

Abstract
Cassava bacterial blight (CBB), incited by Xanthomonas axonopodis pv. manihotis (Xam), is the most important bacterial disease of cassava, a staple food source for millions of people in developing countries. Here we present a widely applicable strategy for elucidating the virulence components of a pathogen population. We report Illumina-based draft genomes for 65 Xam strains and deduce the phylogenetic relatedness of Xam across the areas where cassava is grown. Using an extensive database of effector proteins from animal and plant pathogens, we identify the effector repertoire for each sequenced strain and use a comparative sequence analysis to deduce the least polymorphic of the conserved effectors. These highly conserved effectors have been maintained over 11 countries, three continents, and 70 y of evolution and as such represent ideal targets for developing resistance strategies.
Keywords: innate immunity, type three effectors, next-generation sequencing
The most effective, environmentally sound, and widely used strategy for providing disease resistance to crop plants is through the deployment of resistance (R) proteins. This method of crop protection has been actively used by geneticists, breeders, and farmers for over 100 y. However, whereas some R proteins have been effective in the field for decades, others were defeated in a single season (1, 2). Our inability to a priori predict which R proteins will confer broad spectrum, durable resistance is a major limitation to crop improvement efforts and stems from an incomplete understanding of the genomic diversity present in a pathogen population. The advent of high-throughput sequencing technologies makes population genomics studies practical (3, 4).
R proteins trigger resistance responses upon recognition of pathogen-associated molecular patterns (PAMPs) at the host cell periphery or type three effector (T3E) molecules inside the host cell (5). The latter are delivered via the type three secretion system (T3SS) and collectively act to promote virulence in the host. PAMP-triggered and effector-triggered immunities have historically been divided into different classes of resistance. Resistance triggered by PAMPs tends to be weaker than resistance triggered by effectors; however, the characterization of additional PAMPs has made this type of classification less clear (5, 6). Effector repertoires from important plant pathogens have been characterized and the results have shown that pathogen genomes encode variable T3E repertoires (7). However, such studies have been limited by the use of techniques such as PCR and hybridization, neither of which easily identifies the presence of frameshift or truncation mutations, and consequently important polymorphisms may have been missed. The respective role of several individual effectors has also been deduced (7). With a few exceptions (8–12), the results have shown that effector protein functions are widely redundant and loss of a single effector does not result in a significant detriment to pathogen fitness (7). However, our ability to fully understand the role of effector proteins in pathogenesis is limited by the experiments we can perform in the laboratory, generally considering a single inoculation method with pathogen growth measured over a relatively small timescale.
Behind rice and corn, cassava is the third largest source of calories eaten by people living in the tropics yet it is a comparatively understudied crop (13). Since its domestication 5,000–7,000 y ago in South America, cassava has been spread across the tropical regions of the world. Cassava bacterial blight (CBB) incited by the bacterial pathogen, Xanthomonas axonopodis pv. manihotis (Xam) can result in severe crop losses (14). Crop improvement efforts have aimed at attaining CBB resistance; however, these efforts have been largely unsuccessful, most likely due to genetic diversity among Xam strains (15). Introduction of R proteins into cassava through genetic engineering or breeding is possible (16, 17); however, both processes are time consuming and laborious, highlighting the importance of first identifying the most promising R proteins. Here we report full-genome sequencing, PAMP, and effector identification from 65 geographically and temporally diverse Xam strains. Our data pinpoint highly conserved components of the Xam genome as ideal targets for developing resistance strategies.
Results
We assembled a collection of 65 Xam strains covering three continents and 11 countries and spanning 70 y of evolution (Fig. 1 and Table S1). We used Illumina next-generation sequencing (NGS) technology to generate 100-bp paired-end datasets for each Xam strain. Reads were de novo assembled into draft genomes (assembly statistics: Table S1). A comparison of our assembly statistics with those published previously (19) confirms the overall high quality of our assemblies. An important goal of this research was to accomplish large-scale sequencing at relatively low cost (Table S2). Notably, our high-quality draft genomes were attained without the use of a costly secondary technology such as Sanger or 454 sequencing. Our datasets ranged from 37× to 598× coverage. We did not find a significant correlation between assembly quality and coverage, suggesting that other factors, such as library quality or percentage of repetitive DNA in each genome, may dictate assembly quality.
Fig. 1.

Geographical/temporal representation of sequenced Xam strains. Represented countries are colored green. NR: no record. Map was created with the maps platform of R (18). Specific collection location of older strains is not known and therefore, for the purpose of this study, origin is limited to country.
Full-Genome SNP-Based Phylogeny.
Horizontal gene transfer from distantly translocated pathogen strains can aid the ability of local pathogen populations to overcome local resistance phenotypes. To determine the level of global movement, we constructed a phylogeny for the 65 sequenced Xam strains. Neighbor joining and maximum-likelihood phylogenetic analysis yielded highly similar trees and were done essentially as previously described (3). Genome assemblies and SNP analyses were performed using CLC Genomics Workbench. Identified SNPs were concatenated, aligned, and used in tree building. This method has the advantage of a high level of resolution even among closely related strains. Among Xam strains (12,802 SNPs from 65 strains) we observe a strong clustering by geographic origin with only a few outliers (Fig. 2A and Fig. S1). Our phylogeny shows a distinct Brazil clade that shares a common ancestor with both the Colombia and the African clades. Although CBB was first reported in Brazil in 1912, it was not reported in Africa until the 1970s (20, 21), but is thought to have been spread to Africa during the slave trade. Importantly, our results suggest that whereas Xam is primarily evolving independently in the surveyed areas, some global movement does occur, which is consistent with previous reports (22) and highlights the need for a globally effective resistance strategy.
Fig. 2.

