

Abstract
Background
Refractory anemia with ring sideroblasts associated with marked thrombocytosis was proposed as a provisional entity in the 2001 World Health Organization classification of myeloid neoplasms and also in the 2008 version, but its existence as a single entity is contested. We wish to define the clinical features of this rare myelodysplastic/myeloproliferative neoplasm and to compare its clinical outcome with that of refractory anemia with ring sideroblasts and essential thrombocythemia.
Design and Methods
We conducted a collaborative retrospective study across Europe. Our database included 200 patients diagnosed with refractory anemia with ring sideroblasts and marked thrombocytosis. For each of these patients, each patient diagnosed with refractory anemia with ring sideroblasts was matched for age and sex. At the same time, a cohort of 454 patients with essential thrombocythemia was used to compare outcomes of the two diseases.
Results
In patients with refractory anemia with ring sideroblasts and marked thrombocytosis, depending on the Janus Kinase 2 V617F mutational status (positive or negative) or platelet threshold (over or below 600×109/L), no difference in survival was noted. However, these patients had shorter overall survival and leukemia-free survival with a lower risk of thrombotic complications than did patients with essential thrombocythemia (P<0.001) but better survival (P<0.001) and a higher risk of thrombosis (P=0.039) than patients with refractory anemia with ring sideroblasts.
Conclusions
The clinical course of refractory anemia with ring sideroblasts and marked thrombocytosis is better than that of refractory anemia with ring sideroblasts and worse than that of essential thrombocythemia. The higher risk of thrombotic events in this disorder suggests that anti-platelet therapy might be considered in this subset of patients. From a clinical point of view, it appears to be important to consider refractory anemia with ring sideroblasts and marked thrombocytosis as a distinct entity.
Keywords: refractory anemia, ring sideroblasts, thrombocytosis, essential thrombocythemia, clinical course
Introduction
In the third edition of the World Health Organization (WHO) classification (2001), refractory anemia with ring sideroblasts associated with marked thrombocytosis (RARS-T) was proposed as a provisional entity. This entity was created to group together patients who have the clinical and morphological features of refractory anemia with ring sideroblasts (RARS), a myelodysplastic syndrome (MDS), and thrombocytosis that is a mark of myeloproliferative neoplasms (MPN) such as essential thrombocythemia (ET). At that time, the threshold to define thrombocytosis in RARS-T and ET was a platelet count equal to or above 600×109/L, and most studies performed since then have used this threshold. In the 2008 WHO classification of myeloid neoplasms,1,2 RARS-T was classified in the so-called myelodysplastic/myeloproliferative neoplasms (MDS/MPN), which also include chronic myelomonocytic leukemia,3 atypical chronic myeloid leukemia (BCR-ABL1 negative)4 and juvenile myelomonocytic leukemia.5 The threshold to define RARS-T was lowered to a platelet count equal to or above 450×109/L for two reasons. Firstly, studies showed that patients with a platelet count above or below 600×109/L, for the JAK2 V617F mutation in particular, share common biological features. Secondly, it is coherent with the threshold of 450×109/L now used for the diagnosis of ET patients.
Ring sideroblasts are the distinctive feature of sideroblastic anemias.6 In particular, their presence in the bone marrow (15% or more) is a marker of the myelodysplastic syndrome defined as RARS which is characterized by both morphological dysplasia in only the erythroid lineage and a favorable clinical outcome.7,8
Mutations in the Janus Kinase 2 gene (JAK2) and/or myeloproliferative leukemia protein gene (MPL) have been detected in 50–60% of patients with RARS-T.9–22 Some of us recently showed that RARS-T is indeed a myeloid neoplasm with both myelodysplastic and myeloproliferative features at the molecular and clinical level, and that it may develop from RARS through the acquisition of somatic mutations of JAK2, MPL or other as yet unknown genes.14 This conclusion was recently confirmed by an independent observation.23 However, the very presence of these mutations has also raised the question of whether RARS-T is a form of essential thrombocythemia rather than a separate condition.24
Since RARS-T is a rare disorder, we conducted a collaborative study in several European countries to define the clinical features of this myelodysplastic/myeloproliferative neoplasm, and to compare its clinical outcome with that of RARS and ET.
