Fig. S9).
It is important to note that both the OSU input and OSU cofactor PrPSc samples were produced by propagation of the original OSU seed (containing POPG and RNA) in sPMCA reactions using recPrP substrate. The only experimental difference between these two samples was a change in the cofactor component from POPG/RNA to the purified phospholipid cofactor preparation of the sPMCA substrate mixture. Therefore, in this case, the dramatic differences in neurotropism between the OSU input and OSU cofactor PrPSc samples (Fig. 4, red squares; compare Fig. 4 A and B with F and E) can be attributed unambiguously to the change in cofactor composition and not to the use of recPrP substrate or to the sPMCA technique per se in the experiment.
Because the only component of the purified cofactor preparation able to facilitate PrPSc propagation is PE, we hypothesized that PE alone might be responsible for producing and maintaining the characteristics of the cofactor PrPSc strain, which differ markedly from those of each of the three input strains. To test this hypothesis, we propagated all three sets of cofactor PrPSc molecules into a substrate mixture containing only recPrP and synthetic PE for 18 rounds, yielding a set of “PE PrPSc” molecules also ∼18 kDa in size (Fig. S7B). When inoculated into C57BL mice, all three sets of PE PrPSc molecules caused scrapie with long incubation periods comparable to those induced by cofactor PrPSc molecules (Table 2). Neuropathological studies showed that all three sets of PE PrPSc inocula induced similar patterns of vacuolation and PrP deposition in the brains of inoculated mice (Fig. 4 C and G). Moreover, these patterns were similar to those induced by cofactor PrPSc molecules (compare vacuolation profiles in Fig. 4 C and B and PrP deposition profiles in Fig. 4 F and G).
Statistical analysis confirmed that vacuolation patterns of the three cofactor PrPSc samples were significantly different (Wilcoxon rank-sum test P < 0.05) from their corresponding three input strains in 18 of 21 comparisons (seven brain regions and three output inocula). Similarly, PrP immunodeposition of OSU input- versus OSU cofactor PrPSc-inoculated animals showed statistically significant differences in seven of eight brain regions, Me7 input- versus Me7 cofactor PrPSc-inoculated animals showed statistically significant differences in two of eight brain regions, and 301C input- versus 301C cofactor PrPSc-inoculated animals showed statistically significant differences in five of eight brain regions. Additional statistical analysis showed no evidence to reject the hypothesis that the vacuolation and PrP deposition profiles of the six output (cofactor and PE) strains came from a single distribution. In contrast, when the analysis was repeated after including the three input strains and six output strains, the null hypothesis was rejected for six of the eight brain regions (all P < 0.04).
Cofactor-Induced Modulation of Strain-Dependent PrPSc Conformation.
In some instances, differences in the conformation of PrPSc molecules associated with different prion strains can be detected by biochemical assays. We therefore compared biochemical characteristics of PrPSc molecules in the brains of infected mice by SDS/PAGE/Western blotting and urea denaturation assays. Western blotting showed that all three sets of cofactor PrPSc inocula induced the formation of protease-resistant PrPSc molecules with similar glycoform profiles (dominated by diglycosylated PrPSc) and migration after enzymatic deglycosylation (Fig. 5, lanes 1–3). Similarly, the protease-resistant PrPSc molecules in the brains of animals infected with all three sets of PE PrPSc inocula had glycoform profiles and migration patterns that were similar to those of PrPSc molecules in the brains of animals infected with cofactor PrPSc inocula (Fig. 5, compare lanes 7–9 with lanes 1–3). In contrast, protease-resistant PrPSc molecules induced by input 301C prions were ∼2 kDa smaller in size (Fig. 5, lane 4), and PrPSc molecules induced by input OSU recombinant prions had a characteristic glycoform profile in which diglycosylated PrPSc was the least abundant species (Fig. 5, lane 6).
Fig. 5.

Glycoform distribution and electrophoretic mobility of PrPSc molecules in the brains of infected mice. (Upper) Western blots of brain homogenate samples prepared from animals inoculated with samples containing input prions, cofactor PrPSc, and PE PrPSc molecules derived from different prion strains, as indicated. All samples were subjected to limited proteolysis. (Lower) Samples also were deglycosylated by treatment with PNGase F, as indicated (+), before SDS/PAGE.
