

Abstract
Background
The clinical and hematologic features of β-thalassemia are modulated by different factors, resulting in a wide range of clinical severity. The main factors are the type of disease-causing mutation and the ability to produce α-globin and γ-globin chains. In the present study we investigated the respective contributions of known modifiers to the prediction of the clinical severity of β-thalassemia as assessed by the patients’ age at first transfusion.
Design and Methods
We studied the effect of seven loci in a cohort of 316 Sardinian patients with β0-thalassemia. In addition to characterizing the β-globin gene mutations, α-globin gene defects and HBG2:g.−158C>T polymorphism, we genotyped two different markers in the BCL11A gene and three in the HBS1L-MYB intergenic region using single nucleotide polymorphism microarrays, imputation and direct genotyping. We performed Cox proportional hazard analysis of the time to first transfusion.
Results
According to the resulting model, we were able to explain phenotypic severity to a large extent (Harrell’s concordance index=0.72; Cox & Snell R2=0.394) and demonstrated that most of the model’s discriminatory ability is attributable to the genetic variants affecting fetal hemoglobin production (HBG2:g.−158C>T, BCL11A and HBS1L-MYB loci: C-index=0.68, R2=0.272), while the remaining is due to α-globin gene defects and gender. Consequently, significantly distinct survival curves can be described in our population.
Conclusions
This detailed analysis clarifies the impact of genetic modifiers on the clinical severity of the disease, measured by time to first transfusion, by determining their relative contributions in a homogeneous cohort of β0-thalassemia patients. It may also support clinical decisions regarding the beginning of transfusion therapy in patients with β-thalassemia.
Keywords: beta-thalassemia, genetic modifiers, fetal hemoglobin, thalassemia major, thalassemia intermedia
Introduction
β-thalassemia is characterized by decreased or absent β-globin chain synthesis due to a variety of mutations; this decrease in β-globin chain synthesis results in an excess of α-globin chains which precipitate in red blood cell precursors in the bone marrow, causing their premature death.1 In Sardinia, the most common type of β-thalassemia is due to a nonsense mutation at codon 39 of the β-globin gene (HBB:c118C>T).2
The majority of patients develop a severe form of anemia (thalassemia major) and are transfusion-dependent from the first years of life. When performed regularly, red blood cell transfusions prevent anemia-related complications and compensatory marrow expansion, and, therefore, extend the survival of patients. Approximately 5–10% of patients live without requiring periodic blood transfusions and are said to have thalassemia intermedia.3 These two forms of the disease are the extreme ends of a broad range of clinical variability: patients might need to start transfusions after days, months or even years of life, demonstration of a great variation in disease severity.
This remarkable phenotypic diversity of thalassemia patients is associated with a great variety of genotypes including mild/silent β-thalassemia alleles, coinheritance of α-thalassemia or the presence of genetic determinants associated with increased production of γ-globin chains and consequent ability to produce functional fetal hemoglobin (Hb F) in adult life.4 All these conditions reduce α/β-globin chain imbalance and ineffective erythropoiesis. The level of Hb F is regulated by three major loci: HBG2:g.−158C>T on 11p15.4, HBS1L-MYB intergenic region on 6q23.3, and BCL11A on 2p16.1. Together these three loci are responsible for 20 to 50% of the Hb F trait variance in patients with β-thalassemia or sickle cell disease, and in healthy Europeans.5–10
Here we evaluated the effect of the HBG2:g.−158C>T, BCL11A, and HBS1L-MYB variants, together with coinheritance of α-thalassemia and gender, on the severity of β-thalassemia phenotype, measured by age at first transfusion, in Sardinian patients.