Full-genome SNP identification allows phylogenetic analysis of closely related Xam strains. A vs. B shows difference in phylogenetic relationships when INDELs are excluded (A) or included (B). (A) CLC Genomics Workbench was used for reference-based assemblies, SNP identification, and neighbor-joining analysis (1,000 bootstraps; Fig. S1). Xam strains cluster primarily by geographic region with a few outliers. Colored groups are supported by 1,000/1,000 bootstrap replicates. Yellow, Brazil; blue, Colombia; purple, Africa; white, other/geographic outliers. (B) SNP-based phylogeny of Brazilian strains including INDELs. A total of 55,927 variable positions were identified, concatenated, and applied to neighbor-joining phylogenetic analysis (100 bootstraps, * denotes less than 100-bootstrap support for node). Date of collection for each strain is shown.
Brazil is the best-represented country in our collection (32 strains, 1941–2010). The SNP-based phylogeny described above ignores insertions/deletions (INDELs) as well as strain-specific plasmid sequences. It has previously been shown that plasmids as well as genes encoding effector molecules are often polymorphic in pathogen populations (7, 23). To investigate how inclusion of these sequences would affect phylogeny, we used Mauve (24) to align de novo contigs from the Brazil strains and compared the results to the Brazil clade from the above analyses. Variable positions were identified, concatenated, and used in tree building. With the inclusion of INDELs we observed a fourfold increase in total number of variable positions (12,802–55,927), suggesting that insertions and deletions constitute a significant proportion of the variability among Xam strains. Whereas some phylogenetic relationships were maintained with the inclusion of INDELs, many relationships were altered (compare Fig. 2A with 2B). These results could be explained by horizontal gene transfer through mobile plasmids or other sequences. Indeed, Xam strains contain diverse plasmid profiles (Fig. S2).
The phylogenetic relationship between Xam and other xanthomonads has been previously analyzed (25). As full-genome SNP analysis allows a more in-depth characterization of relatedness, we used Mauve and SNP concatenation to determine the phylogenetic relatedness of representative Xam strains compared with other fully sequenced xanthomonads. Our results show that Xam is phylogenetically most closely related to Xanthomonas euvesicatoria, Xanthomonas perforans, Xanthomonas axonopodis pv. citri (Xac306), and Xanthomonas axonopodis pv. citrumello F1 (on the basis of 1,442,868 SNPs from 20 strains, Fig. S3), consistent with previous reports (25).
Potential R Protein Targets Encoded in the Xam Genome.
R proteins recognize specific microbial molecules such as PAMPs and T3Es. The primary goal of this research was to identify the most highly conserved potential R-protein targets among the Xam pan-genome. To this end we began our analyses with two of the most well-characterized PAMPs from plant pathogenic bacteria: flg22 (from the FliC gene) and Ax21. Genes homologous to FliC and Ax21 were, unsurprisingly, present in all 65 Xam strains, suggesting that corresponding R proteins may be able to trigger resistance in response to Xam (Fig. 3). FLS2, an R protein from Arabidopsis capable of recognizing flg22, may be a good candidate. However, it is important to note that previous work has demonstrated the requirement of PAMP posttranslational modifications for triggered immunity. Ax21, for example, is a novel quorum-sensing PAMP from Xanthomonas oryzae pv. oryzae (Xoo) and requires sulfation by the putative sulfotransferase RaxST to be recognized by the rice R protein Xa21 (5, 26, 27). All tested Xam strains contain homologous sequences to the Ax21 and RaxST genes; however, 28 of 65 Xam strains contain a premature stop codon at the 5′ end of the latter coding sequence. These results suggest that whereas it may be possible to find an R protein capable of recognizing Xam Ax21, Xa21 would likely be capable of recognizing only approximately one-half the surveyed Xam strains.
Fig. 3.

Xam strains contain diverse effector repertoires. Effector repertoires were deduced on the basis of homology to known animal and plant type three effector proteins. (Upper) Eighteen geographically/temporally diverse strains are shown (see Fig. S4 for all strains). Sixty-five Xam strains contain nine core effectors (red box). TAL effector copy number was estimated from Southern blot analysis after genomic DNA digestion with EcoRI (Fig. S6). Note that these data are estimates as this technique would not resolve multiple copies contained in a single EcoRI fragment or multiple copies with conserved EcoRI sites. Symptom development was assessed 10 d postinoculation. Symptoms are representative of six leaves and were scored on the basis of level of water soaking on a scale of 1–3. +, presence of homolog; /, absence of homolog; Ψ, premature stop codon or frameshift mutation. (Lower Left) Growth assay comparing select Xam strains. Day 0, average of two inoculations; days 6 and 10, SD of at least four inoculations. (Lower Right) Bacterial growth 10 d postinoculation. Effector arsenal size, TAL effector count, and symptom development are compared. Additional virulence assays are in Fig. S6.
Identification of Xam Type Three Effectors.