We used a urea denaturation assay to compare PrPSc stability in the brains of mice infected with the various sets of inocula. The results revealed significant differences in the conformational stability of PrPSc molecules in the brains of animals inoculated with the three input strains (Fig. 6A). PrPSc molecules induced by the OSU input strain were the most resistant to urea denaturation [(Urea)1/2 = 3.8 M], whereas input Me7-induced PrPSc molecules were the most susceptible to denaturation, with [(Urea)1/2 = 2.0 M]. In contrast, the PrPSc molecules in the brains of mice inoculated with the three sets of cofactor PrPSc molecules (Fig. 6B) as well as the three sets of PE PrPSc inocula (Fig. 6C) all displayed similar denaturation profiles [(Urea)1/2 = 1.5–2.2 M]. It is interesting that the relatively weak resistance to denaturation exhibited by PrPSc molecules in cofactor PrPSc- and PE PrPSc-inoculated mice was unexpected, given their long scrapie incubation times (Table 2), because it had been suggested previously that long incubation times in mice usually are correlated with a high level of PrPSc conformational stability (27).
Fig. 6.

Analysis of PrPSc conformational stability. Urea denaturation assay showing PrPSc levels in samples of brain homogenates prepared from animals inoculated with samples derived from different prion strains. Inocula were (A) input prions; (B) cofactor PrPSc; (C) PE PrPSc; (D) serial-passage PE PrPSc. OSU, red squares; Me7, blue circles; 301C, green triangles. Mean values ± SEM of three replicates are shown for each point.
Strain Adaptation upon Serial Passage in Vivo.
Finally, we investigated whether the unique strain properties of cofactor PrPSc and PE PrPSc molecules would be maintained upon serial passage in mice. The results of these analyses showed that all six sets of serially passaged prions (i.e., the brains of cofactor PrPSc- and PE PrPSc-inoculated animals for all three strains) displayed incubation times (Table 2), patterns of neurotropism (Fig. 4 D and H), and PrPSc biochemical characteristics (Fig. 6 and Fig. S10) that were similar to each other. These results confirm that all the cofactor PrPSc and PE PrPSc prions had converged into a single strain. The biochemical characteristics of the PrPSc molecules in the brains of animals inoculated with each of the serially passaged prions also were indistinguishable from those of the PrPSc molecules in the brains of animals directly inoculated with cofactor PrPSc and PE PrPSc prions (Fig. 6 and Fig. S10). However, the prion incubation times and patterns of neurotropism of the second-passage prions differed from those of the cofactor PrPSc and PE PrPSc prions (Table 2 and Fig. 4; compare vacuolation profiles in Fig. 4 C and D and PrP deposition profiles in Fig. 4 G and H), indicating that additional strain adaptation occurred during in vivo propagation, presumably because of the availability of additional cellular cofactors in the intact brain.
Discussion
In this paper we used a minimal in vitro prion propagation system to study directly the effect of cofactor molecules on PrPSc conformation, infectivity, and strain properties. Our results show that withdrawal of cofactor during serial propagation of purified recombinant prions caused adaptation of PrPSc conformation, manifest as an ∼2-kDa difference in the size of the protease-resistant core. Moreover, a direct comparison between samples of cofactor-containing and protein-only PrPSc molecules (produced in parallel from the same seed and substrate mixture) using an end-point titration bioassay revealed that phospholipid molecules play a quantitatively large role in maintaining the infectivity of in vitro-generated prions. In contrast, RNA molecules are not required to maintain infectivity in the presence of copurified lipids and therefore can be considered nonessential (15). Our results may help explain why relatively modest levels of infectivity are produced when purified recPrP (presumably containing little or no bacterial lipid) has been used as a solitary substrate to generate prions by a variety of protocols (10–12, 28). Although we cannot rule out the possibility that a yet unidentified protocol could produce highly infectious prions from recPrP alone, the results of our internally controlled experiment in which an alternative, noninfectious PrPSc conformation is propagated after cofactor withdrawal suggest that this scenario is not likely to occur and that the infectious conformation of PrPSc is structurally dependent on physical interactions between PrP and essential cofactor molecules.
