

Abstract
Genetic analysis of von Willebrand disease by von Willebrand factor gene sequencing has not yet become routine practice. Nevertheless, the prospects for molecular diagnosis have changed dramatically in recent years with the unveiling of next-generation sequencing platforms. With the goal of applying this technology to von Willebrand disease, we designed a strategy for von Willebrand factor gene enrichment and multiplexing based on short polymerase chain reactions. Forty patients were simultaneously analyzed enabling the identification of 43 mutations, including 36 substitutions, 2 intronic splice site mutations, 2 indels, and 3 deletions. By pooling patient genomic DNA before polymerase chain reaction enrichment, indexing samples with barcode tags, and re-sequencing on the next-generation sequencing instrument, at least 350 patients and relatives per run can be simultaneously analyzed in a fast, inexpensive manner. This is one of the first reports in which this technology has been shown to be feasible for large-scale mutation screening by single gene re-sequencing.
Keywords: von Willebrand disease, von Willebrand factor gene, von Willebrand factor, next generation sequencing, massive sequencing, mutational analysis
Introduction
Systematic identification of the causal von Willebrand disease (VWD) mutations has been hampered by the large size of the von Willebrand factor gene (VWF) that is highly polymorphic and has a greatly homologous partial pseudogene.1 Because of these difficulties, molecular studies of VWD have been confined mainly to basic investigation. Current sequencing technology has allowed only some expert laboratories to perform VWF sequencing in patients with VWD.2–4
The cost of traditional sequencing based on the Sanger method remains a serious obstacle for most diagnostic laboratories. However, perspectives in the molecular diagnosis of monogenic diseases have radically changed over recent years with the introduction of new massively parallel next-generation sequencing (NGS) platforms that are considered alternatives or complementary to traditional sequencing.5 Systematic re-sequencing of genes that have been implicated in related Mendelian diseases is a promising strategy for identifying risk factors for complex diseases.6 Furthermore, next generation sequencing (NGS) technology could also facilitate the identification of mutations in patients with monogenic diseases. Although NGS is undoubtedly superior in throughput to Sanger sequencing, it has so far had little impact on monogenic disease diagnostics.7–9 To take advantage of this new technology for molecular diagnosis of VWD, we designed a procedure for fast, low-cost characterization of a large number of samples, derived from a highly optimized method developed in our laboratory.4 A proof-of-concept study was performed using an NGS platform. The results demonstrate a high performance DNA sequencing of VWF in a large number of VWD patients and their families, and establishes the basis for performing a large-scale sequencing study in an affected population.
Design and Methods
Patients
Samples were collected and clinical data compiled from 40 unrelated patients of several origins with different types of VWD. Most patients had been diagnosed at the Hemophilia Unit of Vall d’Hebron Hospital (Barcelona, Spain); ISTH criteria were met in all cases.10,11 Peripheral blood samples were collected in tubes of 4 mL containing ACD or EDTA. Genomic DNA was isolated from 300 μL of blood with the FlexiGene DNA Kit (Qiagen, Duesseldorf, Germany). The study was approved by the ethics committee of Vall d’Hebron Hospital, and all patients provided their informed consent.
Polymerase chain reaction enrichment and normalization
Genomic DNA in equimolar concentrations from every 5 patients was mixed and subjected to the conventional short PCR amplification according to our previously described procedure.4 All the coding regions and flanking intronic regions of VWF (~10 Kb) are included (Figure 1A). Normalization of amplicons was carried out by gel quantification performed with ImageJ (Version 1.43u), a public domain Java image processing program (http://rsbweb.nih.gov/ij/). By taking a sample with a known concentration as the control, it was possible to extrapolate the concentration of each sample to create a normalized pool of the 47 PCR products in one tube. This pooling process produced 8 tubes and each tube was the result of simultaneous amplification of VWF of 5 patients.
Figure 1.