R proteins can target a second class of pathogen molecules that are collectively known as T3Es. Type three effectors are delivered to host cells via the T3SS and previous research has shown that pathogenic bacteria have diverse sets of effector proteins that collectively contribute to overall pathogen virulence levels (19, 28). For the purpose of this study, we define T3Es as any proteins that have been shown to be translocated into the plant cell via the T3SS and use the recently described effector class nomenclature (29–31). To date, a definitive type three secretion signal consensus has not been identified and consequently, de novo computational prediction of T3Es remains a challenge. We used BLAST and keyword searches in the National Center for Biotechnology Information (NCBI) to generate a comprehensive database of 1,019 known and putative type three effector sequences from plant and animal pathogens (Dataset S1). We then used BLAST to identify matches in the Xam genomic contigs (Fig. 3, Figs. S4 and S5, and Materials and Methods). This initial effector identification step was conducted with loose stringency (45% amino acid identity over 50% of the effector coding sequence) as previous research has suggested that functionally homologous effector domains can be polymorphic between different pathogens (8). In addition to effector homologs from other xanthomonads, we identified several potential coding sequences with homology to known effectors from diverse pathogens, including Pseudomonas syringae and Ralstonia solanacearum. In-depth characterization of these genes will be an intriguing direction for future research. However, for the next step of this study we chose to focus our analysis on xanthomonad effectors as these are most likely to have a conserved virulence function and were generally highly similar (>70% identity) to the putative Xam homologs. Notably, homology searches may not identify frameshift or premature stop codon mutations. Consequently, we extracted the corresponding nucleotide sequence for each potential effector hit from each Xam strain, translated the coding sequence into amino acids, and performed amino acid alignments to identify alternate gene models. The results were used to modify the original BLAST output as to the presence of pseudogenes. Total effector gene content ranged from 14 to 22 (Fig. 3 and Fig. S4). Interestingly, in contrast to previous reports that attempted to assess effector content among bacterial strains on the basis of hybridization techniques, we were able to conclusively identify the presence of pseudogenes and note that this type of mutation (opposed to complete loss of coding sequence) is relatively high in Xam.
Does Size Matter?
To investigate virulence levels among Xam strains, we inoculated in vitro plantlets from an African model cassava cultivar TMS60444 (16) with a selection of strains from Africa, South America, and Asia, using an adapted stem puncture method and measuring symptoms with the area under the disease progression curve (AUDPC) scoring system (32) (Fig. S6A). In addition, we conducted bacterial growth assays by monitoring bacterial populations in planta over a 10-d period (Fig. 3). With neither of these inoculation methods did we find a significant correlation between effector repertoire size and virulence. Nor did we find evidence that the number of effector genes increased over time on the basis of comparing strains from the same location but collected during different years. Significantly, our results identified nine core effector genes that have been conserved among Xam strains across three continents, 11 countries, and 70 y of evolution (Fig. 3 and Fig. S4).
The identified conserved effectors are likely to have a role in bacterial fitness, as they have been maintained in the Xam genome for over 70 y. We can speculate about potential function of some effectors on the basis of effector characterization research from other systems. Hpa2, HpaA, HrpF, and XopAE (a.k.a. HpaF) are all located within or near the T3SS (Hrp) gene cluster (33). Hpa2 is homologous to the lysozyme-like family of proteins and is thought to form a complex with HpaA to facilitate secretion of T3Es (34, 35). HrpF is a putative T3E translocon protein although its role in virulence appears to be pathogen specific (36). XopAE and XopL both contain a leucine-rich repeat (LRR) domain and the former has been shown to be required for full virulence in Xanthomonas glycines (37, 38). XopE1 is a member of the HopX family of effectors that contain a conserved putative N-myristoylation motif (38). XopN has been shown to play a role in virulence and is thought to suppress basal defenses (39). Functional studies of XopAK are currently lacking but it contains homology to the secreted HopK1 effector from Pseudomonas strains (19) and is a putative transglutaminase (31). XopV is of unknown function (29, 40).
Sequence Comparisons Identify the Most Static Effectors.
R genes recognize the presence of pathogen molecules directly or indirectly (41–43). Recent work has shown that the virulence and avirulence function of some T3Es can be uncoupled; that is, it is possible to abolish effector recognition while maintaining effector virulence functions through site-specific mutations (8). We hypothesized that domains necessary for bacterial fitness would be those that are most highly conserved in a population. To assess the level of allelic polymorphisms among the Xam strains for each PAMP and effector, we generated nucleotide and amino acid sequence alignments for each effector. Several techniques for measuring nucleotide diversity have previously been compared (44). Although very powerful, these techniques are limited in the number of sequences that can be compared, are accompanied by assumptions about phylogeny and mutation rate that may be inappropriate for analysis of effector sequences, or do not fully encompass sequence variation. To fully encompass the variance present in these alignments as simply as possible, we chose to represent these data as a set of multidimensional vectors. Doing so allowed us to calculate variance at every position in the alignment across the length of each coding sequence without any accompanying assumptions of mutation rate (Materials and Methods). Although several genes display relatively high levels of polymorphism, others, Ax21, FliC, XopAE (HrpF), XopAK, XopL, XopV, and XopN in particular, were remarkably static across all strains (Fig. 4 and Fig. S7).
Fig. 4.

Sequence comparison among Xam effectors identifies the least polymorphic effectors. Effector sequences were obtained from Xam genome assemblies as described in Materials and Methods and Fig. S5. Clustal sequence alignments were created for each effector and sequence variance was calculated by representing each sequence as a set of multidimensional vectors. A standard mathematical measure of variance was then applied (Materials and Methods). Gene models (solid black line) are displayed directly above the x axis and the number of strains with the given gene model is displayed to the right of each gene model. Additional effectors are displayed in Fig. S7.
TAL Effectors.
In addition to the type three effectors mentioned previously, Xam strains contain an additional important class of T3Es: the transcription activator-like (TAL) effectors. TAL effectors are delivered into the host cell and then translocated to the nucleus where they bind host promoter elements and up-regulate transcription (45–48). TAL effectors contain a central repeat domain in which amino acids 12 and 13 [known as the repeat variable diresidue (RVD)] of each repeat dictate binding to a specific nucleotide in the promoter of the target plant gene. The bound sequence is known as the effector binding element (EBE) (49). Although important to pathogen virulence (50), TAL DNA sequences (tal genes) are inaccessible to assembly using short-read technology because of this highly repetitive central domain and consequently were not revealed during our Illumina assemblies and effector prediction. To gain additional information about Xam TAL effectors, we conducted Southern blot analysis to estimate the approximate copy number of TAL effector proteins across our Xam isolate collection (Fig. 3 and Fig. S8). Conveniently, because of the high level of sequence conservation across all TAL effectors, it was possible to use a single probe, to identify multiple TAL sequences. All Xam strains tested contain at least one TAL effector sequence and most strains contain multiple copies. Studies in several other Xanthomonas spp. suggest that TAL effectors play a major role in symptom development (51–54). Correspondingly, we found a moderate correlation (R2 = 0.64) between symptom severity and tal gene copy number. However, bacterial growth was only weakly correlated with tal gene copy number, suggesting that TAL effectors may have unequal contributions to overall virulence (Fig. 3).