Design and Methods
Patient selection
A total of 200 RARS-T cases according to the WHO 2008 criteria from 16 centers in 6 European countries were retrospectively enrolled. Patients were diagnosed with RARS-T if they fulfilled the following criteria: i) refractory anemia (hemoglobin level <125g/L for females and <135g/L for males) with erythroid dysplasia and more than 15% ring sideroblasts; ii) thrombocytosis of more than 450×109 platelets/L; iii) less than 5% blast cells in the bone marrow; iv) the presence of large atypical megakaryocytes similar to those observed in BCR-ABL1 negative MPN; v) no secondary cause of ring sideroblasts; and vi) no karyotype abnormalities such as del (5q), t(3;3)(q21;q26) or inv(3)(q21;q26). Only patients with sustained elevation of platelet count without any other cause of thrombocytosis were recorded.
For each patient with RARS-T, we collected data for one patient with RARS from the same center, matched for year of birth (±3 years) and sex. The different centers provided us with 123 cases that we took up to a total of 200 with RARS patients matched for year of birth (±3 years) from the Dijon registry. After review of the 200 medical records of the collected RARS patients, 35 RARS patients with multilineage dysplasia were excluded because they fulfilled the criteria of refractory cytopenia with multilineage dysplasia and ring sideroblasts which is another entity of the WHO classification associated with a shorter survival than RARS. Finally, 165 RARS patients were recorded. One hundred and forty-three patients with essential thrombocythemia gathered from the different centers were added to 311 patients with the same disease from the Dijon Registry, since their survival was comparable, and these were used as controls (Online Supplementary Table S1).
Data record
The diagnosis of acute myeloid leukemia (AML) was made according to the WHO criteria based on a threshold of 20% blasts in the bone marrow. All thromboses occurring at the time of diagnosis or during follow up were recorded. Arterial thrombotic events included cerebral (stroke, transient ischemic attack), cardiac (myocardial infarction) and peripheral arterial thrombosis. Venous thromboses included deep venous thrombosis, pulmonary embolism or splanchnic (hepatic, portal, splenic or mesenteric) venous thrombosis. Superficial venous thromboses were not considered. The JAK2V617F allele burden was analyzed according to the method published by Lippert et al.25 MPL mutations were detected using sequencing analysis.
Statistical analyses
Comparison between categorical variables was performed by χ2 statistics. Fisher‘s exact test was used when the hypothesis of the χ2 test was not confirmed. Categorical and continuous variables were compared using the Kruskal-Wallis test. All probability values were two-tailed. P<0.05 was considered statistically significant.
Standardized overall survival was estimated by the Kaplan-Meier method and based on the time from the date of diagnosis to death or last contact and with weighted data with frequency according to sex and age. Survival curves for the different groups were compared using the log rank test. Multivariate analysis was performed using Cox’s proportional hazards models. A similar analysis was performed for leukemia free survival (LFS). Time to leukemia transformation or death was used to calculate LFS. Analyses were performed using STATA.10 software. We performed Poisson’s regression analysis to compare the incidence of thrombotic events and AML transformation between the three groups of patients. Median follow up was determined using the reverse Kaplan-Meier estimator. The median follow-up survival was 102, 79 and 61 months for ET, RARS and RARS-T patients, respectively. The median follow up for thrombotic events was 102, 81, and 61 months for ET, RARS and RARS-T patients, respectively. The median follow up for AML transformation was 100, 73 and 59 months for ET, RARS and RARS-T patients, respectively.
Approval for the study was obtained from the ethics committee of each institution and the procedures were carried out in accordance with the Helsinki Declaration of 1975, as revised in 2000.
Results
Clinical and biological features
A total of 200 RARS-T (105 females and 95 males), 165 RARS (84 females and 81 males) and 454 ET (252 females and 202 males) patients were included in the study. At the time of diagnosis, ET patients were significantly younger than RARS-T (P<0.001) and RARS patients (P<0.001) (median age of 68.4, 73.6 and 77.1 years, respectively), which led us to standardize for age and sex in the overall survival analyses. The demographic, clinical and biological features of each group are described in Table 1.
Table 1.
Demographic, clinical and biological characteristics of patients with RARS-T compared with RARS and ET.