We also found that cofactor molecules seem to exert an influence on prion strain properties in our minimal prion propagation system. Most importantly, we found that the strain properties of recombinant prions initially formed from recPrP substrate by sPMCA with POPG and RNA molecules could be altered during subsequent serial propagation by changing only the cofactor component (from POPG/RNA to PE) to form a unique output strain characterized by a long scrapie incubation period and unique neuropathological and biochemical characteristics. These results directly show that a change in cofactor, rather than the absence of cellular processes, is sufficient to cause a change in prion strain properties. Additional experiments revealed that that two different native prion strains also adapted into the same unique output strain when propagated in vitro with PE as the only available cofactor. Given the clear differences among three input prion strains, our results indicate that a single cofactor can selectively pressure multiple prion strains to converge into a single, phenotypically distinct strain.
Prior studies have shown that strain properties are not altered either randomly or as a result of cross-contamination in seeded sPMCA reactions (26, 29). These potential pitfalls also are very unlikely to explain the results of our experiments because (i) a completely unique strain was produced reproducibly in all six independent samples containing PE; (ii) each sample was propagated in its own sonicator horn; (iii) each sample was propagated at a different time; (iv) the reaction tubes were sealed with Parafilm, a maneuver that eliminates cross-contamination during sPMCA (30); and (v) the OSU input and OSU cofactor PrPSc samples exhibit different strain characteristics, but both samples are products of sPMCA reactions with recPrP substrate seeded with OSU prions.
It is interesting to speculate that individual prion strains may propagate most efficiently with their own unique set of endogenous cellular cofactors and that the levels of these particular cofactors may vary among cell types and animal species. Various types and combinations of cofactors easily could account for the natural diversity of prion strains. This “cofactor selection” hypothesis provides a potential molecular mechanism for the generation of multiple PrPSc conformations (31) (potentially because of the interaction of PrP with multiple potential cofactors) that can adapt in response to selective pressure in cell cultures (19) or cross-species transmission (18) as well as for the phenomenon of strain-specific neurotropism (if strain-specific cofactors are enriched selectively in different brain regions). This hypothesis also is consistent with the generation of unique strain phenotypes that have been reported when infectious prions are produced in the absence of endogenous cofactors (11, 12, 28). Cofactors capable of maintaining the properties of native murine prion strains in vitro are present in crude brain homogenate, because sPMCA in brain homogenate substrate has been shown to preserve the strain-specific characteristics of 301C and other murine prions (26, 29).
Taken together, our results show that cofactor molecules such as PE modulate PrPSc structure in infectious prions, enabling the formation of the infectious conformer and restricting strain properties. These findings suggest that cofactor molecules are likely integral and essential components of infectious prions. It is possible that cofactor molecules also may play a critical role in the pathogenesis of other neurodegenerative diseases in which protein misfolding can spread through the brain, such as Alzheimer’s disease, Parkinson disease, and amyotrophic lateral sclerosis (32).
Experimental Procedures
Reagents.
The Me7 (mouse adapted scrapie originally from sheep) and 301C (mouse adapted bovine spongiform encephalopathy prions) prion strains used in this study were kindly provided by Stanley Prusiner (University of California, San Francisco) and Claudio Soto (University of Texas, Houston), respectively. The recombinant strain designated “OSU” is the recPrPSc sample originally produced de novo by F.W. as previously described (22) and subsequently propagated with purified cofactor by N.R.D. The pET-22b(+) expression plasmid (catalog no. 69744), Overnight Express Autoinduction System (catalog no. 71300–3), Bug Buster 10× plus Lyso catalog no. nase Kit (catalog no. 71370), and Ni-NTA His-Bind Superflow Resin (catalog no. 70691) were purchased from EMD Chemicals. Micrococcal (S7) nuclease (catalog no. 107921) was purchased from Roche. Thermolysin (catalog no. 88303) was purchased from Sigma. Synthetic plasmalogen phosphatidylethanolamine (PE) (catalog no. 852758P) was purchased from Avanti Polar Lipids.