To further characterize Xam TAL effectors, we chose to focus our studies on Xam668 and CIO151. We used Sanger sequencing from cosmid libraries and subclones to determine the TAL effector sequences for each strain. CIO151 encodes two TAL effectors (with 14 and 21 RVDs) whereas Xam668 encodes five distinct TAL effectors (with 13, 14, 15, 18, and 22 RVDs). CIO151 and Xam668 share one TAL effector of common size but differing sequence and consequently predicted EBEs (Table 1). The two largest TAL effectors from each strain (TAL21CIO151 and TAL22Xam668) are most similar to each other with mostly HD and NG RVDs, suggesting a common origin. Notably, no two sequenced TAL effectors were identical, suggesting that TAL effector sequences within and among strains may be very diverse. Effective TAL effector-based resistance strategies necessitate a more complete understanding of the TAL effector diversity on a population scale. Unfortunately, subcloning and Sanger sequencing are not high-throughput. Pacific Biosciences now offers a third-generation sequencing technology with the advantage of very long reads (up to 20 kb) but the problem of a high error rate (∼20%). We generated PacBio data for strain Xam668; however, the TAL effector sequences were still not resolved via commercial platforms such as CLC Genomics Workbench and LaserGene. Of ∼160,000 PacBio reads, we identified ∼200 reads containing tal gene sequences and many of these reads spanned the repeat region; however, because of the incredibly high error rate, it was not possible to unambiguously resolve the tal gene sequences. Our results suggest that in the near future, as long-read sequencing technologies improve their error rate, it may be possible to resolve tal gene sequences.
Table 1.
TALE RVDs
| TALE | RVD Sequence |
| TAL13XAM668 | NI NS NN HD NG HD NI NG HD NN NI NI NG |
| TAL14XAM668 | NI NG NI NN NG HD NS NS NN NG HD NN NI NG |
| TAL14CIO151 | NI NG NI NN NI HD NS NS NN NG HD NN NI NG |
| TAL15XAM668 | NI NG NI NN HD HD NS NS NS HD HD NS HD NG NG |
| TAL18XAM668 | NI NG NI NN NI HD NS NS NN NS HD NN HD HD HD NI NG NG |
| TAL21CIO151 | NI NG HD NG HD N* NG NG HD HD HD NG N* NG HD NG NG NG HD NG NG |
| TAL22XAM668 | NI NG HD NG NG NG HD HD NG NG HD NG HD HD NG NG HD NG NG HD NG NG |
Discussion
The level of dependence that people have on cassava was demonstrated during the devastating cassava mosaic disease epidemic in Uganda in the 1990s when cassava crop losses led to widespread starvation. CBB is the most important bacterial disease of cassava with losses in Africa alone totaling 7.5 million tons annually (15). Especially because Xam is a vascular pathogen and cassava is generally propagated through stem cuttings, CBB is difficult to control. To date, the most effective means of disease control in crop species are through the introduction of specific plant R proteins that recognize conserved pathogen molecules. The fact that cassava genome modification through breeding or engineering is time consuming and laborious necessitates a priori identification of the most promising resistance proteins. A fundamental limitation of most resistance proteins is that they are often defeated quickly as the cognate pathogen mutates to avoid detection. This limitation stems from an incomplete understanding of pathosystems and the factors that determine the durability of a given resistance protein.
In this study we have combined next-generation sequencing with computational biology to rapidly gain a vast amount of information about the Xam–cassava pathosystem. SNP-based phylogeny allowed high-resolution comparisons between closely related strains and suggested that although Xam strains are mostly geographically isolated, some amount of genetic drift occurs. These results highlight the need for a broadly effective strategy. In planta virulence assays were done on the cassava cultivar TMS60444, which originates from Nigeria. Our results indicate that African strains are more virulent than South American strains (P < 0.01, Fig. S6) on this cultivar, suggesting adaptation of Xam strains against regional cassava varieties; however, this observation requires further investigation to determine whether this trend applies to other African and South American varieties.
Central to the goal of this research is a fundamental concept of plant innate immunity first proposed by H. H. Flor in the 1950s: that resistance responses are traceable to a single gene from the pathogen and a single gene from the host (55). Whereas our understanding of the molecular components governing host–pathogen interactions has expanded, the underlying concept proposed by Flor has held true for innate immunity in both plants and animals. This “gene-for-gene” concept is key to the success of the resistance strategies proposed here. We identified the most globally and temporally conserved PAMP and effector molecules from the sequenced Xam genomes as ideal targets for R gene mining from cassava germplasm and cassava’s wild relatives. In addition, it may be possible to use nonhost R genes as recent research has shown that R proteins can retain function in diverse hosts (56). To identify potentially useful R genes, our future research will aim at transiently expressing the conserved PAMPS, effectors, and effector domains in diverse hosts in search of R-gene–triggered resistance responses. Once a resistance response is identified, the causal R gene must be cloned. Cloning plant genes is not an insignificant task, highlighting the importance of first identifying R-protein targets that are widespread in a pathogen population. Although still a considerable amount of work, the speed at which R genes can be cloned has increased significantly during the last decade, in large part because of creative genomic techniques (57). Once identified, R proteins will be transferred to farmer-preferred varieties of cassava that are currently susceptible to Xam infection.