Conventional short PCR approach. (A) Representative agarose gel electrophoresis of the 47 PCRs comprising all 52 exons of VWF from 5 patients amplified simultaneously. Correspondence between each PCR product and the specific exon/s amplified is indicated above each lane. (B) Agarose gel electrophoresis showing heat incubation time-course experiment for short PCR fragmentation. Size distribution of the fragments after 9 h checked with a DNA 100 chip on an Agilent 2100 Bioanalyzer is also shown. (C) Sequence coverage diagram representative of the Illumina GA readings for conventional short PCR samples. The x axis represents the position on the reference sequence comprised of concatenated sequences of VWF amplified regions. The y axis shows the fold coverage (number of reads per bp). Regions of very high coverage represent the ends of the short PCR amplicons.
Polymerase chain reaction fragmentation, massively parallel sequencing and bioinformatic analysis
In NGS, the fragmentation step is critical for the success of the sequencing process and must be performed according to stringent parameters. Because the commonly used mechanical systems for genomic DNA fragmentation such as sonication are not effective for short DNA fragments (e.g., <300–500 bp),12 conventional short PCRs were fragmented by heat incubation at 95ºC for 9 h (Figure 1B) followed by 1 h of re-annealing at 72°C. This provided efficient DNA fragmentation that was relatively unbiased, as required for Illumina GA sequencing procedures.12 All samples were purified using the QIAquick PCR Purification Kit (Qiagen) and the size distribution of the fragments was checked using a DNA 100 chip on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Subsequent massively parallel sequencing and bioinformatic analysis are described in the Online Supplementary Design and Methods section.
Mutation assignment
To confirm the mutations identified by NGS and assign them to patients, the specific region including the mutation was PCR-amplified and sequenced by dideoxynucleotide method, as described.4 The sequences obtained were assembled and aligned against the consensus wild-type VWF sequence using SeqScape software (v2.7) (Applied Biosystems, Foster City, CA, USA).
Results and Discussion
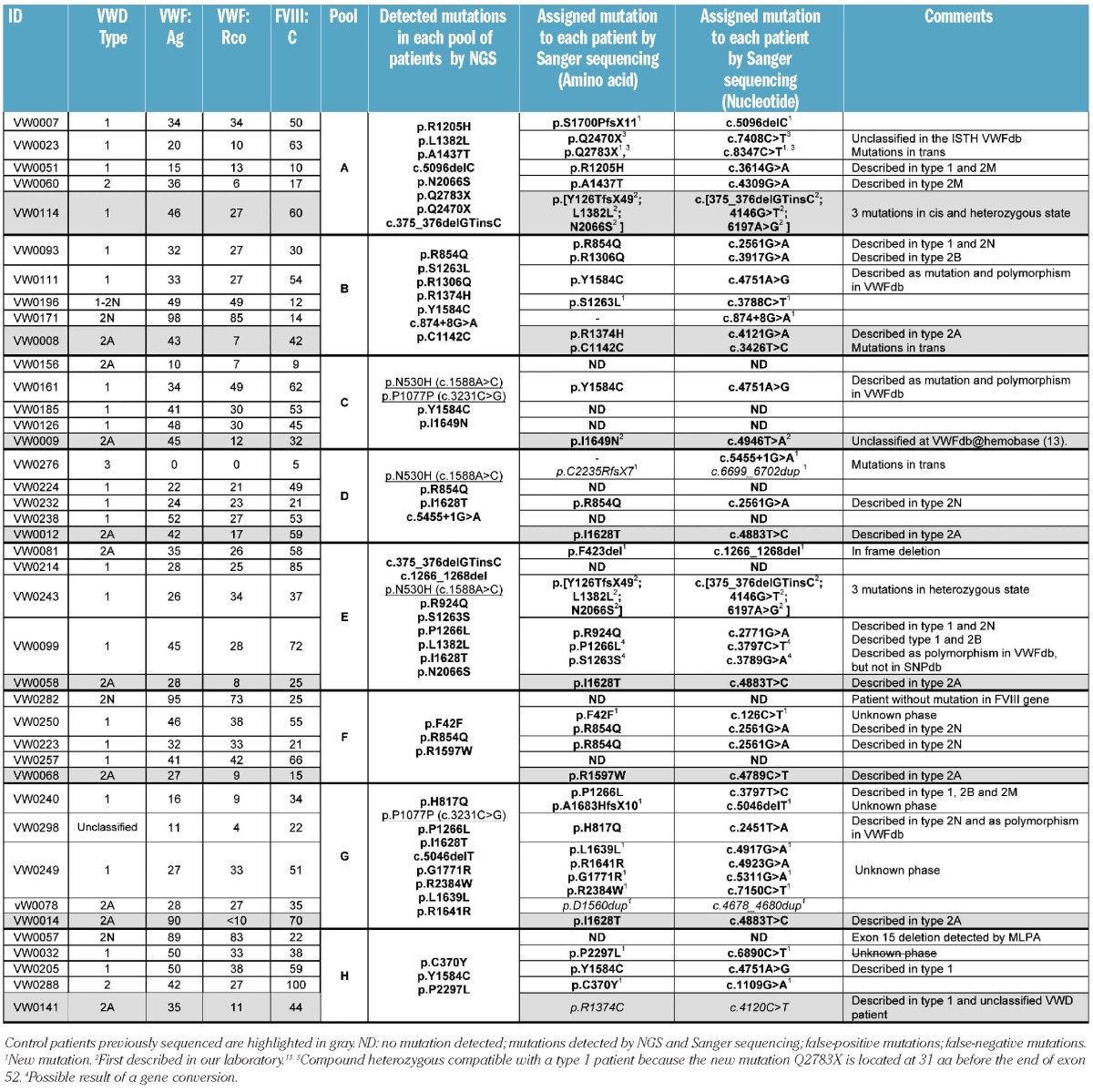