Animal Care.
Female C57BL mice were purchased from Charles River Laboratories (Wilmington, MA). Mice were housed in microisolation cages and handled in strict accordance with good animal practice, as defined by the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The Dartmouth College Institutional Animal Care and Use Committee approved the animal work (assurance number A3259-01). Inoculations were performed under isoflurane anesthesia, and all efforts were made to minimize suffering.
Recombinant Mouse PrP Expression and Purification.
Amplified DNA sequences coding for mouse PrP 23–231 were ligated into the pET-22b(+) expression vector (EMD Chemicals), and sequences were verified. The expression vector then was transformed into Escherichia coli Rosetta Cells (EMD Chemicals). Cells were grown overnight in 1 L of LB medium (5 g yeast extract, 10 g Bacto tryptone, 10 g NaCl) supplemented with the Overnight Express Autoinduction System (EMD Chemicals). The next day the cells were centrifuged at 8,000 × g for 10 min, and the supernatant was discarded. Pellets were resuspended in a solution of 1× Bug Buster and 10 μL Lysonase (EMD Chemicals) containing EDTA-free Complete protease inhibitors (Roche). Cells then were incubated on ice and lysed using intermittent sonication for 20 min. The lysate was centrifuged at 16,000 × g for 20 min and was washed twice with 0.1× Bug Buster. The resulting inclusion bodies were solubilized using 8 M guanidine HCl and physical agitation, and insoluble material was removed by centrifugation at 8,000 × g for 15 min. PrP then was purified as described previously (22).
Cofactor Preparation.
The protocol for isolating the cofactor preparation and details about its composition have been described previously (21). All centrifugation was done at 4 °C unless otherwise noted. A 10% (wt/vol) brain homogenate was made by processing 0.5 g normal mouse brain in 4.5 mL of 20 mM 3-(N-morpholino)propanesulfonic acid (Mops) (pH 7.0), 150 mM NaCl with a Potter homogenizer. Debris was removed by centrifugation at 200 × g for 30 s. The postnuclear supernatant was centrifuged for 30 min at 10,000 × g, and the resulting pellet was rehomogenized in 4.5 mL of 20 mM Mops (pH 7.0), 150 mM NaCl containing 3% (wt/vol) N-octyl-β-d-glucopyranoside (NOG) (Anatrace) and incubated at room temperature for 30 min. Next, the homogenate was centrifuged at 100,000 × g for 60 min. The resulting supernatant was adjusted to 2 mM CaCl2 and 150 U/mL S7 nuclease (Roche) and was incubated at 37 °C for 30 min using an end-over-end rotator. Thermolysin (Sigma) was added at a final concentration of 25 μg /mL, and the sample was incubated at 70 °C for 60 min with intermittent mixing. Next, the sample was cooled on ice, adjusted to 5 mM EDTA, and centrifuged for 1 h at 100,000 × g. The supernatant then was placed in cellulose ester dialysis tubing with a 20,000 Molecular Weight Cutoff (Spectrum Laboratories) and dialyzed at 4 °C against water. Following dialysis, the sample centrifuged for 3 h at 100,000 × g. The supernatant was discarded, and the pellet was resuspended in 1 mL of deionized water by trituration (one-fifth of the original homogenate volume).
sPMCA.
For experiments comparing cofactor PrPSc with protein-only PrPSc molecules, reconstituted sPMCA reactions were conducted as previously reported (20), with the following modifications. Sonication pulses were 15 s every 30 min with power output ∼215 W, and 100 μL reactions contained 6 μg /mL recombinant mouse PrP (MoPrP), 20 mM Tris (pH 7.5), 135 mM NaCl, 5 mM EDTA (pH 7.5), 0.15% (vol/vol) Triton X-100 supplemented with either cofactor or water as indicated. The original recPrPSc seed used for these experiments was generated de novo by F.W. as previously described (22) and then was propagated by sPMCA with recPrP and purified cofactor by N.R.D. Cofactor PrPSc and protein-only PrPSc samples were propagated by sPMCA (18 rounds) using the same recPrPSc seed and base substrate mixture (with the addition of either cofactor for cofactor PrPSc samples or water for protein-only PrPSc samples, as indicated) and were processed in parallel in concentric positions relative to the center of the same microplate horn.