Our results identify candidate targets for new resistance strategies as well as alert us to potential pitfalls. For example, the rice Xa21 gene confers strong resistance to strains of Xoo expressing the sulfated bacterial PAMP, Ax21 (5). Sulfation of Ax21 is accomplished via a second Xoo protein, RaxST (27). Ax21 is highly conserved among Xam strains but many Xam alleles of raxST contain a premature stop codon at the 5′ end of the coding sequence. These results suggest that Xa21 would confer resistance only to some strains of Xam. Notably, this important result would not have been identified through hybridization studies such as Southern analysis or PCR.
Xam strains contain TAL effectors. TAL effector proteins have previously been implicated in virulence and are thus of interest here (45). A previous study of 189 Xam strains from Colombia used Southern analysis to show that all tested strains contained at least one TAL sequence, further highlighting the high level of conservation of this group of proteins among Xam strains (58). Unfortunately, Illumina data alone were not sufficient to decipher the complicated and repetitive nature of the TAL effector coding sequences. Through traditional molecular biology techniques we confirm that the Xam genome encodes multiple members of the TAL effector class of proteins of variable size, sequence, and copy number. Our desire to further understand the genomic diversity among Xam TAL effectors directed us to the long, error-prone PacBio sequencing reads. Our hope was that the length of the PacBio reads would allow us to span the repetitive region of TAL effectors and deduce their respective RVDs. We found that it was possible to make use of these reads through manual error correction, however, and not through the tested commercial platforms. The unique structure and function of TAL effector proteins invites novel resistance strategies as described by others (46); however, our results highlight the need for a more complete understanding of the variation among TAL effectors on a pathogen population scale. This need is especially noteworthy as previous research has shown that TAL effectors induce expression of susceptibility genes in their respective hosts, facilitating pathogen growth. An outstanding question of particular interest is whether distinct TAL effectors target distinct susceptibility genes, potentially correlating with the geographic origin of different host cultivars. Technology surrounding genomic sequencing is improving essentially at lightning speed and consequently we believe that it will soon be possible to overcome the challenges currently posed by repetitive DNA sequences.
At least nine effectors, additional members of the TAL effector proteins as well as the bacterial PAMP genes FliC and Ax21, have been maintained in the Xam genome across 11 countries, three continents, and 70 y. We acknowledge that a high level of genomic conservation does not necessarily mean that the corresponding genes are impervious to loss or mutation, especially in the face of strong selective pressure. We propose that several R genes be stacked in the genome of one crop plant, as has been proposed by others (59), and that our approach be used to identify those R genes.
Notably, our studies have been inexpensive (Table S2). As prices continue to fall, in the near future it will be possible to continually sample pathogen populations collected in the field to identify potentially problematic mutations before positive selection and spread in the field. Although our experiments have focused on the cassava–Xam interaction, the methodology described herein is widely applicable to diverse pathosystems.
In summary, we identified the most highly conserved potential R-gene targets in our pathogen population. In addition, our data provide an unparalleled analysis of the evolution of a plant pathogen. Combination of NGS and computational biology represents a paradigm shift in the way resistance strategies are and will be developed against pathogens and is an essential step in the goal of feeding the rapidly increasing global human population.
Materials and Methods
Strains.
CBB was identified in cassava fields on the basis of the presence of CBB-like symptoms. Bacterial isolations were performed essentially as previously described (60). All bacterial isolates were determined to be Xam on the basis of the 16S sequence analysis (61). Spontaneous rifampicin-resistant mutations were generated for Xam strains and used in all downstream analyses.
Illumina Sequencing.
Genomic DNA was isolated using a modified CTAB (hexadecyltrimethyl ammonium bromide) protocol. Briefly, 2-mL cultures were grown in NYGB (nutrient yeast extract glycerol broth) for 2 d at 28 °C under constant shaking. Cells were pelleted and resuspended in TE (Tris/ethylenediaminetetraacetic acid). Cells were lysed in SDS/CTAB extraction buffer before phenol:chloroform:isoamylalcohol extraction. DNA was precipitated in isopropanol and washed with 70% (vol/vol) EtOH. DNA quantity and quality were assessed by a combination of nanodrop and gel electrophoresis. Ten micrograms of DNA was sheared to ∼400 bp, using a Covaris S-series instrument. Illumina paired-end libraries were prepared on an APOLLO 324 automated library prep robot (IntegenX) followed by 15 cycles of PCR amplification to add index primers for multiplexing. Before sequencing, libraries were quality tested on a bioanalyzer. Between 19 and 30 samples were multiplexed per lane of the Illumina HiSEq 2000 sequencer.
Assembly and Computation Analysis.
Assemblies were performed on CLC Genomics Workbench v 4.9, using the default parameters for reference assemblies to Xac306 and a length fraction of 0.9 and a similarity of 1.0 for reference assemblies to Xam668 and de novo assemblies.
At the time of analysis, full-genome sequences were available from NCBI for 12 xanthomonads. We used Mauve3.2.1 to align these genomes with 7 geographically and temporally diverse Xam strains and to identify genome-wide SNPs among all strains. SNPs were concatenated for use in a maximum-likelihood analysis (3). Unfortunately, Mauve was not able to process all 65 Xam strains, for unknown reasons. Consequently, we used CLC to identify SNPs from reference-based assemblies (Xam668 or Xac306) to compare all 65 Xam strains. Concatenated SNPs were fed into PHYLIP3.69 dnaml for maximum-likelihood analysis or the neighbor-joining tree-building program of CLC genomics workbench. Default parameters were used (transition/transversion ratio 2.0000, with constant rate variation among sites). Resulting trees were formatted using FigTree.