Based on the conclusions from a pilot study (Online Supplementary Appendix), a proof-of-concept study was performed for the simultaneous analysis by NGS of 40 VWD patients previously diagnosed and classified according to their clinical characteristics (Table 1). The mutations responsible for the disease were known in 8 of the selected patients who had been characterized by Sanger sequencing4 and were used as controls. Patients included in this study were randomly selected from all those who met the criteria for VWD, to obtain a representative picture of the types, percentages, and variability of patients usually seen at the Hemophilia Unit.
Table 1.
Summary of clinical and molecular data of sequenced VWD patients.
NGS technologies are economically extremely advantageous when the sequencer runs completely full which, in the case of short region sequencing (e.g., VWF), means including the maximum number of samples14 and the preparation of one sequencing library per sample. However, library construction is one of the most expensive steps in the overall NGS process. Therefore, when the study was conceived, a key concern was to minimize the number of libraries constructed per flow cell lane. Consequently, genomic DNA in equimolar concentrations from every 5 patients, including one control patient per pool, was mixed and subjected to the conventional short PCR amplification described above.4 After amplification, normalization, and pooling of amplicons, the process yielded 8 tubes, each representing 5 patients. Illumina’s library construction protocol was performed for each of the 8 tubes, inserting 8 different pentanucleotide sequences as index tags (i.e. 1 tag for every 5 patients). Sequence tagging allows software-mediated segregation and independent analysis of the sequences. Therefore, 40 patients could be analyzed in 1 of 8 of the Illumina flow cells, representing a considerable reduction in costs. As a preliminary approach, we amplified genomic DNA from 5 patients mixed together, considering that for a single heterozygous variant allele in a pool of 5 diploid individuals one would expect 10% of the reads to carry the variant allele. A drawback to this strategy is that pooling genomic DNA before PCR amplification means the information on SNPs for each patient obtained with the Sanger sequencing method is not used. However, it is possible to save this information with NGS using novel, innovative approaches such as microfluidics technology15 that can also be explored by introducing slight modifications to our current protocol.