For experiments comparing different prion strains as seed, 100-μL reactions contained 6 μg /mL recombinant MoPrP, 20 mM Tris (pH 7.5), 135 mM NaCl, 5 mM EDTA (pH 7.5), 0.15% (vol/vol) Triton X-100, and either 25 μL purified cofactor or 10 mM plasmalogen PE [resuspended in 0.05% (vol/vol) Triton X-100]. Day 1 reactions were seeded with 10 μL of scrapie brain homogenate diluted 1:10 in PBS or with 10 μL recPrPSc. Samples were sonicated with 15-s pulses every 30 min for 24 h at 37 °C. After each 24-h period, 1/10th of the reaction volume was transferred to a different tube containing fresh substrate mixture, and the 24-h cycle of sonication was repeated. To prevent cross-contamination between samples, all tubes were sealed with Parafilm (Pechiney Plastic Packaging Company) and clamped shut using a plastic holder (30). Separate strains were propagated at different times in separate sonicator horns (either new or presoaked in 100% bleach) to avoid the possibility of cross-contamination between strains. Each sample was propagated for 18 rounds to eliminate the original seed by serial dilution. The effectiveness of this dilution was confirmed for each sample by performing mock propagation reactions of seeded samples lacking PrP substrate for 18 rounds. The lack of infectivity in these mock-propagated samples confirmed the adequacy of serial dilution to eliminate the original seed as well as our ability to prevent cross-contamination. To confirm that our substrate materials and inocula were not contaminated, we inoculated unseeded substrate mixture that was not subjected to sPMCA.
Seeded sPMCA experiments using normal mouse brain homogenate as substrate were performed as previously described (33).
PrPSc Detection.
To detect PrPSc molecules in either the products of PMCA reactions or brain homogenates (5% wt/vol in PBS 0.5% (vol/vol) Triton X-100), samples were digested with 25 μg/mL proteinase K for 30 min at 37 °C. All samples were processed for SDS/PAGE and Western blotting as previously described (29), substituting Towbin transfer buffer (34) and using mAb 6D11 as the primary antibody. SDS/PAGE signals were quantified using Image Gauge v4.22 (Fujifilm). PrP levels were determined semiquantitatively by comparison with a dilution series of recPrP as standard.
Scrapie Inoculation and Diagnosis.
Samples containing PMCA products were prepared and serially diluted in sterile PBS 1% (wt/vol) BSA. PMCA samples were diluted directly 1:10 (i.e., without prior concentration) to generate the 10−1 titer sample. Samples of brain homogenates were adjusted to 1% (wt/vol) final concentration in PBS 1% (wt/vol) BSA for inoculation. Intracerebral inoculations (using a volume of 30 μL) and scrapie diagnosis in C57BL female mice were performed as described previously (20).
Neuropathology.
Mice were killed, and brains were removed rapidly using new, sterile-packaged dissection instruments and disposable surfaces to avoid cross-contamination. Brains were immersion-fixed in 10% (wt/vol) buffered formalin for 2–30 d, cut into ∼3-mm-thick sagittal sections, and placed in a tissue-processing cassette. Cassettes were treated with 88% formic acid for 1 h and then were stored in PBS. The tissue was processed for paraffin embedding, and representative slides were stained with H&E. Immunohistochemistry was performed on deparaffinized slides using 2 μg/mL 27/33 anti-PrP mAb for 30 min at room temperature after citrate antigen retrieval and a Biocare Mouse on Mouse development kit. Blinded scoring for vacuolation and PrP deposition was performed using standard scales as described previously (20).
Atomic Force Microscopy.