Effector Prediction.
All known type three effectors from animal and plant pathogens were collated into a spreadsheet with NCBI accessions, annotations, and amino acid sequence. This database is available for download as Dataset S1. Effector prediction in the newly sequenced Xam strains followed a three-stage process: (i) Potential effectors were identified by BLAST with a 45% amino acid homology as the lower limit. (ii) In-house python scripts were used for gene prediction and sequence extraction from Illumina contigs. Nucleotide sequences were translated to amino acids. (iii) Hits were inspected for truncations and frameshift mutations. A confirmed hit represents an amino acid sequence that is at least 80% of the length of the target sequence and at least 45% homologous. Most analyses were performed on de novo assemblies from CLC genomics workbench; however, in some cases, coding sequences extended past the end of contigs. In this case, reference-based assemblies to Xac306 or Xam668 were also considered.
Sequence Variation Analysis.
We represent a position in a sequence as a five-dimensional unit vector of the form si = (A, T, C, G, –), where the values of A, T, C, G, and – are 1 if the sequence has that nucleotide or gap at a given position and 0 otherwise. The variance of the set of N vectors is
where c is the average or “consensus” sequence, 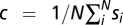 , and
, and 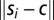 is the Euclidean distance between si and c. For amino acid variance, the same concept was used with vectors long enough to encompass all possible amino acids.
is the Euclidean distance between si and c. For amino acid variance, the same concept was used with vectors long enough to encompass all possible amino acids.
Growth Assays.
Xam was suspended in 10 mM MgCl2 at an OD600 = 0.01. A razor blade was used to create a small nick on the underside of fully expanded leaves and a 1-mL syringe was used to introduce a small amount of the Xam solution to the leaf (cassava cultivar TMS60444). At each time point, leaf punches (1 cm2) were ground in 10 mM MgCl2 with one 3-mm glass bead in a beadbeater. Serial dilutions were plated on NYGA (nutrient yeast extract glycerol agar) plates supplemented with rifampicin (100 μg/mL) to estimate bacterial populations.
AUDPC Assays.
Area under the disease progression curve was measured essentially as previously described (32).
Supplementary Material
Acknowledgments
We thank A. Bogdanove, P. Ronald, S. Goritschnig, and M. Scharlach for critical reading of the manuscript; Drs. K. Murch and H. Haggard for guidance on measuring sequence variation; and Prof. J. Taylor for guidance on phylogenetic analysis. This research was supported by a National Science Foundation/BREAD (Basic Research to Enable Agricultural Development) grant (Award 0965418 to B.J.S.) and a NIFA (National Institute of Food and Agriculture) postdoctoral fellowship (Award 2011-67012-30662 to R.B.).
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AKCW00000000–AKFI00000000).
See Author Summary on page 11082 (volume 109, number 28).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1208003109/-/DCSupplemental.
References
- 1.Fu D, et al. A kinase-START gene confers temperature-dependent resistance to wheat stripe rust. Science. 2009;323:1357–1360. doi: 10.1126/science.1166289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunkeaw S, Tan S, Coaker G. Molecular and evolutionary analyses of Pseudomonas syringae pv. tomato race 1. Mol Plant Microbe Interact. 2010;23:415–424. doi: 10.1094/MPMI-23-4-0415. [DOI] [PubMed] [Google Scholar]
- 3.Lieberman TD, et al. Parallel bacterial evolution within multiple patients identifies candidate pathogenicity genes. Nat Genet. 2011;43:1275–1280. doi: 10.1038/ng.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris SR, et al. Whole-genome analysis of diverse Chlamydia trachomatis strains identifies phylogenetic relationships masked by current clinical typing. Nat Genet. 2012;44:413–419, S1. doi: 10.1038/ng.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee S-W, et al. A type I-secreted, sulfated peptide triggers XA21-mediated innate immunity. Science. 2009;326:850–853. doi: 10.1126/science.1173438. [DOI] [PubMed] [Google Scholar]
- 6.Thomma BPHJ, Nürnberger T, Joosten MHAJ. Of PAMPs and effectors: The blurred PTI-ETI dichotomy. Plant Cell. 2011;23:4–15. doi: 10.1105/tpc.110.082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunnac S, et al. Genetic disassembly and combinatorial reassembly identify a minimal functional repertoire of type III effectors in Pseudomonas syringae. Proc Natl Acad Sci USA. 2011;108:2975–2980. doi: 10.1073/pnas.1013031108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, Dahlbeck D, Krasileva KV, Fong RW, Staskawicz BJ. Computational and biochemical analysis of the Xanthomonas effector AvrBs2 and its role in the modulation of Xanthomonas type three effector delivery. PLoS Pathog. 2011;7(12):e1002408. doi: 10.1371/journal.ppat.1002408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metz M, et al. The conserved Xanthomonas campestris pv. vesicatoria effector protein XopX is a virulence factor and suppresses host defense in Nicotiana benthamiana. Plant J. 2005;41:801–814. doi: 10.1111/j.1365-313X.2005.02338.x. [DOI] [PubMed] [Google Scholar]
- 10.Kim J-G, et al. Xanthomonas T3S effector XopN suppresses PAMP-triggered immunity and interacts with a tomato atypical receptor-like kinase and TFT1. Plant Cell. 2009;21:1305–1323. doi: 10.1105/tpc.108.063123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Underwood W, Zhang S, He SY. The Pseudomonas syringae type III effector tyrosine phosphatase HopAO1 suppresses innate immunity in Arabidopsis thaliana. Plant J. 2007;52:658–672. doi: 10.1111/j.1365-313X.2007.03262.x. [DOI] [PubMed] [Google Scholar]
- 12.Kay S, Bonas U. How Xanthomonas type III effectors manipulate the host plant. Curr Opin Microbiol. 2009;12:37–43. doi: 10.1016/j.mib.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Food and Agriculture Organization 2008. Cassava [Food and Agriculture Organization of the United Nations (FAO), Rome]
- 14.Lozano JCS, L. Bacterial blight of cassava in Colombia: Epidemiology and control. Phytopathology. 1974;64:83–88. [Google Scholar]
- 15.Hillocks RJ, Thresh JM, Bellotti AC. Cassava: Biology, Production and Utilization. New York: CABI; 2002. [Google Scholar]
- 16.Bull SE, et al. Agrobacterium-mediated transformation of friable embryogenic calli and regeneration of transgenic cassava. Nat Protoc. 2009;4(12):1845–1854. doi: 10.1038/nprot.2009.208. [DOI] [PubMed] [Google Scholar]
- 17.Ceballos H, Iglesias CA, Pérez JC, Dixon AGO. Cassava breeding: Opportunities and challenges. Plant Mol Biol. 2004;56(4):503–516. doi: 10.1007/s11103-004-5010-5. [DOI] [PubMed] [Google Scholar]
- 18.Becker RA, Wilks AR, Brownrigg R, Minka TP. 2012. maps: Draw Geographical Maps. R package version 2.2-5.