Design and Methods
Patients and phenotypic assessment
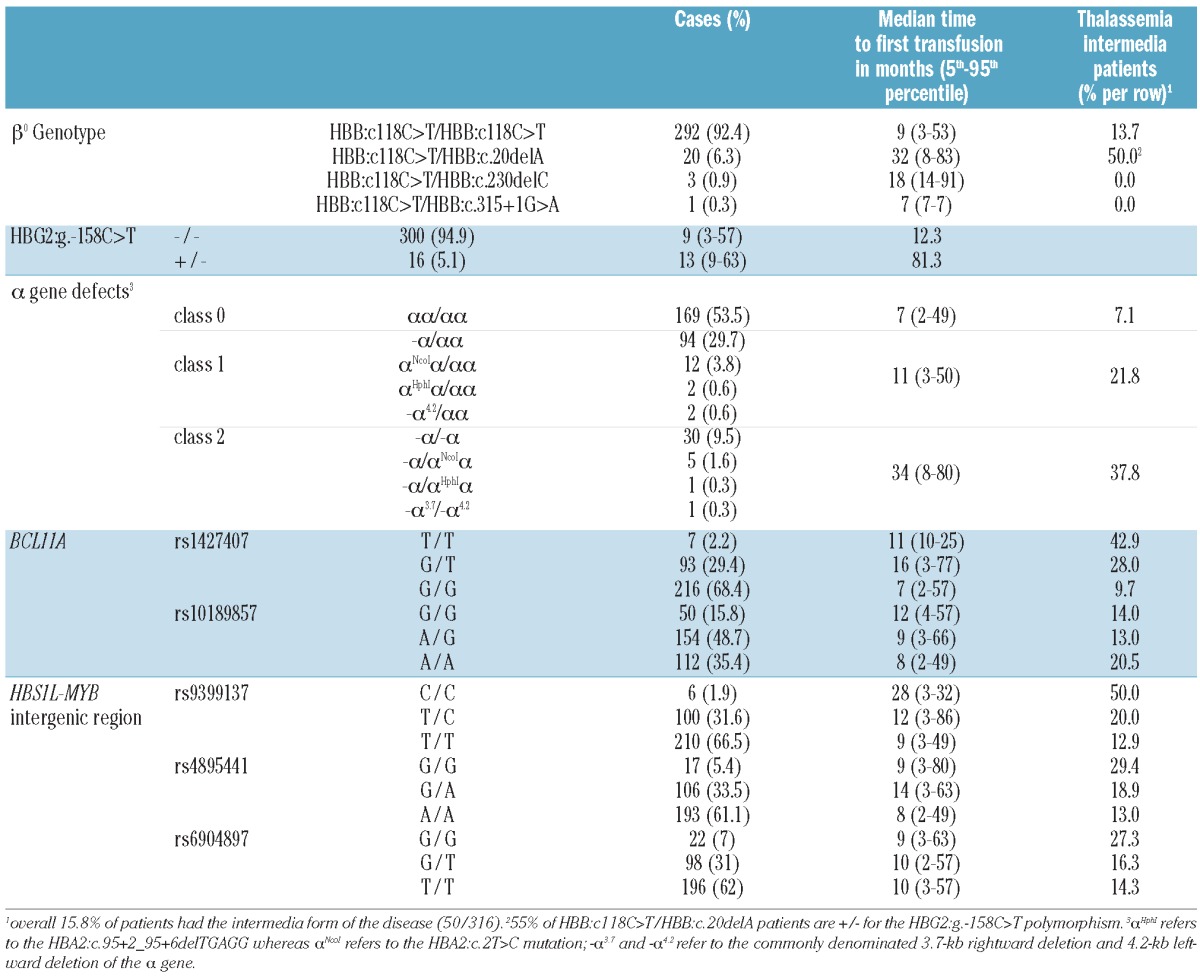
We retrospectively studied 316 β0-thalassemia patients (168 males and 148 females) from Sardinia, all followed at the Microcythemia Hospital of Cagliari. Of these patients, 266 had thalassemia major (median age 33 years; 5th and 95th percentiles, 13 and 38 years, respectively) and 50 had thalassemia intermedia (median age 43 years; 5th and 95th percentiles, 17 and 61 years, respectively); 125 had been enrolled in a previous study on phenotype amelioration.11 Thalassemia intermedia patients were defined as patients who had never been transfused, or had only been transfused sporadically during infections or surgery (<10 blood units in total).3 The β-thalassemia mutations were of HBB:c118C>T/HBB:c118C>T type in 92.4% of cases and HBB:c118C>T/HBB:c.20delA type in 6.3% of the studied sample; the remaining mutations are reported in Table 1. The continuous distribution of the phenotypic severity among thalassemia patients was measured by the time at which they started transfusion therapy. Criteria for starting transfusion were persistent (i.e. more than 2 weeks) hemoglobin level lower than 7 g/dL in the absence of infections, moderate to severe spleen enlargement and poor growth. The time to event was calculated as the time between birth and the first red blood cell transfusion or between birth and the last follow-up (January 2011) for patients who were not on transfusion therapy. Age at first transfusion was retrospectively collected through the WebTHAL computerized clinical records database (http://www.thalassemia.it), in use for the daily management of patients in our center.