Analysis of the sequences obtained by NGS for each group of 5 patients allowed a variable number of potential mutations to be detected. These were further corroborated and assigned to the right patient by sequencing the corresponding exons in all patients included in the pool. The results of this analysis are shown in Table 1. Excluding pool H (control patient VW0141 carrying the mutation c.4120C>T), all mutations from the control patients (last row of each pool) were detected by NGS. This pool had an unidentified problem somewhere in the sequencing process that resulted in low overall coverage. Although we did not systematically study the cause, differences in tag performance have been described previously16 and could be the likely explanation. Therefore, VWF from patients in this pool were entirely sequenced by the Sanger method and the mutations detected were identical to those detected by NGS, except for the above-mentioned c.4120C>T. With the current analytical parameters used in the CLC Genomics Workbench (see Online Supplementary Design and Methods section), we detected 2 mutations by NGS (c.1588A>C and c.3231C>G) in several pools (C, D, E and G) that could not be confirmed by Sanger sequencing and were, therefore, false-positive. Probably such false-positive calls were the result of systematic bias since they were present in more than one sample. This limitation has been reported previously for NGS and is mainly associated (>70% of the cases) with repetitive elements, a homopolymer stretch of 6 bases or more, simple repeats, and/or the presence of an indel in the vicinity.17 Nevertheless, analysis of sequence contexts for mutation c.1588A>C (p.N530H) and c.3231C>G showed that they do not have such elements associated. Additional controls ruled out potential contamination, so we consider these mutations artifacts, although we cannot suggest a likely explanation for this occurrence at this time.
Furthermore, after mutation assignment, we were unable to identify any genetic variations in 11 (27.5%) of the 40 VWD patients studied. VWF of these patients was entirely sequenced by the Sanger method and a mutation was detected in only 2 cases; there was no detectable mutation in the remaining 9 cases. Multiplex ligation-dependent probe amplification (MLPA)18 was applied to all these 9 cases and only patient VW0057 showed an abnormal MLPA pattern, revealing a heterozygous deletion of exon 15. The point mutations identified by Sanger sequencing and not by NGS (false-negative mutations) were a substitution in patient VW0141 (c.4120C>T) and the duplication of an amino acid (c.4678_4680dup, p.D1560dup) in patient VW0078. Furthermore, in VWF from the type 3 patient VW0276, only one mutation that did not explain the severe phenotype was detected by NGS, so complete sequencing by Sanger was also performed. Remarkably, the second mutation detected in this patient was the duplication of 4 bases (c.6699_6702dup). Accordingly, we achieved encouraging results with high efficiency in mutation detection. The only problem we encountered was detecting insertions of three or more bases. These results are likely related to the software analysis rather than the sequencing procedure, because all point mutations were positively detected in pools with optimal coverage conditions. It is known that indel detection using NGS platforms is technically challenging. Misalignments and/or low coverage can lead to missed frameshift insertion calls17 which occurred in 2 of the patients we studied. To avoid this, the use of paired-end sequencing libraries rather than single read runs can be used to detect large and small insertions, deletions, inversions, and other rearrangements. Nevertheless, the rate of false-positive and false-negative mutations is still a significant drawback for these platforms. Therefore, it is still essential to validate the mutations by Sanger sequencing.
All variations identified in the patients were consistent with the VWD type and laboratory values. Furthermore, most of the patients with no mutation identified had slightly decreased levels of VWF or little hemorrhagic symptoms, consistent with the percentage of VWD type 1 without mutations in VWF. In this single NGS experiment, positive identification of 43 VWF mutations was achieved, including 36 substitutions (27 missense, 2 nonsense and 7 synonymous), 2 intronic splice site mutations, 2 indels, and 3 deletions. Based on all the data from NGS and Sanger sequencing, we now report 46 mutations (43 considering only NGS) from 40 unrelated VWD patients of different types, 31 of whom have been previously described and the other 15 who were new. The deleterious mechanisms of these mutations are, in several cases, obvious given that they create premature termination codons or a frameshift leading to truncated VWF protein (5 frameshift mutations). However, without the help of functional studies, it is often difficult to decide whether a particular sequence variant is associated with disease or represents a rare polymorphism with no deleterious effect. In this regard, we have used dbSNP (Build 133) to rule out the presence of the putative mutations in an unaffected population since access to polymorphism databases is a fundamental first step in the investigation of new variants of unknown effect. Furthermore, software tools can provide useful information before in-depth mutation expression analysis is started. We have performed the in silico analysis (Online Supplementary Design and Methods) for all new missense, synonymous and potential splice site mutations and the obtained results (Online Supplementary Tables S3 and S4) show that 5 were expected to cause disease (4 missense and 1 PSSM) while 5 of them have unknown significance (1 missense and 4 PSSM).
