

Abstract
NADPH-dependent flavoreductases are important drug targets. During their enzymatic cycle thiolates and selenolates that have high affinity for transition metals are generated. Auranofin (AF), a gold-containing compound, is classified by the World Health Organization as an antirheumatic agent and it is indicated as the scaffold for the development of new anticancer and antiparasitic drugs. AF inhibits selenocysteine-containing flavoreductases (thioredoxin reductase and thioredoxin glutathione reductase) more effectively than non Se-containing ones (glutathione reductase); this preference has been ascribed to the high affinity of selenium for gold. We solved the 3D structure of the Se-containing Thioredoxin Glutathione Reductase from the human parasite Schistosoma mansoni complexed with Au and our results challenge this view: we believe that the relative velocity of the reaction rather than the relative affinity, depends on the presence of Sec residues, which appear to dictate AF selectivity.
Keywords: Thioredoxin Glutathione Reductase, Gold–cysteine complex, Gold–selenocysteine complex, Auranofin
1. Introduction
Gold, as well as other transition metals and metalloids (e.g. silver, mercury and antimony), is a selective inhibitor of thiol-dependent flavoreductases, notably of thioredoxin reductase [1,2,3]. Metal transfer to target enzymes constitutes the molecular basis of gold-based drugs (e.g. auranofin, AF), that are widely employed to treat chronic rheumatoid arthritis and are under in vitro study in such diverse conditions as cancer and parasitic infections [4,5,6]. Given that the metal salts are usually poorly soluble, most gold (and other metal) preparations are coordination complexes from which the metal is transferred to its binding sites on the protein targets (Fig. 1).
Fig. 1.
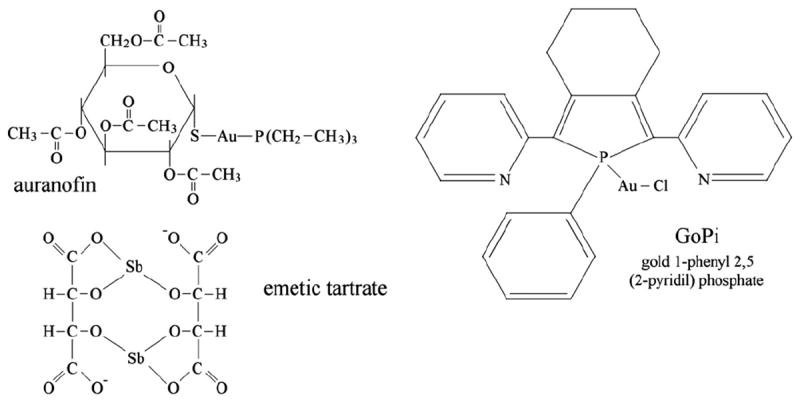
Formulas of some metal-based TrxR inhibitors: auranofin (acetoxy-thioglucose gold triethyl phosphine; gold 3,4,5-triacetyloxy- 6-acetyloxymethyl oxane 2-thiolate triethylposphanium, upper left); GoPI (gold 1-phenyl 2,5-di (2-pyridyl) phosphate, upper right); emetic tartar (potassium antimony tartrate, lower left).
The observation that the preferred biological targets of gold-based drugs are the selenium-containing thioredoxin reductases (TrxR), together with the known high affinity of Au for Se, has been taken as an indication that selenocysteine (Sec) residues are the preferred binding sites for the metal. Sec residues, however, cannot be the only target for the metals, since: (i) the gold complex GoPI (Fig. 1) is capable of transferring the metal also to the Sec-lacking glutathione reductase (GR), in keeping with the high affinity of gold for the FAD/Cys active site [7, see below]; (ii) AF is an effective inhibitor of the Selacking TrxR from Plasmodium falciparum [5]; and (iii) a study on gold(I)–TrxR interactions by mass spectrometry indicated that up to four Au(Pet)3 moieties can be attached to each protein monomer, that contains only one Sec residue [8].
We became particularly interested in the binding of gold (released by AF) to thioredoxin reductases after the discovery that this drug is effective against schistosomes. Schistosomes are helminth parasites of humans that cause a serious disease widespread in tropical and subtropical countries [9]. Since we had already solved the structure of the Se-containing Thioredoxin Glutathione Reductase (TGR) from Schistosoma mansoni [10], a validated drug target whose inhibition by AF kills the parasite [6], it seemed straightforward to crystallize the AF–TGR complex. After crystallization we solved the 3D structure of the complex [11], which revealed some puzzling features, and prompted us to challenge commonly held views on the mechanism of inhibition of thiol-reductases by Au. In particular, as detailed below, we could demonstrate that Se plays by-and-large a catalytic role, and that the Au can be transferred from Se to the sulfur of two Cys couples [11]. In this article we review the thermodynamic, kinetic and stereochemical bases of gold binding to TGR and related flavoreductases.