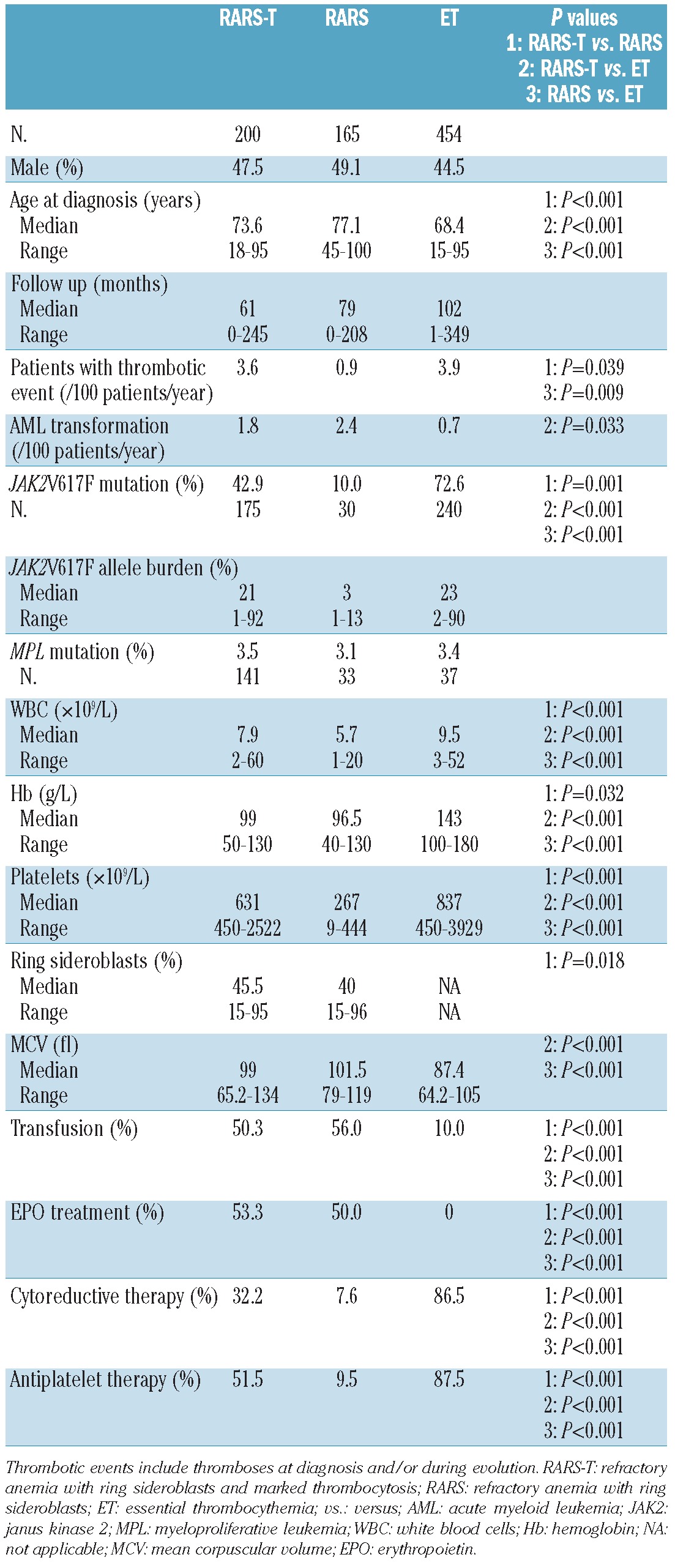
Compared with RARS patients, RARS-T individuals had a higher hemoglobin level (P=0.032), white blood cell count (WBC) (P<0.001), platelet count (by definition) (P<0.001) and percentage of ring sideroblasts (median 45.5% vs. 40%, P=0.018), with similar mean corpuscular volume (MCV). In contrast, RARS-T patients showed lower hemoglobin levels (P<0.001), and WBC (P<0.001) and platelet counts (P<0.001), but higher MCV (P<0.001) than did ET patients. Of note, 6 patients classified as having RARS at diagnosis had a subsequent increase in platelet count with RARS-T features after a median delay of 63 months, and were thus recorded as RARS-T. Two of them carried the JAK2V617F mutation at the time of diagnosis of RARS, with an increase in the allele burden in one patient when disease evolved to RARS-T. For 2 others, the evolution toward the RARS-T diagnosis was associated with the detection of the JAK2V617F mutation that had not been observed at the RARS state. The platelet count ranged from 55 to 397×109/L at the RARS state, then from 458 to 958×109/L when these patients transformed to RARS-T.