PMCA reactions were pooled to a total volume of 1,250 μL. Each tube received 100 μL of a 50% (vol/vol) slurry of immobilized l-1-tosylamido-2-phenylethyl chloromethyl ketone (TPCK)-treated trypsin (catalog no. 20230; Thermo Scientific) and was incubated overnight at 37 °C while rotating end over end. The next morning each tube was centrifuged briefly to pellet the agarose beads. After the supernatant was removed, each tube was centrifuged at 100,000 × g for 1 h. A 250-μL pellet was resuspended to 1,200 μL in 3% (wt/vol) NOG and then was centrifuged at 100,000 × g for 1 h. Again, a 250-μL pellet was washed with 3% (wt/vol) NOG followed by two washes in 1 mL of water. The final pellet was vortexed for 20 s, sonicated for 20 s at power 65, and vortexed again for 20 s. A small amount then was treated with 25 μg/mL of proteinase K, and the concentration was determined by Western blot and densitometry.
Samples were scanned by atomic force microscopy as previously described (33). Briefly, images were captured in tapping mode using two different tips [TAP300AL cantilever with a tip radius <10 nm and a force constant of 40 N/m (Budget Sensors) or SSS-NCHR cantilever with a tip radius <2 nm and a force constant of 42 N/m (Nanosensors)]. Each sample (15 μL) was laid down on a piece of freshly cleaved mica and allowed to dry under nitrogen. Samples were rinsed three times with 150 μL of water. Data were collected as 512 × 512 pixel images, using multiple samples and several scanning sessions. Images were exported using Gwyddion 2.19 software (Czech Metrology Institute).
Fluorescence Double-Color Assay.
Two hundred microliters of recPrPSc [prepared by three rounds sPMCA using 0.5 mM NBD-PE (Avanti Polar Lipids) as a cofactor] was adjusted to 1.5-mL total volume with PBS 1% (vol/vol) Triton, centrifuged at 100,000 × g for 1 h, and washed three times with PBS 3% (wt/vol) NOG and once with PBS. The pellet was resuspended in 100 μL 10 mM acetate buffer, pH 5.0, and incubated for 1 h at room temperature and then overnight at 4 °C in a Permanox eight-well Lab-Teks chamber slide system (Nunc) in 10 mM sodium acetate, pH 5.0. The following day, the samples were fixed to the slide by the addition of 150 μL of PBS, 8% (vol/vol) formaldehyde and incubation at room temperature for 15 min. Unfixed sample was removed by aspiration, and each slide well was washed once with 150 μL of PBS. Then 150 μL of mAb D13 diluted 1:250 in PBS + 4% (wt/vol) BSA was applied to each slide well and allowed to incubate overnight at 4 °C or for 2 h at room temperature in a humidified chamber. After primary antibody incubation, the slide well was washed three times with 150 μL of PBS + 4% (wt/vol) BSA and was allowed to incubate for 15 min at room temperature during each wash. Then150 μL of secondary antibody, sheep anti-mouse conjugated with Alexa Fluor 568 (Invitrogen), diluted 1:250 in PBS + 4% (wt/vol) BSA, was applied to each slide and allowed to incubate for 2 h in the dark at room temperature in a humidified chamber. The slide well then was washed three times with 150 μL of PBS + 4% (wt/vol) BSA and was allowed to incubate for 15 min at room temperature during each wash. ProLong Antifade solution (15 μL) (Invitrogen) was added to each sample and 18-mm2 glass coverslips (catalog no. 1.5; Corning) were mounted on each slide and allowed to dry overnight in the dark in a desiccating chamber. We also prepared and analyzed a control slide coated with reaction buffer only and then stained with D13 mAb and Alexa Fluor 568 secondary antibody to rule out nonspecific binding of either antibody to the slide. A slide coated only with NBD-PE in reaction buffer was prepared to rule out nonspecific binding of NBD-PE to the slide. Finally, a slide coated with PrPSc in reaction buffer that did not contain NBD-PE was prepared to rule out autofluorescence of PrPSc in the NBD excitation wavelength. Samples were analyzed visually using a Zeiss Axioplan 2 wide-field fluorescence microscope, and digital images were captured with Phylum Live 4.2.1 software (Improvision).
Statistical Methods.