Table 1.
Genotypic frequencies of genetic markers and clinical characteristics.
This retrospective study was conducted in accordance with the Declaration of Helsinki and the patients gave informed consent to analysis of their DNA.
Selection of single nucleotide polymorphisms
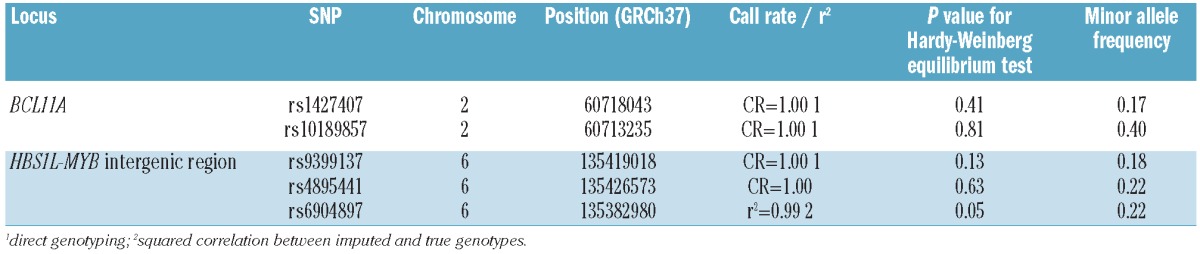
We selected five single nucleotide polymorphisms (SNP) from the HBS1L-MYB intergenic region and the BCL11A locus known to be associated with Hb F levels (Table 2):
Table 2.
Characteristics of single nucleotide polymorphisms used in the study.
rs1427407: the most significant SNP associated with Hb F levels within BCL11A, as reported by Menzel et al.12 This SNP is in high linkage disequilibrium (LD) with rs766432 (r2=0.98) in our sample, and with rs4671393 (r2=0.88/D’=1) in the CEU samples based on the 1000 Genomes Project pilot phase 1 (CEU.1kG), for which effects on Hb F levels are also well-documented;13,14
rs10189857: within BCL11A, documented to have an independent effect on Hb F levels;15
rs9399137: the most significant SNP for Hb F levels within the HBS1L-MYB intergenic region in different populations,5,12 in complete LD with a 3-bp deletion located in close proximity to four erythropoiesis-related transcription factor binding sites;15,16
rs4895441: a SNP within the HBS1L-MYB intergenic region, widely reported to be associated with Hb F levels5,17 and in complete LD with rs9402686 (r2=1/D’=1 from CEU.1kG data), also reported to be independently associated with Hb F levels;15
rs6904897: within the HBS1L-MYB intergenic region, this SNP is in complete LD with rs28384513 (r2=1/D’=1 from CEU.1kG data), reported to be independently associated with Hb F levels.15
Genotyping
DNA was extracted from venous peripheral blood with standard methods. Mutations of the β-globin gene were analyzed by direct DNA sequencing. The HBG2:g.−158C>T polymorphism was determined as described elsewhere.18 α-globin gene defects were determined using gap-polymerase chain reaction or restriction enzyme digestion for deletional and non-deletional defects, respectively.19
SNP were directly genotyped except for rs4895441 which was genotyped using the Affymetrix Genome-Wide Human SNP Array 6.0 according to the manufacturer's protocol and rs6904897 which was imputed with MACH software, version 1.0.16, using a combined panel of Utah Residents of Northern and Western European Ancestry (CEU) and Tuscan samples (TSI) from the International Hapmap consortium as reference samples.20
Sixteen samples (from patients selected for being positive for the HBG2:g.−158C>T polymorphism) for which SNP array data were not available, were genotyped using TaqMan SNP genotyping assay (Applied Biosystems, Warrington, UK) for each of the five SNP.