When the RARS-T JAK2V617F positive and negative patients were compared, JAK2V617F positive patients had higher hemoglobin levels (P=0.002), and WBC (P=0.023) and platelet counts (P<0.001).
Mutational status
The JAK2V617F mutation was screened for 175 RARS-T patients, 30 RARS patients and 240 ET patients, and MPL mutations were screened for 141 RARS-T patients, 33 RARS patients and 37 ET patients. The JAK2V617F mutation was found in 42.9%, 10.0% and 72.6% in RARS-T, RARS and ET patients, respectively (P≤0.001), whereas an MPL mutation was found in 3.5%, 3.1% and 3.4%, respectively. The JAK2V617F allele burden was available for 160 RARS-T patients, 30 RARS patients and 57 ET patients. Among the JAK2 positive patients, the JAK2V617F allele burden was 50% or over in 16% of RARS-T.
Survival
Using sex and age-standardized survival analyses, survival in RARS-T patients was shorter than in ET patients (median survival 76 months vs. 115 months, P<0.001) but longer than in RARS (median survival 76 months vs. 63 months, P<0.001) (Figure 1). Similar results were noted for LFS.
Figure 1.
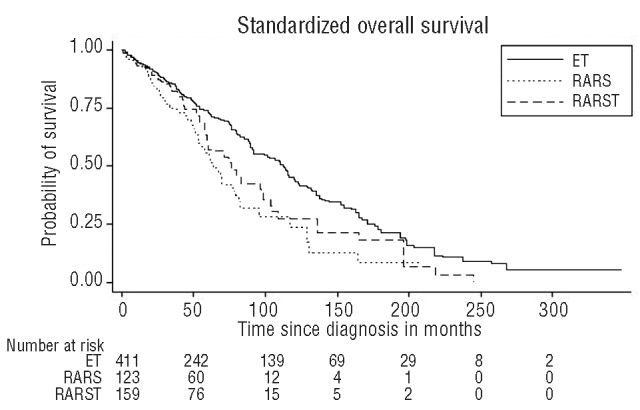
Kaplan-Meier standardized overall survival curves of three patient groups. Standardized survival of RARS-T is shorter than ET patients (median survival 76 months vs. 115 months, log rank test: P<0.001) but also longer than RARS patients (median survival 76 months vs. 63 months, P<0.001). ET: essential thrombocythemia; RARS: refractory anemia with ring sideroblasts; RARS-T: refractory anemia with ring sideroblasts and marked thrombocytosis.
In RARS-T patients, no difference in survival was noted regardless of the JAK2 mutational status or platelet threshold (i.e. > or <600×109/L) (Figures 2 and 3).
Figure 2.
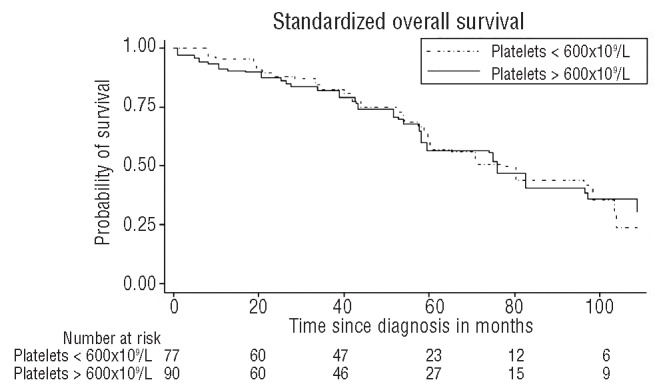
Kaplan-Meier standardized overall survival curves of RARS-T patients according to platelet count (> or < 600×109/L). No difference is noted between the two groups.
Figure 3.
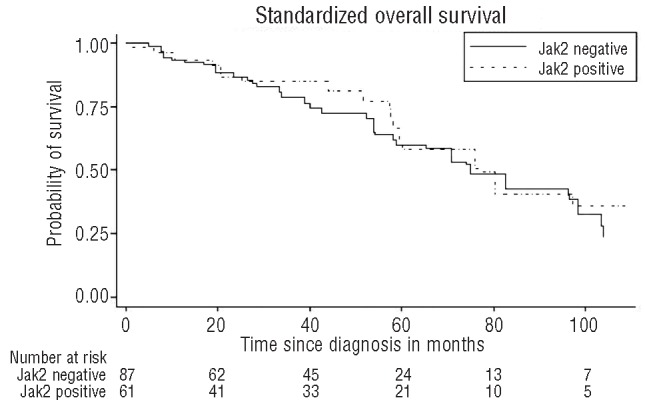
Kaplan-Meier standardized overall survival curves of RARS-T patients according to the JAK2V617F mutation. No difference is noted in JAK2 positive and negative patients. JAK2: Janus Kinase 2.