Our proof-of-concept study showed simultaneous sequencing of 40 patients per flow cell lane. However, we also have results showing that 50 patients per lane instead of 40 can give sufficient coverage to obtain good results in mutation identification (data not shown). Therefore, at least 350 VWD patients could be analyzed per run, taking up 7 lanes. The hands-on time and instrument run times for sequencing 40 patients was measured in days or weeks rather than months, while the cost was less than 100 euros per sample; this price is at least 10 times lower than that for traditional sequencing. Obviously, this procedure is not suitable for use in diagnostic services to identify the mutations responsible for VWD, since they usually receive and analyze small batches of patients together. In contrast, large multicenter epidemiological studies requiring massively parallel VWF sequencing of patients and relatives are only affordable at the lower price of NGS. Along these lines, there is currently an ongoing initiative which is being encouraged by the Spanish Society of Thrombosis and Hemostasis with the objective of establishing a National VWD Registry for Spain: the clinical and molecular profile of VWD in Spain (PCM-EVW-ES) project. The genetic analysis of this study will be simplified and the costs drastically reduced with the introduction of NGS technology. The results presented here could have singular relevance for type 1 VWD (70–80% of cases). Type 1 VWD requires evaluation of the entire VWF and that is very expensive and time consuming by means of traditional sequencing methods. In addition, frequently no mutation is detected or the identified variations cannot be confirmed as causative mutations. For these reasons, the value of genetic studies in type 1 families is currently questioned in a routine health care environment in which cost is an issue. However, the reduction in costs to be achieved with NGS could change this picture: the combination of massively parallel mutation analysis together with phenotypic and familial data in a well-defined population will be very useful for a better understanding of the mechanisms involved in the pathophysiology of type 1 VWD.
In conclusion, our study shows that, even with the existing difficulties, individual investigators can use NGS as a research tool with confidence and ease. To our knowledge, this is the first report in which NGS re-sequencing is examined in a genetic study of multiple patients with a congenital monogenic disease. Therefore, this approach can serve as an example for other coagulopathies or disorders linked to large and complex genes and help NGS to become a routine diagnostic reality in the near future.