Quality controls
Microarray data from the samples underwent quality control procedures, including: sample call rate (exclusion when the call rate was <95%), cryptic relatedness (exclusion of first degree relatives), inbreeding coefficient (exclusion if negative with a call rate <98%) and reported gender versus heterozygosity of X chromosome SNP (exclusion if discordant). Principal component analysis, as implemented in EIGENSTRAT, was performed for the detection of outliers.21 Quality control attributes for the SNP used in the present study are described in Table 2.
Statistical analysis
All genome-wide quality control measures were performed using the PLINK software package, version 1.07,22 while the SPSS statistical software package, version 18.00 (SPSS, IBM, Somers, NY, USA), was used for subsequent analysis. All markers selected for the present study were entered in a backward stepwise Cox proportional hazard model to characterize their effect on time to first transfusion, together with gender, α-globin gene defects and status for HBG2:g.−158C>T polymorphism (only −/− and +/− genotypes were observed for this SNP). For each SNP a variable was defined with the value of 0, 1, or 2 according to the number of copies of the less frequent allele, except for rs6904897: since there was no difference between the G/G and G/T genotype survival curves for this SNP, it was codified 0 for both these genotypes and 1 otherwise. α-globin gene defects were classified as 0, 1, or 2 according to the number of deleted or mutated copies of the HBA gene (see Table 1 for details). Gender was codified 0 when female and 1 when male. Covariates were excluded from the model when their P value was greater than 0.10. Patients were considered uncensored when blood transfusion occurred during the study and censored when blood transfusion did not occur. We report Cox and Snell R2 as well as Harrell’s concordance index (C-index) to assess how well the model performed.
Results
Results from the stepwise Cox proportional hazard model are presented in Table 3. We refer below to predictors for later time to transfusion as positive values and predictors for earlier time to transfusion as negative values.
Table 3.
Results of the Cox proportional hazards model.
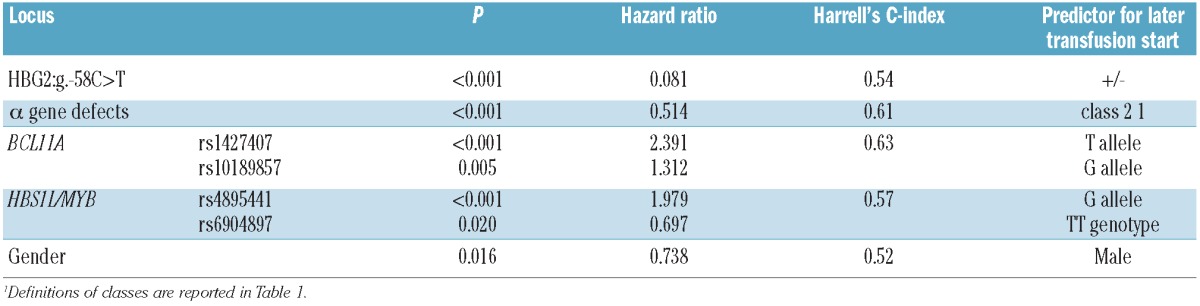
The HBG2:g.−158C>T polymorphism had the strongest effect on the severity of β-thalassemia phenotype [hazard ratio (HR)=0.08; P<0.001], followed by rs1427407 (BCL11A) (HR=2.37; P<0.001), α-globin gene defects (HR=0.52; P<0.001), rs4895441 (HBS1L-MYB) (HR=1.94; P<0.001), rs10189857 (BCL11A) (HR=1.31; P=0.004), rs6904897 (HBS1L-MYB) (HR=0.79, P=0.047) and gender (HR=0.73; P=0.013). The SNP rs9399137 (HBS1L-MYB), in high LD with rs4895441 (r2=0.90 from CEU.1kG data), was the only predictor removed from the model (HR=1.29; P=0.298). Among all two-way interactions tested, the only significant one was between rs1427407 and rs10189857 (HR=1.66; P=0.036).