2. The geometries of Gold–Cys complexes
Sulfur is a strong ligand of gold and other transition metals, provided that the sulfur containing molecular scaffold allows a geometry suitable for metal chelation. In the case of gold and other monovalent transition metals, the ideal coordination geometry is linear bidentate, thus it should be possible to predict which Cys couples in a polypeptide chain of known structure may constitute a high affinity binding site.
Gold complexes formed by a single Cys residue have been described and characterized, also in view of their possible biotechnological applications (e.g. immobilization of proteins onto gold surfaces or complexation with gold nanoparticles). In these complexes gold is usually coordinated by a Cys residue and by an external ligand (e.g. cyanide): this occurs in rhodanese, whose gold complexes have been characterized by X-ray crystallography [12], cathepsin B [13], and albumin [14]. Sec may form similar complex as observed in the case of glutathione peroxidase [15].
Complexes of gold with a single Cys or Sec are reported to be reversible, usually in the sense that the metal can be transferred to other acceptors, often free Cys or GSH; however, in the case of cathepsin B, gold displacement by the substrate has been observed [13].
Much more stable complexes are formed when the polypeptide chain provides two ligands with linear geometry, in the form of Cys–Cys or Cys–Sec couples. Since the stereochemical requirements for gold binding to these couples partially overlap with those for the formation of disulfide (or sulfide–selenide) bonds, and since gold can only combine with reduced S or Se, not all Cys–Cys or Cys–Sec couples are suitable for gold complexation. Based on crystallography studies, we propose that the C(X)nC motif (with 3<n<8) is intrinsically apt to bind gold and other monovalent metals. Moreover, the same motif may bind divalent or trivalent metals, with non linear geometry, if other residues of the polypeptide chain provide additional ligands [16].
The motif is characteristic of proteins belonging to the metal regulatory family (Mer), whose function is to bind metal ions and to activate protein transcription in bacteria. CueR, a member of this protein family, binds metal ion with zeptomolar affinity (10−21 M). CueR accomplishes his task taking advantage on a couple of Cys on a loop between a long helix and a 2-turn helix spaced by 7 amino acidic residues, which traps Ag(I), Cu(I) and Au(I) between the two sulfurs arranged in a linear geometry [16]. The evolutionarily unrelated flavoenzyme mercuric ion reductase [MerA, 17], which protects cells from mercury(II), also presents the C(X)nC motif. This enzyme is able to bind and to reduce mercuric ion between the two cysteines of the same motif at the FAD binding site, which is comprised in a secondary structure region analogous to CueR, i.e. helix/loop/1-turn helix. It is important to remind that Hg2+ behaves as a monovalent metal given its preference for a linear coordination geometry.
The C(X)4C motif is present in the active site of several flavoreductases, including glutathione reductase, thioredoxin reductase, and thioredoxin glutathione reductase [7,10]. These enzymes are evolutionarily and structurally related to mercuric ion reductase and share same fold, same cofactors and same residues at the active sites. TrxR and TGR, having a second redox active Cys–Sec couple are most reactive towards gold containing drugs, and have been identified as their preferred macromolecular targets. The two active site Cys close to the FAD occur in a helix-turn-helix secondary structure region similar to that of Mer and MerA, that maintain the linear arrangement of the S atoms.
Trypanothione reductase, another flavoreductase of the same family, whose structure and whose complexes with gold(I), silver (I) and other metals have been characterized [18,19,20] presents a C(X)4C in a helix-loop-helix structure, that proved particularly suitable to accommodate metal ions.
Given that the active site of flavoreductases possesses the stereochemical features required for tight metal binding, the reasons of the specificity of gold containing drugs for Sec-containing flavoreductases must be searched in properties other than the high metal affinity of Se; the hypothesis that the role of Sec in Au combination is catalytic is discussed below.
3. Our model protein, Thioredoxin Glutathione Reductase from S. mansoni, and its similarity to TrxRs and GRs
TGR is a homodimeric flavoenzyme, each monomer resulting from the fusion of an N-terminal glutaredoxin (Grx)-like domain and a much larger C-terminal TrxR-like domain. The sequence of the wild-type enzyme comprises 598 residues, the last two representing a stretch of the glutathione moiety. The molecule has an overall W shape (Fig. 2), the subunit interface being contributed by the TrxR domain [10]. The FAD binds in a cleft situated between the NADPH binding site and the redox active Cys couple formed by Cys 154 and Cys 159. Since in most of our structures the last 6 residues are disordered, the position of the catalytically important Sec residue cannot be ascertained; after extensive search of different crystallization conditions however, we solved one structure in where one of the two C-termini of the dimer could be located, and found to point in the direction of the Cys154–Cys159 couple [21]. Starting from this structure, modeling showed that the C-terminus could approach the Grx domain as well as the redox site of a bound Trx molecule [21,22]. The binding site of Trx on TGR was modeled using the similar Trx–TrxR complex from Escherichia coli [23] as a template; whereas the complexes with the other physiological substrates, NADPH and GSH, could be obtained experimentally by crystallography [21].
Fig. 2.