Thrombotic events and acute myeloid transformation
Thrombotic events expressed as thrombotic events per 100 patient years were similar in ET (3.9) and in RARS-T (3.6) but more frequent than in RARS (0.9) patients (P=0.009 and P=0.039, respectively). Acute myeloid transformation per 100 patient years was similar in RARS-T (1.8) and RARS (2.4), and higher in RARS-T than in ET (0.7, P=0.033).
Treatments
There was no difference in supportive care, including transfusion and the use of erythropoietin, between RARS-T and RARS patients (50.3% and 56.0% were transfused, whereas 53.3% and 50.0% received erythropoietin, respectively). However, RARS-T patients were more frequently treated with antiplatelet drugs (51.5%) and cytoreductive therapy (32.2%) than were RARS patients (P<0.001 in both cases).
Discussion
RARS-T was first proposed in the WHO 2001 classification as a provisional entity and was included again as a provisional entity in the 2008 version since it was not clear whether this form was a distinct entity or whether it represented a group of MDS or MPN that acquired secondary ring sideroblasts.1 However, it has been proposed that this category should be discarded since it is a rare disease with similar features to those observed in ET and with no differences in therapeutic management.24
In fact, the lack of any previous large series of patients prevented us from drawing any clear and definitive conclusions on the best classification of this entity. The present study included 200 RARS-T cases which is, to the best of our knowledge, the largest series of RARS-T patients ever studied.
In our study, as previously noted,2,3 RARS-T patients had higher hemoglobin levels and WBC and platelet counts than did RARS patients, as well as RARS-T JAK2V617F positive patients compared with JAK2V617F negative patients. On the other hand, this myeloproliferative advantage of the JAK2V617F mutation has been reported both for hemoglobin and WBC but not for the platelet count in ET, since mutated ET classically has a lower platelet count than the non-mutated types.26,27 Of note, unlike a previous study that did not find a JAK2V617F mutation in RARS patients with mild thrombocytosis (platelets 400–600×109/L),22 in our study, 28% of our RARS-T patients with platelets less than 600×109/L harbored JAK2 mutations.
The JAK2V617F allele burden was more than 50% in 16% of our RARS-T patients while Schmitt-Graeff et al. reported a higher percentage.18 Nevertheless, this proportion of homozygous forms was clearly higher than the commonly reported low rate of homozygous JAK2V617F mutated ET.27 Finally, MCV was high in both RARS-T and RARS patients (99 and 101.5 fl, respectively) whereas it was normal in ET (87.4 fl).
Surprisingly, in our work, we found higher percentages of RARS patients with a mutation of JAK2 and/or MPL than those usually reported.10,14,21 This finding is related to the small number of RARS patients tested, since these tests were performed in only 30 and 33 RARS patients, respectively. Three were mutated for the JAK2V617F associated with a low allele burden (1%, 3% and 13%). Only one patient carried an MPL mutation (to the best of our knowledge, this has never been reported in RARS) and had a high platelet count (391×109/L) with a normal WBC count. These cases can be related to recent findings: Malcovati et al. reported 2 cases of initial RARS that subsequently acquired the JAK2V617F mutation associated with thrombocytosis, leading to the diagnosis of RARS-T.14 The authors hypothesized that RARS-T may develop as a consequence of the acquisition of a JAK or MPL mutation. In fact, the presence of an MPL mutation in RARS, mainly noted in MPN and less frequently in RARS-T, reinforces this hypothesis. Furthermore, in our series of patients, 6 who were initially considered as having RARS (2 with a JAK2V617F mutation) went on to develop RARS-T after a median delay of 63 months (associated with an appearance or an increase in the JAK2 allele burden in 3 cases), suggesting that RARS-T may develop from a pre-existing RARS through the acquisition of a JAK or MPL mutation.