The discriminatory power of the model was high (C-index=0.72; R2=0.394) and most of it was attributable to Hb F production modulators (HBG2:g.−158C>T, BCL11A and HBS1L-MYB loci: C-index=0.68, R2=0.272), while the remaining was attributable to α-globin gene defects and gender.
According to our model prediction, 50% of patients with all negative predictors would undergo their first transfusion within the first 100 days of life and 99% of them would need regular transfusions before the first year of life. On the other hand, with all positive predictors, the probability of undergoing transfusion by 10 years was only 6‰.
We evaluated survival curves for time to first transfusion for four groups defined by the quartiles of the distribution of the linear predictor score (i.e. the sum of the product between covariate values and their corresponding parameter estimates). Lower values (first quartile) corresponded to different combinations of mostly positive predictors (82 cases - linear predictor score values below 1.45), while higher values (fourth quartile) corresponded to different combinations of mostly negative predictors (76 cases - linear predictor score values above 2.70). Intermediate groups included 78 cases with linear predictor score values between 1.45 and 2.05 (second quartile) and 80 cases with linear predictor score values between 2.05 and 2.70 (third quartile).
Following this classification, 50% of patients in the fourth quartile group underwent their first transfusion within 6 months of life, whereas only 3% of patients in the first quartile group had started transfusions by the same age. In this group it took more than 6 years for 50% of the patients to start transfusions, whereas by the same age all patients in the fourth quartile group had undergone their first transfusion. In the first quartile group, 47% of patients never started red blood cell transfusion (Figure 1).
Figure 1.
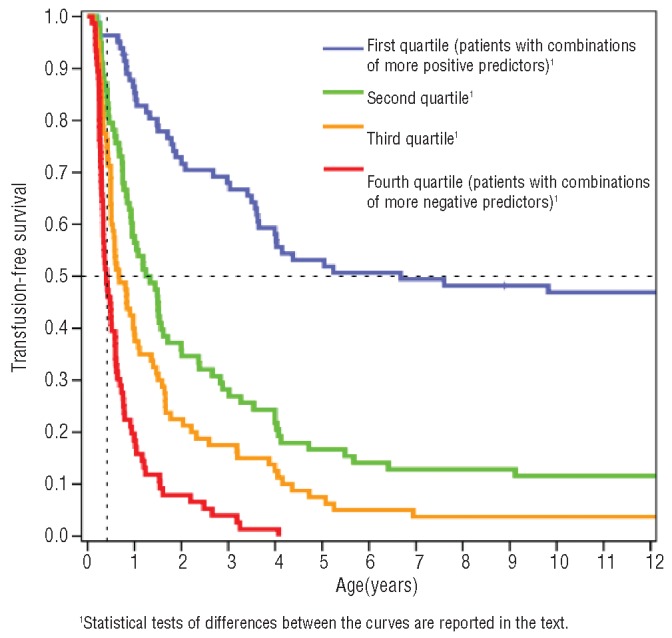
Kaplan-Meier survival curves for patients with different combinations of predictors for later or earlier transfusion need.