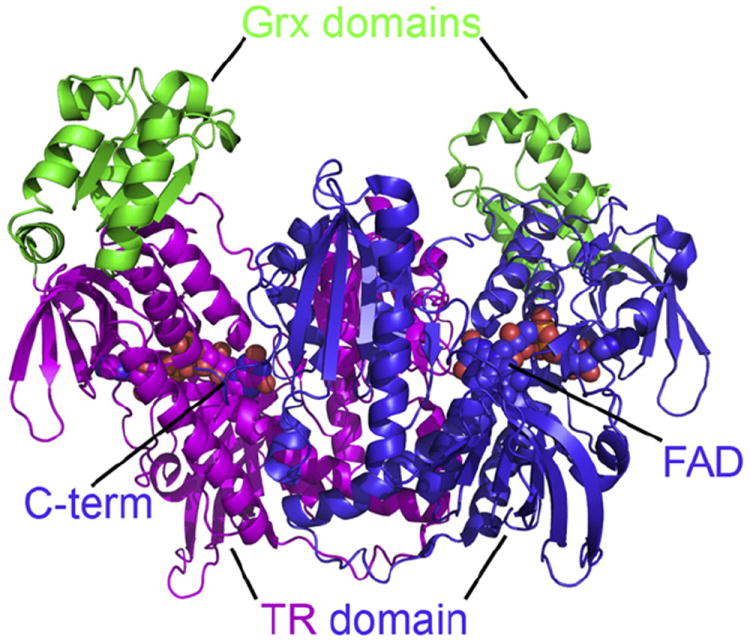
Overall structural architecture of the biological unit of TGR from Schistosoma mansoni. The enzyme is a W-shaped homodimer, each monomer being the fusion of a Grx domain (in green) and a TrxR domain (in magenta and in blue). At the bottom of a large cavity lies the FAD cofactor (shown as a sphere). The C-terminal, Sec-containing arm of each TrxR subunit points towards the FAD binding site of the other.
The first structure we solved was a truncated TGR derivative lacking the last two C-terminal residues (i.e. Sec597 and Gly 598). This enzyme was unable to reduce either Trx or its internal Grx domain, as expected given that internal electron transfer demands movement of the C-terminal arm that carries the redox active Cys–Sec couple. Nevertheless the truncated enzyme could accept electrons from NADPH and could reduce DTNB and GSSG. This led us to hypothesize that the site of GSSG reduction was located on the Cys154–Cys159 couple, homologous to that of GRs [10]. Further experiments carried out on the Sec598–Cys mutant, as well as on the wild-type enzyme, convinced us that the principal site for reduction of GSSG is located on the Grx domain [21,24].
4. The known structures of gold (and other metal) complexes of GR, TryR and TGR
Given the efficacy of gold-based drugs, several functional and structural studies have been published on the inhibition of thiol-dependent reductases by this metal and its complexes. In all known structures, gold is incorporated in the target enzyme as the ion complexed with two sulfur atoms from Cys residues (with the exception of site 3 in TGR). In no case does the complex involve the metal bound to its original ligands (see Fig. 1); thus we conclude that the Au complexes are pro-drugs, whose role is that of delivering the metal to its final target.
Fig. 3 shows the complexes of Au (released by AF) with two Cys couples of S. mansoni TGR (panels A and B), and that of Au (released by GoPI) with the active site of human GR (panel C). In panel D the structure of the active site of the related enzyme trypanothione reductase (TryR) from Leishmania infantum complexed with antimony (released by emetic tartrate) is shown [20].
Fig. 3.
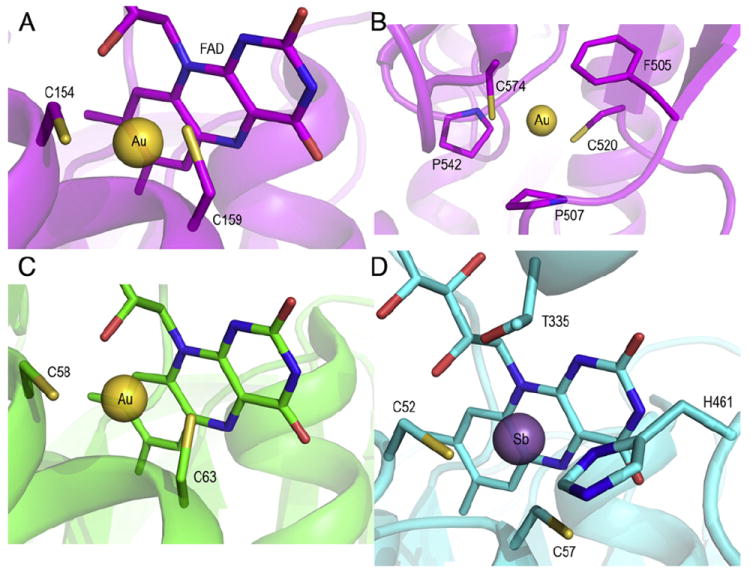
Panel A: Structure of the gold complex with C154-C159 of SmTGR (site 1); panel B: structure of the gold complex with C2520-C574 of SmTGR (site 2); panel C: gold binding site of human GR [7]; panel D: antimony binding site of TryR. Notice that while gold is two-coordinated, antimony is three- or four-coordinated [20].