Conflicting results have been reported on the impact of the JAK2V617F mutation on survival in RARS-T patients.15,18 Our results clearly show that no difference was observed in the survival rate between JAK2V617F positive and negative RARS-T patients.
In the 2008 WHO classification, the threshold platelet count for the definition of RARS-T was decreased from 600 to 450×109 platelets/L. One could ask whether the outcome in patients with 450–600×109 platelets/L is similar to that in those patients with more than 600×109 platelets/L. Raya et al. have already answered this question in a previous study in which they show that there was no difference in survival between the two groups.15 Our study confirmed this finding in a large series since no difference was noted in the outcome according to the platelet threshold.
Since the median age of RARS-T patients was high at diagnosis, control ET and RARS patients were also old. As a consequence, the median survival of RARS and ET patients was shorter than that usually reported. On the other hand, to rule out sex and age differences between the three groups, we performed age and sex standardized survival analyses. Our results showed better outcomes in RARS-T than RARS patients related to survival and LFS but worse outcomes than those in ET. This confirms previous findings in a large series of patients.22,28,29 In these three studies, median overall survival in the RARS-T group was lower than in the ET group. However, in the present study, RARS-T patients were more likely than RARS patients to have thrombosis, suggesting that anti-platelet therapy might be considered in this subset of patients.
Finally, from a therapeutic point of view, the recent report of the efficacy of lenalidomide, a drug that has been used successfully in MDS, in 2 RARS-T patients is also in keeping with an MDS rather than MPN origin of RARS-T.30
Figure 4.
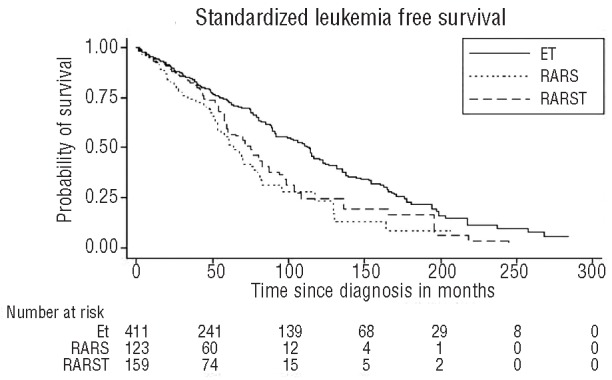
Kaplan-Meier standardized leukemia free survival curves of three patient groups. Standardized leukemia free survival of RARS-T is shorter than that of ET patients (median survival 74 months vs. 110 months, log rank test: P<0.001). Standardized leukemia free survival of RARS patients was lower than that of ET patients (P<0.001). ET: essential thrombocythemia; RARS: refractory anemia with ring sideroblasts; RARS-T: refractory anemia with ring sideroblasts and marked thrombocytosis.
In conclusion, taking all of the parameters into account, and despite the fact that RARS-T shares several features with both ET and RARS, the findings of this study show that survival and LFS in RARS-T are lower than those in ET. This confirms previous observations. Survival and LFS in RARS-T are better than in RARS, but with a higher risk of thrombosis, justifying anti-platelet therapy to prevent thrombotic events. RARS-T seems to be a particular disease potentially deriving, at least in some cases, from a previous RARS but with a better outcome. Therefore, from a clinical point of view, it appears to be important to consider RARS-T as a distinct entity.
Acknowledgments
The authors would like to thank Pr Radek Skoda for his advice and Philip Bastable for revising the manuscript.
Footnotes
Funding: this work was supported by grants from Tulipes contre le cancer Châlon s/Saône to FG, from AIRC (Associazione Italiana per la Ricerca sul Cancro) and Fondazione Cariplo to MC, from IS Carlos III, Ministerio de Sanidad y Consumo (Spain): PI 07/1009. 2008, AEHH, and Generalitat de Catalunya SGR 2009 541 to LF.