All survival curves were significantly different from each other (P<0.01, Breslow’s test). In particular, the third quartile group was significantly different from the fourth quartile group (P<0.001) and the second quartile group was significantly different from both the first and third quartile risk-groups (P<0.001 and P=0.007, respectively).
Discussion
The purpose of this study was to measure the influence of known genetic modifiers of β0-thalassemia on phenotype severity, assessed as time to first transfusion. To this aim, SNP in the BCL11A gene and HBS1L-MYB intergenic region were selected based on previous studies and genotyped in a group of 316 patients, as were α-globin gene defects and the HBG2:g.−158C>T polymorphism. All these variables, together with gender, were included in a Cox proportional hazard model for time to first transfusion, and their respective effects were measured. The results showed that Hb F production variants and α-globin gene defects had a substantial impact on the severity of β-thalassemia phenotype, allowing prediction of the risk of patients to start transfusion at different times of their life.
In this study we assumed that the time to first transfusion accurately reflects variations in β-thalassemia phenotype severity. This hypothesis seems to be supported by our results, as all variables and the hierarchy of their effects agree with previous studies on genetic modifiers of both Hb F levels and the clinical severity of β-thalassemia, even though other unknown genetic factors and clinical conditions might be co-responsible for the need for transfusions.3,11,15,23,24
To the best of our knowledge, the present study is the first to analyze the severity of β-thalassemia in a quantitatively defined manner and to include such a complete set of known predictors. In a previous study, Galanello et al.11 studied the effect of two SNP (rs11886868 in BCL11A and rs9389268 in the HBS1L-MYB intergenic region) and α-globin gene defects on the phenotypic expression (defined as major versus intermedia status) of Sardinian patients with β0-thalassemia. A recent study by Badens et al.24 further extended this analysis accounting for the HBG2:g.−158C>T polymorphism and β0/β+ status, in addition to the previously mentioned markers, in a heterogeneous cohort of 106 patients with 30 different β-globin gene mutations. The present analysis expands these results by including the effect of different independent predictors in each gene, selected to be the strongest reported to date, in a homogeneous cohort of β0-thalassemia patients. This, we believe, enables a better definition of the respective effects of each predictor. Above all this work relates genetic modifiers to time to first transfusion, a key event that characterizes disease severity regardless of patients’ major or intermedia phenotype, thus notably increasing our knowledge on the specific effects of genetic modifiers of the clinical severity of β-thalassemia.
While it is likely that future whole genome sequencing studies will better define the genetic polymorphisms that modulate the effect of the BCL11A and HBS1L-MYB loci, the results from the present study could already be of support in clinical settings, by providing clear probabilities for the need to start transfusion at different ages as a function of the personal genetic background of individual patients.
Acknowledgments
We thank Serena Sanna for her support and constructive comments and Francarosa Demartis for her technical assistance.
Footnotes
Funding: supported by grants from Regione Autonoma Sardegna L.R. 11/90 and a Research Grant from Apopharma (Ontario – Canada).