In human GR Au is complexed to the two Cys residues of the active site in a typical linear S–Au–S geometry (Fig. 3C); the same applies to Au bound to site 1 of S. mansoni TGR (Fig. 3A). Antimony bound to TryR (Fig. 3D) is coordinated by the two active site Cys residues and by Thr335 and His461. The Cys couples involved in these three cases are homologous and comprise the residues numbered 58–63 in human GR, 154–159 in S. mansoni TGR, and 52–57 in L. infantum TryR. These cysteines, located in the neighborhood of the bound FAD (see above), constitute the principal redox site of all three enzymes. In the case of GR and TryR, the FAD–Cys couple is the only redox center of the protein, shuttling electrons along the path: NADPH → FAD ↔ Cys2–GSSG/Try. At low concentration of NADPH, the coupling between the FAD and Cys residues is so strong that the site accepts only two electrons forming the so called EH2 state [25]. In EH2 electrons are shared between the FAD and the Cys couple, the visible spectrum of this redox intermediate suggesting partial reduction of the flavine. Even, in the presence of excess NADPH, the derivative EH4 (with fully reduced FAD and Cys couple) is slowly populated, although it is believed to be a major species during the catalytic cycle under physiological conditions [25]. In TGR/TrxR a more complex pathway operates since EH2 is suggested to deliver its electrons to the C-terminal Sec-Cys couple forming a second type of EH2. The C-terminal arm, which is long and flexible, is required to deliver electrons to the Grx domain of the enzyme or to Trx. Since the oxidized FAD can accept other electrons from NADPH, in TGR/TrxR both the redox states EH4 and EH6 can be populated, depending on the redox balance of the cell [21,24,26,27].
Gold in complex with Cys520–Cys574 at site 2 in TGR, though basically similar to site 1, is remarkable because of two facts: (i) the Cys residues involved are somewhat farther apart from each other so that the metal exhibits a fluxional behavior, moving in between two different conformers; and (ii) the metal is greatly stabilized in its binding site by a charge transfer complex with Phe505 [11].
In our maps of TGR we also found a metal ion, identified as Au, at an additional site that we called site 3 [11]. This has been puzzling since the metal seems trapped in a hydrophobic pocket but is not covalently bound to any residue and we could not imagine how it had been stripped from its ligands, which are either acetoxythioglucose (Tagl) and triethyl phosphine (P(Et)3) or Sec and Cys.
Since in our structures of S. mansoni TGR the last 6C-terminal residues (that include Cys 596 and Sec 597) are disordered, we cannot exclude nor confirm a further Au binding site with a S–Au–Se adduct.
5. On the presumed affinity of the Cys–Au–Cys complex
The structures of Au–GR and Au–TGR, though strongly consistent with each other, contrast sharply with some widely held conceptions on gold binding to thiol-reductases [3]. Indeed gold is assumed to preferentially combine with Sec residues and the reported affinities of the metal complexes for Sec-containing TrxRs are approximately three orders of magnitude greater than the corresponding affinities for Se-lacking GRs [3]. However, in the structures described above the metal is bound to Cys couples both in the Se-lacking GR and in the Sec-containing TGR, and the geometry of the complexes is very similar. Moreover the estimated affinity of the linear S–Au–S complex is extremely high, on the order of 55 kcal/mole [28], very reasonable for a coordination complex, but completely inconsistent not only with the high IC50 of AF for GR, but also with the relatively low IC50 for TrxR and TGR [3,6].
In our hands the Au–TGR complex never dissociated (by dialysis) and whatever degree of inhibition we were able to achieve by AF could not be reversed with even partial restoration of enzyme activity. Thus to the best of our estimates, the binding of Au to TGR is essentially irreversible and the true IC50 of AF, defined as the free drug concentration required to reduce the enzyme activity to 50%, is essentially zero. How can we reconcile this observation with the published IC50 values? How can we explain the large difference in the IC50s of GR and TrxR, given that both enzymes form the same type of (irreversible) complex with the metal? We suggest that the data published are vitiated by insufficient incubation time and that a correct analysis of the time course of inhibition of thiol reductases by gold (Fig. 4) is the key to explain these discrepancies. Before discussing this point, and the pertinent test experiments we carried out, we would like to point out some theoretical problems inherent to the very definition of the IC50 of Au-based compounds, like AF or GoPI.
Fig. 4.