- 19.Potnis N, et al. Comparative genomics reveals diversity among xanthomonads infecting tomato and pepper. BMC Genomics. 2011;12:146. doi: 10.1186/1471-2164-12-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verdier V, Restrepob S, Mosquerab G. Genetic and pathogenic variation of Xanthomonas axonopodis pv. manihotis in Venezuela. Plant Pathol. 1998;47:601–608. [Google Scholar]
- 21.Williams RJ, Agboola SD. Bacterial wilt of cassava in Nigeria. Plant Dis Reporter. 1973;57:824–827. [Google Scholar]
- 22.Verdier V, Dongo P, Boher B. Assessment of genetic diversity among strains of Xanthomonas campestris pv. manihotis. Microbiology. 1993;139:2591–2601. [Google Scholar]
- 23.Sundin GW, Demezas DH, Bender CL. Genetic and plasmid diversity within natural populations of Pseudomonas syringae with various exposures to copper and streptomycin bactericides. Appl Environ Microbiol. 1994;60:4421–4431. doi: 10.1128/aem.60.12.4421-4431.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darling AE, Mau B, Perna NT. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010 doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parkinson N, Cowie C, Heeney J, Stead D. Phylogenetic structure of Xanthomonas determined by comparison of gyrB sequences. Int J Syst Evol Microbiol. 2009;59:264–274. doi: 10.1099/ijs.0.65825-0. [DOI] [PubMed] [Google Scholar]
- 26.Han SW, et al. Small protein-mediated quorum sensing in a Gram-negative bacterium. PLoS ONE. 2011;6:e29192. doi: 10.1371/journal.pone.0029192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Lee SW, Han SW, Bartley LE, Ronald PC. From the Academy: Colloquium review. Unique characteristics of Xanthomonas oryzae pv. oryzae AvrXa21 and implications for plant innate immunity. Proc Natl Acad Sci USA. 2006;103:18395–18400. doi: 10.1073/pnas.0605508103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai R, et al. The plant pathogen Pseudomonas syringae pv. tomato is genetically monomorphic and under strong selection to evade tomato immunity. PLoS Pathog. 2011 doi: 10.1371/journal.ppat.1002130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.White FF, Potnis N, Jones JB, Koebnik R. The type III effectors of Xanthomonas. Mol Plant Pathol. 2009;10:749–766. doi: 10.1111/j.1364-3703.2009.00590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dean P. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiol Rev. 2011;35:1100–1125. doi: 10.1111/j.1574-6976.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- 31.Koebnik R. 2011. The Xanthomonas Resource. Available at http://xanthomonas.org/
- 32.Restrepo S, Duque M, Verdier V. Characterization of pathotypes among isolates of Xanthomonas axonopodis pv. manihotis in Colombia. Plant Pathol. 2000;49:680–687. [Google Scholar]
- 33.Lu H, et al. Acquisition and evolution of plant pathogenesis-associated gene clusters and candidate determinants of tissue-specificity in xanthomonas. PLoS ONE. 2008 doi: 10.1371/journal.pone.0003828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li YR, et al. Hpa2 required by HrpF to translocate Xanthomonas oryzae transcriptional activator-like effectors into rice for pathogenicity. Appl Environ Microbiol. 2011;77:3809–3818. doi: 10.1128/AEM.02849-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lorenz C, et al. HpaA from Xanthomonas is a regulator of type III secretion. Mol Microbiol. 2008;69:344–360. doi: 10.1111/j.1365-2958.2008.06280.x. [DOI] [PubMed] [Google Scholar]
- 36.Büttner D, Nennstiel D, Klüsener B, Bonas U. Functional analysis of HrpF, a putative type III translocon protein from Xanthomonas campestris pv. vesicatoria. J Bacteriol. 2002;184:2389–2398. doi: 10.1128/JB.184.9.2389-2398.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JG, et al. Characterization of the Xanthomonas axonopodis pv. glycines Hrp pathogenicity island. J Bacteriol. 2003;185:3155–3166. doi: 10.1128/JB.185.10.3155-3166.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang W, et al. Identification of six type III effector genes with the PIP box in Xanthomonas campestris pv. campestris and five of them contribute individually to full pathogenicity. Mol Plant Microbe Interact. 2009;22:1401–1411. doi: 10.1094/MPMI-22-11-1401. [DOI] [PubMed] [Google Scholar]
- 39.Kim JG, et al. Xanthomonas T3S effector XopN suppresses PAMP-triggered immunity and interacts with a tomato atypical receptor-like kinase and TFT1. Plant Cell. 2009;21:1305–1323. doi: 10.1105/tpc.108.063123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Furutani A, et al. Identification of novel type III secretion effectors in Xanthomonas oryzae pv. oryzae. Mol Plant Microbe Interact. 2009;22:96–106. doi: 10.1094/MPMI-22-1-0096. [DOI] [PubMed] [Google Scholar]
- 41.Mackey D, Holt BF, 3rd, Wiig A, Dangl JL. RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell. 2002;108:743–754. doi: 10.1016/s0092-8674(02)00661-x. [DOI] [PubMed] [Google Scholar]