Footnotes
The online version of this article has a Supplementary Appendix:
Funding: this work was supported in part by a grant from the Fondo de Investigaciones Sanitarias (PI080385) and from the Industrial Applied Investigation Subprogram (IAP-580000-2008-20), Spanish Ministry of Science and Innovation. The work carried out at our laboratory received support from the Fundació Privada Catalana de l’Hemofília.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Bernardi F, Marchetti G, Casonato A, Gemmati D, Patracchini P, Legnani C, et al. Characterization of polymorphic markers in the von Willebrand factor gene and pseudogene. Br J Haematol. 1990;74(3):282–9. doi: 10.1111/j.1365-2141.1990.tb02584.x. [DOI] [PubMed] [Google Scholar]
- 2.Hashemi Soteh M, Peake IR, Marsden L, Anson J, Batlle J, Meyer D, et al. Mutational analysis of the von Willebrand factor gene in type 1 von Willebrand disease using conformation sensitive gel electrophoresis: a comparison of fluorescent and manual techniques. Haematologica. 2007;92(4):550–3. doi: 10.3324/haematol.10606. [DOI] [PubMed] [Google Scholar]
- 3.Kakela JK, Friedman KD, Haberichter SL, Buchholz NP, Christopherson PA, Kroner PA, et al. Genetic mutations in von Willebrand disease identified by DHPLC and DNA sequence analysis. Mol Genet Metab. 2006;87(3):262–71. doi: 10.1016/j.ymgme.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 4.Corrales I, Ramirez L, Altisent C, Parra R, Vidal F. Rapid molecular diagnosis of von Willebrand disease by direct sequencing. Detection of 12 novel putative mutations in VWF gene. Thromb Haemost. 2009;101(3):570–6. doi: 10.1160/th08-08-0500. [DOI] [PubMed] [Google Scholar]
- 5.McPherson JD. Next-generation gap. Nat Methods. 2009;6(11 Suppl):S2–5. doi: 10.1038/nmeth.f.268. [DOI] [PubMed] [Google Scholar]
- 6.Ropers HH. New perspectives for the elucidation of genetic disorders. Am J Hum Genet. 2007;81(2):199–207. doi: 10.1086/520679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raca G, Jackson C, Warman B, Bair T, Schimmenti LA. Next generation sequencing in research and diagnostics of ocular birth defects. Mol Genet Metab. 2010;100(2):184–92. doi: 10.1016/j.ymgme.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonnal RJ, Severgnini M, Castaldi A, Bordoni R, Iacono M, Trimarco A, et al. Reliable resequencing of the human dystrophin locus by universal long polymerase chain reaction and massive pyrosequencing. Anal Biochem. 2010;406(2):176–84. doi: 10.1016/j.ab.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 9.Lim BC, Lee S, Shin JY, Kim JI, Hwang H, Kim KJ, et al. Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology: comprehensive mutational search in a single platform. J Med Genet. 2011;48(11):731–6. doi: 10.1136/jmedgenet-2011-100133. [DOI] [PubMed] [Google Scholar]
- 10.Sadler JE. A revised classification of von Willebrand disease. For the Subcommittee on von Willebrand Factor of the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. Thromb Haemost. 1994;71(4):520–5. [PubMed] [Google Scholar]
- 11.Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4(10):2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 12.Rogaev EI, Grigorenko AP, Faskhutdinova G, Kittler EL, Moliaka YK. Genotype analysis identifies the cause of the “royal disease”. Science. 2009;326(5954):817. doi: 10.1126/science.1180660. [DOI] [PubMed] [Google Scholar]
- 13.Corrales I, Ramirez L, Ayats J, Altisent C, Parra R, Vidal F. Integration of molecular and clinical data of 40 unrelated VWD families in a Spanish locus-specific mutation database. First release including 58 mutations. Haematologica. 2010;95(11):1982–4. doi: 10.3324/haematol.2010.028977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mardis ER. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008;24(3):133–41. doi: 10.1016/j.tig.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 15.Grossmann V, Kohlmann A, Eder C, Haferlach C, Kern W, Cross NC, et al. Molecular profiling of chronic myelomonocytic leukemia reveals diverse mutations in >80% of patients with TET2 and EZH2 being of high prognostic relevance. Leukemia. 2011;25(5):877–9. doi: 10.1038/leu.2011.10. [DOI] [PubMed] [Google Scholar]
- 16.Craig DW, Pearson JV, Szelinger S, Sekar A, Redman M, Corneveaux JJ, et al. Identification of genetic variants using bar-coded multiplexed sequencing. Nat Methods. 2008;5(10):887–93. doi: 10.1038/nmeth.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10(3):R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cabrera N, Casana P, Cid AR, Moret A, Moreno M, Palomo A, et al. First application of MLPA method in severe von Willebrand disease. Confirmation of a new large VWF gene deletion and identification of heterozygous carriers. Br J Haematol. 2011;152(2):240–2. doi: 10.1111/j.1365-2141.2010.08400.x. [DOI] [PubMed] [Google Scholar]