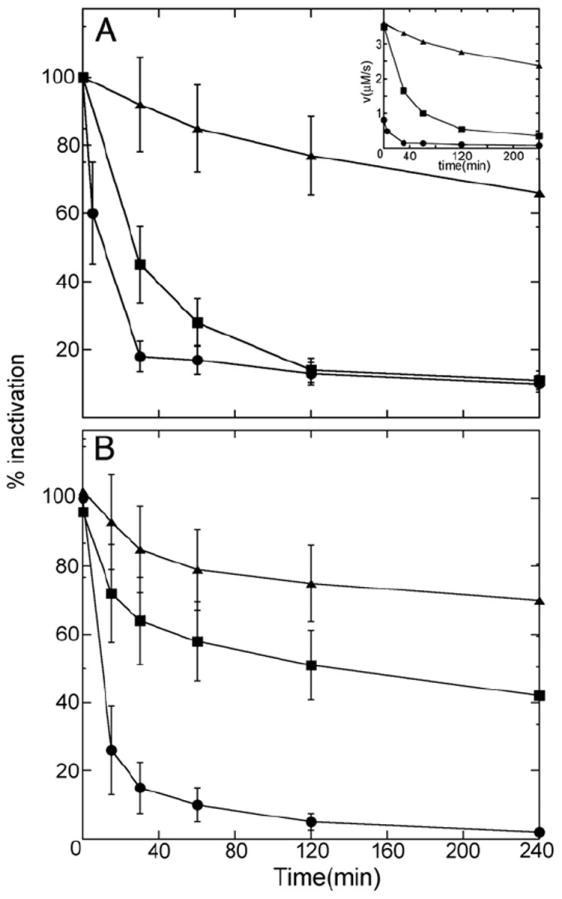
Truncated SmTGR and wild type yeast GR inhibition with AF and the effect of BzSe. Panel A: time course of truncated SmTGR 3 μM in the presence of 8 μM AF(-▲-) or 8 μM AF plus 3 μM BzSe (-■-); wtSmTGR 20 nM exposed to 50 nM AF (-●-). The error bars are drawn to represent ± 1 SD. In the inset the time course of a representative experiments plotted against initial velocities is shown in order to compare the relative velocities between C-terminal truncated and wild type TGR. Panel B: time course of the wild type yeast GR 0.5 μM in the presence of 1 μM AF (-▲-), 4 μM AF (-■-) and 4 μM AF plus 2 μM BzSe (-●-). The error bars are drawn to represent ± 1 SD.
The structures described above make it clear that AF and GoPI are Aucarriers and that the actual inhibitor of thiol reductases is the metal ion itself. The overall reaction of dissociation of the enzyme–inhibitor complex in the case of AF must therefore be schematized as:
| (1) |
If we omit the intermediate, presumably unstable, state in which gold is coordinated to one of its original ligands and one Cys (see Fig. 5), the overall equilibrium constant of the dissociation reaction is:
| (2) |
where E stands for GR, TrxR or TGR. The true thermodynamic equilibrium, whatever the affinity of the E–metal complex, clearly involves re-synthesis of the drug rather than simple dissociation of the metal. The usual concept of a IC50 is inherently unsuitable for this reaction, since the condition required to achieve 50% inhibition (i.e. [Au–E] = [E]) is:
| (3) |
Fig. 5.

Proposed reaction mechanism of AF with TGR. The mechanism of inhibition of AF towards Sec-containing flavoreductases is envisaged as a series of displacement reactions of the ligands of the metal, presumably occurring via a transient three-coordinate state. The acetoxythioglucose (tagl) is lost upon nucleophilic attack of the Sec, accordingly to Shaw [32]. The metal could be either be trapped by the adjacent Cys loosing the P(Et)3 or be transported to the more exposed Cys of the FAD active site. Both of these mechanisms may account for the arrangement of Au observed in the crystal structure of SmTGR in complex with AF (Fig. 3 panel A; pdb code: 3h4k [11]).
Even though the IC50 is an apparent parameter and not strictly a constant, for classical reversible enzyme inhibitors it equals the equilibrium constant of the dissociation of the EI complex; whereas in the present case it depends on the concentration of the original gold ligands under all experimental conditions. Therefore reversal of inhibition might be seen by increasing considerably the concentration of the original gold ligands P(Et)3 and Tagl. Moreover, Eq. (1) suggests that, if the transfer of gold from AF to TGR was reversible, dilution of the mixture should cause a paradoxical increase, rather than a decrease of the fraction of the enzyme complexed with the inhibitor. All of the above is clearly irrelevant to our proposed value of IC50, which is essentially zero, but constitutes theoretical evidence that the values reported in the literature must be considered with caution.
A different interpretation of the IC50, namely that once the Au–E complex has been formed it can equilibrate with free Au in the medium, is totally implausible because free Au(I) is practically insoluble in water; moreover, as already stated, the affinity of gold for its sulfur ligands is expected to be very high. This interpretation would give the usual meaning to the IC50 if when the equilibrium condition is reached the concentration of AF were negligible.
6. Which role for Sec?
How can we reconcile the inconsistencies illustrated above? We noticed that the IC50s for TrxR and GR were estimated after 10 to 15 min incubation of the enzyme with the chosen inhibitor [3]. While this procedure is routinely employed, its relevance or effectiveness is seldom if ever assessed, i.e. the authors usually do not state whether the chosen incubation time is necessary or whether it is long enough. It is unnecessary to remark that if a significant incubation time of the enzyme with the inhibitor is necessary for the complex to be formed before the substrate is added and enzyme activity assessed, then the steady state mixture is not tested under pseudo-equilibrium conditions and a competitive inhibitor may appear to behave non-competitively.