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Vardiman JW, Bennett JM, Bain BJ, Baumann I, Thiele J, Orazi A. Myelodysplastic/myeloproliferative neoplasms, unclassifiable. Lyon: IARC; 2008. [Google Scholar]
- 2.Vardiman JW, Brunning RD, Arber DA, et al. Introduction and overview of the classification of the myeloid neoplasms. Lyon: IARC; 2008. [Google Scholar]
- 3.Orazi A, Bennett JM, Germing U, Brunning RD, Bain BJ, Thiele J. Chronic myelomonocytic leukaemia. Lyon: IARC; 2008. [Google Scholar]
- 4.Vardiman JW, Bennett JM, Bain BJ, Brunning RD, Thiele J. Atypical chronic myeloid leukaemia, BCR-ABL1 negative. Lyon: IARC; 2008. [Google Scholar]
- 5.Baumann I, Bennett JM, Niemeyer CM, Thiele J, Shannon K. Juvenile myelomonocytic leukaemia. Lyon: IARC; 2008. [Google Scholar]
- 6.Cazzola M, Invernizzi R. Sideroblastic anemias. Philadelphia: Mosby Elsevier; 2005. [Google Scholar]
- 7.Hasserjian RP, Gattermann N, Bennett JM, Brunning RD, Thiele J. Refractory anemia with ringed sideroblasts. Lyon: IARC; 2008. [Google Scholar]
- 8.Malcovati L, Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol. 2005;23(30):7594–603. doi: 10.1200/JCO.2005.01.7038. [DOI] [PubMed] [Google Scholar]
- 9.Boissinot M, Garand R, Hamidou M, Hermouet S. The JAK2-V617F mutation and essential thrombocythemia features in a subset of patients with refractory anemia with ring sideroblasts (RARS) Blood. 2006;108(5):1781–2. doi: 10.1182/blood-2006-03-008227. [DOI] [PubMed] [Google Scholar]
- 10.Ceesay MM, Lea NC, Ingram W, Westwood NB, Gaken J, Mohamedali A, et al. The JAK2 V617F mutation is rare in RARS but common in RARS-T. Leukemia. 2006;20(11):2060–1. doi: 10.1038/sj.leu.2404373. [DOI] [PubMed] [Google Scholar]
- 11.Flach J, Dicker F, Schnittger S, Kohlmann A, Haferlach T, Haferlach C. Mutations of JAK2 and TET2, but not CBL are detectable in a high portion of patients with refractory anemia with ring sideroblasts and thrombocytosis. Haematologica. 2010;95(3):518–9. doi: 10.3324/haematol.2009.013631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gattermann N, Billiet J, Kronenwett R, Zipperer E, Germing U, Nollet F, et al. High frequency of the JAK2 V617F mutation in patients with thrombocytosis (platelet count>600x109/L) and ringed sideroblasts more than 15% considered as MDS/MPD, unclassifiable. Blood. 2007;109(3):1334–5. doi: 10.1182/blood-2006-05-022491. [DOI] [PubMed] [Google Scholar]
- 13.Hellstrom-Lindberg E, Cazzola M. The role of JAK2 mutations in RARS and other MDS. Hematology Am Soc Hematol Educ Program. 2008:52–9. doi: 10.1182/asheducation-2008.1.52. [DOI] [PubMed] [Google Scholar]
- 14.Malcovati L, Della Porta MG, Pietra D, Boveri E, Pellagatti A, Galli A, et al. Molecular and clinical features of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Blood. 2009;114(17):3538–45. doi: 10.1182/blood-2009-05-222331. [DOI] [PubMed] [Google Scholar]
- 15.Raya JM, Arenillas L, Domingo A, Bellosillo B, Gutierrez G, Luno E, et al. Refractory anemia with ringed sideroblasts associated with thrombocytosis: comparative analysis of marked with non-marked thrombocytosis, and relationship with JAK2 V617F mutational status. Int J Hematol. 2008;88(4):387–95. doi: 10.1007/s12185-008-0169-1. [DOI] [PubMed] [Google Scholar]
- 16.Remacha AF, Nomdedeu JF, Puget G, Estivill C, Sarda MP, Canals C, et al. Occurrence of the JAK2 V617F mutation in the WHO provisional entity: myelodysplastic/myeloproliferative disease, unclassifiable-refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2006;91(5):719–20. [PubMed] [Google Scholar]