- 42.Tang XY, et al. Initiation of plant disease resistance by physical interaction of AvrPto and Pto kinase. Science. 1996;274:2060–2063. doi: 10.1126/science.274.5295.2060. [DOI] [PubMed] [Google Scholar]
- 43.Abramovitch RB, Anderson JC, Martin GB. Bacterial elicitation and evasion of plant innate immunity. Nat Rev Mol Cell Biol. 2006;7:601–611. doi: 10.1038/nrm1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nei M, Jin L. Variances of the average numbers of nucleotide substitutions within and between populations. Mol Biol Evol. 1989;6:290–300. doi: 10.1093/oxfordjournals.molbev.a040547. [DOI] [PubMed] [Google Scholar]
- 45.Römer P, et al. Promoter elements of rice susceptibility genes are bound and activated by specific TAL effectors from the bacterial blight pathogen, Xanthomonas oryzae pv. oryzae. New Phytol. 2010;187:1048–1057. doi: 10.1111/j.1469-8137.2010.03217.x. [DOI] [PubMed] [Google Scholar]
- 46.Bogdanove AJ, Schornack S, Lahaye T. TAL effectors: Finding plant genes for disease and defense. Curr Opin Plant Biol. 2010;13:394–401. doi: 10.1016/j.pbi.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 47.Mak AN-S, Bradley P, Cernadas RA, Bogdanove AJ, Stoddard BL. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science. 2012;335(6069):716–719. doi: 10.1126/science.1216211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng D, et al. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science. 2012;335(6069):720–723. doi: 10.1126/science.1215670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antony G, et al. Rice xa13 recessive resistance to bacterial blight is defeated by induction of the disease susceptibility gene Os-11N3. Plant Cell. 2010;22:3864–3876. doi: 10.1105/tpc.110.078964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bonas U, Stall RE, Staskawicz B. Genetic and structural characterization of the avirulence gene avrBs3 from Xanthomonas campestris pv. vesicatoria. Mol Gen Genet. 1989;218:127–136. doi: 10.1007/BF00330575. [DOI] [PubMed] [Google Scholar]
- 51.Marois E, Van den Ackerveken G, Bonas U. The xanthomonas type III effector protein AvrBs3 modulates plant gene expression and induces cell hypertrophy in the susceptible host. Mol Plant Microbe Interact. 2002;15:637–646. doi: 10.1094/MPMI.2002.15.7.637. [DOI] [PubMed] [Google Scholar]
- 52.Yang B, White FF. Diverse members of the AvrBs3/PthA family of type III effectors are major virulence determinants in bacterial blight disease of rice. Mol Plant Microbe Interact. 2004;17:1192–1200. doi: 10.1094/MPMI.2004.17.11.1192. [DOI] [PubMed] [Google Scholar]
- 53.Shiotani H, Fujikawa T, Ishihara H, Tsuyumu S, Ozaki K. A pthA homolog from Xanthomonas axonopodis pv. citri responsible for host-specific suppression of virulence. J Bacteriol. 2007;189:3271–3279. doi: 10.1128/JB.01790-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Al-Saadi A, et al. All five host-range variants of Xanthomonas citri carry one pthA homolog with 17.5 repeats that determines pathogenicity on citrus, but none determine host-range variation. Mol Plant Microbe Interact. 2007;20:934–943. doi: 10.1094/MPMI-20-8-0934. [DOI] [PubMed] [Google Scholar]
- 55.Flor HH. Host-parasite interaction in flax rust- its genetics and other implications. Phytopathology. 1955;45:680–685. [Google Scholar]
- 56.Zhao B, et al. The avrRxo1 gene from the rice pathogen Xanthomonas oryzae pv. oryzicola confers a nonhost defense reaction on maize with resistance gene Rxo1. Mol Plant Microbe Interact. 2004;17:771–779. doi: 10.1094/MPMI.2004.17.7.771. [DOI] [PubMed] [Google Scholar]
- 57.Bart RS, Chern M, Vega-Sánchez ME, Canlas P, Ronald PC. Rice Snl6, a cinnamoyl-CoA reductase-like gene family member, is required for NH1-mediated immunity to Xanthomonas oryzae pv. oryzae. PLoS Genet. 2010 doi: 10.1371/journal.pgen.1001123. 6:pii:e1001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Restrepo S, Verdier V. Geographical differentiation of the population of Xanthomonas axonopodis pv. manihotis in Colombia. Appl Environ Microbiol. 1997;63:4427–4434. doi: 10.1128/aem.63.11.4427-4434.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu S, Li Y, Vossen JH, Visser RG, Jacobsen E. Functional stacking of three resistance genes against Phytophthora infestans in potato. Transgenic Res. 2012;21:89–99. doi: 10.1007/s11248-011-9510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Verdier V, Mosquera G, Assigbétsé K. Detection of the cassava bacterial blight pathogen, Xanthomonas axonopodis pv. manihotis, by polymerase chain reaction. Plant Dis. 1998;82:79–83. doi: 10.1094/PDIS.1998.82.1.79. [DOI] [PubMed] [Google Scholar]
- 61.Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703. doi: 10.1128/jb.173.2.697-703.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