We determined the incubation time required by AF to form complexes with yeast GR, wild type S. mansoni TGR and with a truncated variant of S. mansoni TGR lacking the last two residues (the penultimate being Sec in the wild type sequence). The results of this experiment, reported in Fig. 4 (panel A), unequivocally demonstrate that: (i) the formation of the enzyme–gold complex requires longer than 15 min and thus, if the activity were tested before, the IC50 would be greatly overestimated; and (ii) the Se-containing wild type TGR reacts faster with AF than the Se-lacking GR and truncated TGR.
Besides confirming that the reported IC50 values are to be taken with great caution, the results in Fig. 4 clearly indicate that Sec plays a catalytic role, i.e. Se is relevant not only because of its high affinity for gold but also because it speeds up complex formation (minimizing the danger of overestimation of the IC50 for TrxR and TGR than for GR). To substantiate this conclusion the time course of inhibition of yeast GR by AF in the presence of an external source of Se, namely benzene selenol, was followed. As seen from Fig. 4 (panel B), this compound significantly speeds up the time course of inhibition, confirming that Se catalyzes the undressing of gold and its binding to the active site Cys residues.
There is in the literature a controversy about the role of Se in selenoproteins: some maintain that the specific advantage of Se over S is due to its higher nucleophilic character [29]; others, on the contrary, attribute to Se amore electrophilic behavior [30]. Indeed, in Fig. 5 the attack of Sec on AF is nucleophilic, whereas the transfer of Au to other Cys couples would be electrophilic. We can offer some interpretation whereby Se allows incorporation of Au into flavoreductases faster than sulfur, and is capable of these two apparently opposite roles: since Sec has lower pKa than Cys, it is expected to be almost entirely in the anionic state, and thus more reactive and more nucleophilic at neutral pH [31]; a Cys residue with low pKa would be expected to be almost as reactive as Sec [30,32] (this is possibly the case with the Se-lacking TrxR from P. falciparum, which is inhibited by AF at stoichiometric concentration, as suggested by Sannella et al. [5]; preliminary data from this laboratory appear to confirm this hypothesis). Once coordinated to the metal, Se nucleophilicity is decreased, and thereby transfer to Cys is facilitated. The higher nucleophilic character of Se relative to S, being due to its easier ionization, is thus not in contrast with the higher electrophilicity of Se when the complexes Se–1Au (or Se–H) are compared with S–Au (or S–H) [30,33,34]. Moreover, we remark that if the nucleophilic character of the selenolate anion speeds up its reaction with AF by lowering the activation energy of the exchange of gold ligands, i.e. of S–Au (in AF) with Se–Au, it must also speed up the “reverse” reaction, i.e. of Se–Au with S–Au (in Cys–Au). Nucleophilicity is kinetically relevant but does not imply anything on the relative affinities of Se and S for Au. We may speculate that a relevant contribution to the difference in affinity of S and Se for the metal comes from the fact that protons compete with gold more efficiently for S than for Se. The sole difference in the pKas of Sec and Cys would justify a 1,000-fold difference in the affinity for gold (in water). Gas-phase studies on the affinity of the anionic forms of both Sec and Cys for cobalt indicate that the two chalcogens bind this metal with similar affinity [35]. Additional considerations are: (i) the Sec residue, being located on the C-terminal arm, has less steric hindrance and ismore easily accessible than the Cys residues located over the FAD; and (ii) the intermediate complex formed in TrxRs and TGRs Se–Au–P(Et)3 would release triethylphosphine faster than the S–Au–P(Et)3 of GRs [32,14].
Of course our results do not exclude, and all the literature strongly suggests, that gold is also bound at equilibrium to the C-terminal Sec–Cys couple, presumably in a linear S–Au–Se complex, and that this type of complex causes inhibition of the thioredoxin reductase activity.
We demonstrate (Fig. 4 panels A and B) that in Se-lacking thiol reductases (GRs) the activation of gold based drugs occurs slowly, directly at the level of the catalytic Cys, which must be in the reduced state. By contrast, in the Se-containing thiol reductases (TrxRs, TGRs) gold preferentially binds to the selenium atom, from which it is then transferred to its other binding sites (Fig. 5). Gold transfer from its complex to a Cys couple, or between Cys couples, is a Lewis acid–base exchange, and we assume that the reaction goes through a transient unstable three-coordinate state of the metal [32]. Studies on the reaction between AF and albumin indicated that the first gold ligand to be released is acetoxy-thioglucose [32]. Once gold is bound to Sec, the length and the mobility of the C-terminal arm are sufficient to reach the other redox sites of the enzyme, consistent with its physiological role of electron transfer. To substantiate this possibility we systematically explored the crystallization conditions and found a combination in which one C-terminal arm is ordered and clearly visible in the electron density maps [21]. Starting from this structure it was possible to model the C-terminus of TGR over the other redox sites of the macromolecule, thus confirming that gold transfer from Cys596–Sec597 to its binding sites 1 and 2 is possible.