- 17.Renneville A, Quesnel B, Charpentier A, Terriou L, Crinquette A, Lai JL, et al. High occurrence of JAK2 V617 mutation in refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Leukemia. 2006;20(11):2067–70. doi: 10.1038/sj.leu.2404405. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt-Graeff AH, Teo SS, Olschewski M, Schaub F, Haxelmans S, Kirn A, et al. JAK2V617F mutation status identifies subtypes of refractory anemia with ringed sideroblasts associated with marked thrombocytosis. Haematologica. 2008;93(1):34–40. doi: 10.3324/haematol.11581. [DOI] [PubMed] [Google Scholar]
- 19.Schnittger S, Bacher U, Haferlach C, Dengler R, Krober A, Kern W, et al. Detection of an MPLW515 mutation in a case with features of both essential thrombocythemia and refractory anemia with ringed sideroblasts and thrombocytosis. Leukemia. 2008;22(2):453–5. doi: 10.1038/sj.leu.2404909. [DOI] [PubMed] [Google Scholar]
- 20.Szpurka H, Gondek LP, Mohan SR, Hsi ED, Theil KS, Maciejewski JP. UPD1p indicates the presence of MPL W515L mutation in RARS-T, a mechanism analogous to UPD9p and JAK2 V617F mutation. Leukemia. 2009;23(3):610–4. doi: 10.1038/leu.2008.249. [DOI] [PubMed] [Google Scholar]
- 21.Szpurka H, Tiu R, Murugesan G, Aboudola S, Hsi ED, Theil KS, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis (RARS-T), another myeloproliferative condition characterized by JAK2 V617F mutation. Blood. 2006;108(7):2173–81. doi: 10.1182/blood-2006-02-005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang SA, Hasserjian RP, Loew JM, Sechman EV, Jones D, Hao S, et al. Refractory anemia with ringed sideroblasts associated with marked thrombocytosis harbors JAK2 mutation and shows overlapping myeloproliferative and myelodysplastic features. Leukemia. 2006;20(9):1641–4. doi: 10.1038/sj.leu.2404316. [DOI] [PubMed] [Google Scholar]
- 23.Vercauteren SM, Spronk HM, Karsan A, Young S, Nevill T, Coupland R. Transformation of RARS to JAK V617F positive RARS with thrombocytosis (RARS-T) Blood. 2010 Apr 9; e-letter. 2010. [Google Scholar]
- 24.Wardrop D, Steensma DP. Is refractory anaemia with ring sideroblasts and thrombocytosis (RARS-T) a necessary or useful diagnostic category? Br J Haematol. 2009;144(6):809–17. doi: 10.1111/j.1365-2141.2008.07526.x. [DOI] [PubMed] [Google Scholar]
- 25.Lippert E, Boissinot M, Kralovics R, Girodon F, Dobo I, Praloran V, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108(6):1865–7. doi: 10.1182/blood-2006-01-013540. [DOI] [PubMed] [Google Scholar]
- 26.Campbell PJ, Scott LM, Buck G, Wheatley K, East CL, Marsden JT, et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: a prospective study. Lancet. 2005;366(9501):1945–53. doi: 10.1016/S0140-6736(05)67785-9. [DOI] [PubMed] [Google Scholar]
- 27.Girodon F, Schaeffer C, Cleyrat C, Mounier M, Lafont I, Santos FD, et al. Frequent reduction or absence of detection of the JAK2-mutated clone in JAK2V617F-positive patients within the first years of hydroxyurea therapy. Haematologica. 2008;93(11):1723–7. doi: 10.3324/haematol.13081. [DOI] [PubMed] [Google Scholar]
- 28.Atallah E, Nussenzveig R, Yin CC, Bueso-Ramos C, Tam C, Manshouri T, et al. Prognostic interaction between thrombocytosis and JAK2 V617F mutation in the WHO subcategories of myelodysplastic/myeloproliferative disease-unclassifiable and refractory anemia with ringed sideroblasts and marked thrombocytosis. Leukemia. 2008;22(6):1295–8. doi: 10.1038/sj.leu.2405054. [DOI] [PubMed] [Google Scholar]
- 29.Shaw GR. Ringed sideroblasts with thrombocytosis: an uncommon mixed myelodysplastic/myeloproliferative disease of older adults. Br J Haematol. 2005;131(2):180–4. doi: 10.1111/j.1365-2141.2005.05747.x. [DOI] [PubMed] [Google Scholar]
- 30.Huls G, Mulder AB, Rosati S, van de Loosdrecht AA, Vellenga E, de Wolf JT. Efficacy of single-agent lenalidomide in patients with JAK2 (V617F) mutated refractory anemia with ring sideroblasts and thrombocytosis. Blood. 2010;116(2):180–2. doi: 10.1182/blood-2010-01-263087. [DOI] [PubMed] [Google Scholar]