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Cao A, Galanello R. Beta-Thalassemia. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 1993–2000. [updated 2010] [Google Scholar]
- 2.Cao A, Rosatelli C, Pirastu M, Galanello R. Thalassemias in Sardinia: molecular pathology, phenotype-genotype correlation, and prevention. Am J Pediatr Hematol Oncol. 1991;13(2):179–88. [PubMed] [Google Scholar]
- 3.Taher A, Musallam KM, Cappellini MD. Thalassaemia intermedia: an update. Mediterr J Hematol Infect Dis. 2009;1(1):e2009004. doi: 10.4084/MJHID.2009.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao A, Galanello R, Rosatelli MC. Genotype-phenotype correlations in beta-thalassemias. Blood Rev. 1994;8(1):1–12. doi: 10.1016/0268-960x(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 5.Lettre G, Sankaran VG, Bezerra MAC, Araújo AS, Uda M, Sanna S, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA. 2008;105(33):11869–74. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sedgewick AE, Timofeev N, Sebastiani P, So JCC, Ma ESK, Chan LC, et al. BCL11A is a major HbF quantitative trait locus in three different populations with [beta]-hemoglobinopathies. Blood Cells Mol Dis. 2008;41(3):255–8. doi: 10.1016/j.bcmd.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garner C, Tatu T, Reittie JE, Littlewood T, Darley J, Cervino S, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95(1):342–6. [PubMed] [Google Scholar]
- 8.Creary LE, Ulug P, Menzel S, McKenzie CA, Hanchard NA, Taylor V, et al. Genetic variation on chromosome 6 influences F cell levels in healthy individuals of African descent and HbF levels in sickle cell patients. PLoS One. 2009;4(1):e4218. doi: 10.1371/journal.pone.0004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.So CC, Song YQ, Tsang ST, Tang LF, Chan AY, Ma ES, et al. The HBS1L-MYB intergenic region on chromosome 6q23 is a quantitative trait locus controlling fetal haemoglobin level in carriers of beta-thalassaemia. J Med Genet. 2008;45(11):745–51. doi: 10.1136/jmg.2008.060335. [DOI] [PubMed] [Google Scholar]
- 10.Menzel S, Thein SL. Genetic architecture of hemoglobin F control. Curr Opin Hematol. 2009;16(3):179–86. doi: 10.1097/moh.0b013e328329d07a. [DOI] [PubMed] [Google Scholar]
- 11.Galanello R, Sanna S, Perseu L, Sollaino MC, Satta S, Lai ME, et al. Amelioration of Sardinian beta0 thalassemia by genetic modifiers. Blood. 2009;114(18):3935–7. doi: 10.1182/blood-2009-04-217901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197–9. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 13.Solovieff N, Milton JN, Hartley SW, Sherva R, Sebastiani P, Dworkis DA, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood. 2010;115(9):1815–22. doi: 10.1182/blood-2009-08-239517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhatnagar P, Purvis S, Barron-Casella E, Debaun MR, Casella JF, Arking DE, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J. Hum Genet. 2011;56(4):316–23. doi: 10.1038/jhg.2011.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42(12):1049–51. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farrell JJ, Sherva RM, Chen Z-Y, Luo H-Y, Chu BF, Ha SY, et al. A 3-bp deletion in the HBS1L-MYB intergenic region on chromosome 6q23 is associated with HbF expression. Blood. 2011;117(18):4935–45. doi: 10.1182/blood-2010-11-317081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci USA. 2008;105(5):1620–5. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sutton M, Bouhassira EE, Nagel RL. Polymerase chain reaction amplification applied to the determination of beta-like globin gene cluster haplotypes. Am J Hematol. 1989;32(1):66–9. doi: 10.1002/ajh.2830320113. [DOI] [PubMed] [Google Scholar]
- 19.Galanello R, Sollaino C, Paglietti E, Barella S, Perra C, Doneddu I, et al. Alpha-thalassemia carrier identification by DNA analysis in the screening for thalassemia. Am J Hematol. 1998;59(4):273–8. doi: 10.1002/(sici)1096-8652(199812)59:4<273::aid-ajh2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 20.T. I. H. Consortium. The International HapMap Project. Nature. 2003;426(6968):789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 21.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weatherall DJ, Clegg JB. The Thalassemia Syndromes. 4th. Blackwell Science Ltd; 2001. [Google Scholar]
- 24.Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S, et al. Variants in genetic modifiers of beta-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. 2011;96(11):1712–4. doi: 10.3324/haematol.2011.046748. [DOI] [PMC free article] [PubMed] [Google Scholar]