The mechanism proposed for TGR is parallel to the one described for the Hg reduction in the case of mercuric ion reductase (MR) [36]. This enzyme belongs to the same family of TrxR/TGR, sharing same fold, same cofactors and active site residues together with the redox active C-terminal arm composed by two adjacent cysteines. MR is a detoxifying enzyme whose physiological role is the reduction of mercuric ion to metallic mercury. Its catalytic cycle involves the following sequence of events: the C-terminal Cys residues bind Hg2+, Hg2+ is then transferred to the Cys residues close to the FAD, where the reduction and thus detoxification of the metal take place.
7. Conclusions
Selenoproteins are the preferred targets of some gold- and antimony-based drugs used since antiquity. These metals combine with high affinity with S and Se, and form very stable complexes with Cys–Cys or Sec–Cys couples that provide the appropriate coordination number and stereochemistry. This is the case in redox-active Cys couples, notably in thiol-reductases. Our experiments suggest that the preference for selenoproteins may depend on the fact that Se reacts with metals faster than with sulfur, also because of the lower pKa of selenols with respect to thiols. Once bound to the protein, initially to a Sec–Cys couple, the metal can then be transferred to Cys–Cys couples, as observed by X-ray crystallography.
The affinity of Cys–Cys or Sec–Cys couples for the metals is, in any case, extremely high and dissociation can only occur via the transfer to other couples, which may be either physiologically present in the organism (i.e. other selenoproteins) or administered as drugs (e.g. dimercaptopropanol). These features make gold-containing compounds appealing drugs.
Acknowledgments
Grants from Fondazione Roma (project title “Rational Approach to the Specific Inhibition of Plasmodium falciparum and Schistosoma mansoni”); Sapienza University of Rome “Ricerche Universitarie” 2009 e 2010; MIUR of Italy “FIRB/Proteomica 2007-protRBRN07BMCT”; and The National Institute of Allergy and Infectious Diseases (Grant AI065622) are gratefully acknowledged.
Abbreviations
- AF
auranofin
- GoPi
Gold 1-phenyl 2,5 (2-pyridil) phosphate
- GR
glutathione reductase
- P-Et3
triethyl phosphine
- tagl
tetra-acetoxy thioglucose
- Try
Trypanothione
- Trx
Thioredoxin
- TrxR
Thioredoxin reductase
- TryR
Trypanothione reductase
- TGR
Thioredoxin glutathione reductase
References
- 1.Rigobello MP, Messori L, Marcon G, Cinellu MA, Bragadin M, Folda A, Scutari G, Bindoli A. J Inorg Biochem. 2004;98:1634–1641. doi: 10.1016/j.jinorgbio.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 2.Omata Y, Folan M, Shaw M, Messer RL, Lockwood PE, Hobbs D, Bouillaguet S, Sano H, Lewis JB, Wataha JC, Toxicol In Vitro. 2006;20:882–890. doi: 10.1016/j.tiv.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. J Biol Chem. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 4.Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Free Radic Biol Med. 2007;42:872–881. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 5.Sannella AR, Casini A, Gabbiani C, Messori L, Bilia AR, Vincieri FF, Majori G, Severini C. FEBS Lett. 2008;582:844–847. doi: 10.1016/j.febslet.2008.02.028. [DOI] [PubMed] [Google Scholar]
- 6.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arnér ES, Williams DL. PLoS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urig S, Fritz-Wolf K, Réau R, Herold-Mende C, Tóth K, Davioud-Charvet E, Becker K. Angew Chem Int Ed Engl. 2006;45:1881–1886. doi: 10.1002/anie.200502756. [DOI] [PubMed] [Google Scholar]
- 8.Bindoli A, Rigobello MP, Scutari G, Gabbiani C, Casini A, Messori Coor L. Chem Rev. 2009;253:1692–1707. [Google Scholar]
- 9.Hotez PJ, Molyneux DH, Fenwick A, Kumaresan J, Sachs SE, Sachs JD, Savioli L. N Engl J Med. 2007;357:1018–1027. doi: 10.1056/NEJMra064142. [DOI] [PubMed] [Google Scholar]
- 10.Angelucci F, Miele AE, Boumis G, Dimastrogiovanni D, Brunori M, Bellelli A. Proteins. 2008;72:936–945. doi: 10.1002/prot.21986. [DOI] [PubMed] [Google Scholar]