We used nonparametric approaches to compare the vacuolation characteristics in seven brain regions and immunohistochemical (IHC) deposition in eight brain regions of animals inoculated with OSU input (n = 5), Me7 input (n = 6), 301C input (n = 5), OSU cofactor (n = 6), Me7 cofactor (n = 8), 301 cofactor (n = 8), OSU PE (n = 7), Me7 PE (n = 5), or 301C PE (n = 7). Specifically, we used Mann–Whitney tests to compare IHC characteristics in each brain region from each input strain (OSU, Me7, and 301C) with its corresponding cofactor strain. We used Kruskal–Wallis equality-of-populations rank tests to assess the probability that the IHC characteristics seen within each brain region for the six output strains represented a single distribution. We repeated the analysis including all nine strains to test the hypothesis that the IHC patterns for all nine strains represent a single distribution. We defined P < 0.05 as statistically significant. Data were analyzed using Stata 12.0 (Stata Corporation).
Enzymatic Deglycosylation.
Various 10% brain homogenates were normalized for PrP scrapie content by dilution in PrnP0/0 brain homogenate. Seventy-five microliters of each homogenate was added to 25 μL of PBS, 2% (vol/vol) Triton X-100 containing 40 μg/mL proteinase K, and samples were shaken at 750 rpm for 30 min at 37 °C. After incubation, 5 μL of 200 mM PMSF (in 100% EtOH) was added, and the sample was vortexed and incubated at room temperature for 10 min. Next, samples were diluted with 895 μL PBS, 0.5% (vol/vol) Triton X-100 and were centrifuged at 100,000 × g for 60 min at 4 °C, and supernatants were discarded. Pellets then were resuspended in 20 μL of 5× glycoprotein denaturation buffer, subjected to three 30-s bursts of sonication, and boiled at 95 °C for 10 min. Samples then were diluted with 80 μL water, and sonication and boiling were repeated. Next, samples were cooled to room temperature, and 13 μL each of 10× G7 reaction buffer and 10% (vol/vol) Nonidet P-40 and 5 μL of peptide:N-glycosidase F (PNGase F) were added to each sample. Samples were incubated overnight at 37 °C. Reactions were stopped by the addition of 44 μL of 4× SDS sample buffer and boiling at 95 °C for 10 min.
Urea Denaturation Assay.
Thirty microliters of 10% (wt/vol) brain homogenate was mixed with 120 μL of various urea/0.25% (vol/vol) Triton X-100 solutions to obtain final urea concentrations between 0 and 8 M. Samples then were incubated at 60 °C for 3 h with shaking at 750 rpm. Next, 100 μL of 50 mM Mops (pH 7.0) containing 330 mM NaCl, 1% (vol/vol) Triton X-100, and 125 μg/mL proteinase K was added, and samples were incubated at 37 °C for 45–60 min, with shaking at 750 rpm. Then 84 μL of 4× SDS sample buffer was added, and samples were boiled for 10 min at 95 °C. SDS/PAGE signals were quantified using Image Gauge v4.22 (Fujifilm).
Supplementary Material
Acknowledgments
We thank Dr. Charles Daghlian for helpful advice on atomic force microscopy protocols, Drs. Charles Daghlian and Ekaterina Pletneva for helpful advice on Raman spectroscopy protocols, and Ann Lavanway for help with fluorescence microscopy. Financial support for this study was provided by National Institutes of Health Grants 2R01 NS046478, R01 NS055875, R01 NS071035, and R01 NS060729.
Footnotes
Conflict of interest statement: S.S. and N.D. are inventors on a patent held by Dartmouth College on the use of phosphatidylethanolamine as a prion cofactor.
*This Direct Submission article had a prearranged editor.
See Author Summary on page 11074 (volume 109, number 28).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206999109/-/DCSupplemental.