- 11.Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A. J Biol Chem. 2009;284:28977–28985. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lijk LJ, Kalk KH, Branderburg NP, Hol WGJ. Biochemistry. 1983;22:2952–2957. doi: 10.1021/bi00281a026. [DOI] [PubMed] [Google Scholar]
- 13.Gunatilleke SS, Barrios AM. J Med Chem. 2006;49:3933–3937. doi: 10.1021/jm060158f. [DOI] [PubMed] [Google Scholar]
- 14.Hill DT, Isab AA, Griswold DE, DiMartino MJ, Matz ED, Figueroa AL, Wawro JE, DeBrosse C, Reiff WM, Elder RC, Jones B, Webb JW, Shaw CF., 3rd Inorg Chem. 2010;49:7663–7675. doi: 10.1021/ic902335z. [DOI] [PubMed] [Google Scholar]
- 15.Roberts JR, Shaw CF., 3rd Biochem Pharmacol. 1998;55:1291–1299. doi: 10.1016/s0006-2952(97)00634-5. [DOI] [PubMed] [Google Scholar]
- 16.Changela A, Chen K, Xue Y, Holschen J, Outten CE, O’Halloran TV, Mondragón A. Science. 2003;301:1383–1387. doi: 10.1126/science.1085950. [DOI] [PubMed] [Google Scholar]
- 17.Johs A, Harwood IM, Parks JM, Nauss RE, Smith JC, Liang L, Miller SM. J Mol Biol. 2011;413:639–656. doi: 10.1016/j.jmb.2011.08.042. [DOI] [PubMed] [Google Scholar]
- 18.Baiocco P, Ilari A, Ceci P, Orsini S, Gramiccia M, Di Muccio T, Colotti G. ACS Med Chem Lett. 2011;2:230–233. doi: 10.1021/ml1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilari A, Baiocco P, Messori L, Fiorillo A, Boffi A, Gramiccia M, Di Muccio T, Amino G. Acids. 2011 Aug 11; doi: 10.1007/s00726-011-0997-9. Electronic publication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baiocco P, Colotti G, Franceschini S, Ilari A. J Med Chem. 2009;52:2603–2612. doi: 10.1021/jm900185q. [DOI] [PubMed] [Google Scholar]
- 21.Angelucci F, Dimastrogiovanni D, Boumis G, Brunori M, Miele AE, Saccoccia F, Bellelli A. J Biol Chem. 2010;285:32557–32567. doi: 10.1074/jbc.M110.141960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boumis G, Angelucci F, Bellelli A, Brunori M, Dimastrogiovanni D, Miele AE. Protein Sci. 2011;20:1069–1076. doi: 10.1002/pro.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lennon BW, Williams CH, Jr, Ludwig ML. Science. 2000;289:1190–1194. doi: 10.1126/science.289.5482.1190. [DOI] [PubMed] [Google Scholar]
- 24.Huang HH, Day L, Cass CL, Ballou DP, Williams CH, Williams DL. Biochemistry. 2011 doi: 10.1021/bi200107n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Böhme CC, Arscott LD, Becker K, Schirmer RH, Williams CH., Jr J Biol Chem. 2000;275:37317–37323. doi: 10.1074/jbc.M007695200. [DOI] [PubMed] [Google Scholar]
- 26.Bauer H, Massey V, Arscott LD, Schirmer RH, Ballou DP, Williams CH., Jr J Biol Chem. 2003;278:33020–33028. doi: 10.1074/jbc.M303762200. [DOI] [PubMed] [Google Scholar]
- 27.Huang HH, Arscott LD, Ballou DP, Williams CH., Jr Biochemistry. 2008;47:1721–1731. doi: 10.1021/bi702040u. [DOI] [PubMed] [Google Scholar]
- 28.Schröder D, Schwarz H, Hrušák J, Pyykkö P. Inorg Chem. 1998;37:624–632. [Google Scholar]
- 29.Arnér ES. Exp Cell Res. 2010;316:1296–1303. doi: 10.1016/j.yexcr.2010.02.032. [DOI] [PubMed] [Google Scholar]
- 30.Hondal RJ, Ruggles EL. Amino Acids. 2011;41:73–89. doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickey JL, Ruhayel RA, Barnard PJ, Baker MV, Berners-Price SJ, Filipovska A. J Am Chem Soc. 2008;130:12570–12571. doi: 10.1021/ja804027j. [DOI] [PubMed] [Google Scholar]
- 32.Shaw CF., III Chem Rev. 1999;99:2589–2600. doi: 10.1021/cr980431o. [DOI] [PubMed] [Google Scholar]
- 33.Lothrop AP, Ruggles EL, Hondal RJ. Biochemistry. 2009;48:6213–6223. doi: 10.1021/bi802146w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinmann D, Nauser T, Koppenol WH. J Org Chem. 2010;75:6696–6699. doi: 10.1021/jo1011569. [DOI] [PubMed] [Google Scholar]
- 35.Spezia R, Tournois G, Cartailler T, Tortajada J, Jeanvoine Y. J Phys Chem A. 2006;110:9727–9735. doi: 10.1021/jp0614998. [DOI] [PubMed] [Google Scholar]
- 36.Engst S, Miller SM. Biochemistry. 1999;38:3519–3529. doi: 10.1021/bi982680c. [DOI] [PubMed] [Google Scholar]