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 2.Caughey BW, et al. Secondary structure analysis of the scrapie-associated protein PrP 27-30 in water by infrared spectroscopy. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 3.Pan KM, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kocisko DA, et al. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 5.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci USA. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043–1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 7.Supattapone S. Biochemistry. What makes a prion infectious? Science. 2010;327:1091–1092. doi: 10.1126/science.1187790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Surewicz WK, Apostol MI. Prion protein and its conformational conversion: A structural perspective. Top Curr Chem. 2011;305:135–167. doi: 10.1007/128_2011_165. [DOI] [PubMed] [Google Scholar]
- 9.Colby DW, Prusiner SB. De novo generation of prion strains. Nat Rev Microbiol. 2011;9:771–777. doi: 10.1038/nrmicro2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colby DW, et al. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarava N, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim JI, et al. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–14087. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bosque PJ, Prusiner SB. Cultured cell sublines highly susceptible to prion infection. J Virol. 2000;74:4377–4386. doi: 10.1128/jvi.74.9.4377-4386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klöhn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci USA. 2003;100:11666–11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piro JR, Harris BT, Supattapone S. In situ photodegradation of incorporated polyanion does not alter prion infectivity. PLoS Pathog. 2011;7:e1002001. doi: 10.1371/journal.ppat.1002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bruce ME. Scrapie strain variation and mutation. Br Med Bull. 1993;49:822–838. doi: 10.1093/oxfordjournals.bmb.a072649. [DOI] [PubMed] [Google Scholar]
- 17.Carlson GA. Prion strains. Curr Top Microbiol Immunol. 1996;207:35–47. doi: 10.1007/978-3-642-60983-1_4. [DOI] [PubMed] [Google Scholar]
- 18.Angers RC, et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science. 2010;328:1154–1158. doi: 10.1126/science.1187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry. 2010;49:3928–3934. doi: 10.1021/bi100370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deleault NR, et al. Isolation of phosphatidylethanolamine as a solitary cofactor for prion formation in the absence of nucleic acids. Proc Natl Acad Sci USA. 2012;109:8546–8551. doi: 10.1073/pnas.1204498109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atarashi R, et al. Ultrasensitive detection of scrapie prion protein using seeded conversion of recombinant prion protein. Nat Methods. 2007;4:645–650. doi: 10.1038/nmeth1066. [DOI] [PubMed] [Google Scholar]
- 24.Mathur V, Taneja V, Sun Y, Liebman SW. Analyzing the birth and propagation of two distinct prions, [PSI+] and [Het-s](y), in yeast. Mol Biol Cell. 2010;21:1449–1461. doi: 10.1091/mbc.E09-11-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tyedmers J, et al. Prion induction involves an ancient system for the sequestration of aggregated proteins and heritable changes in prion fragmentation. Proc Natl Acad Sci USA. 2010;107:8633–8638. doi: 10.1073/pnas.1003895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castilla J, et al. Cell-free propagation of prion strains. EMBO J. 2008;27:2557–2566. doi: 10.1038/emboj.2008.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Colby DW, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA. 2009;106:20417–20422. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legname G, et al. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci USA. 2005;102:2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Piro JR, et al. Prion protein glycosylation is not required for strain-specific neurotropism. J Virol. 2009;83:5321–5328. doi: 10.1128/JVI.02502-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cosseddu GM, et al. Ultra-efficient PrP(Sc) amplification highlights potentialities and pitfalls of PMCA technology. PLoS Pathog. 2011;7:e1002370. doi: 10.1371/journal.ppat.1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 32.Jucker M, Walker LC. Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol. 2011;70:532–540. doi: 10.1002/ana.22615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piro JR, et al. Seeding specificity and ultrastructural characteristics of infectious recombinant prions. Biochemistry. 2011;50:7111–7116. doi: 10.1021/bi200786p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nishina KA, et al. The stoichiometry of host PrPC glycoforms modulates the efficiency of PrPSc formation in vitro. Biochemistry. 2006;45:14129–14139. doi: 10.1021/bi061526k. [DOI] [PubMed] [Google Scholar]
- 36.Baskakov IV, Legname G, Baldwin MA, Prusiner SB, Cohen FE. Pathway complexity of prion protein assembly into amyloid. J Biol Chem. 2002;277:21140–21148. doi: 10.1074/jbc.M111402200. [DOI] [PubMed] [Google Scholar]
