

Abstract
Background
Shwachman–Diamond syndrome is an autosomal recessive disorder in which severe bone marrow dysfunction causes neutropenia and an increased risk of leukemia. Recently, novel particulate cytoplasmic structures, rich in ubiquitinated and proteasomal proteins, have been detected in epithelial cells and neutrophils from patients with Helicobacter pylori gastritis and several epithelial neoplasms.
Design and Methods
Blood neutrophils from 13 cases of Shwachman–Diamond syndrome – ten with and three without SBDS gene mutation – and ten controls were investigated by confocal microscopy and ultrastructural immunocytochemistry using antibodies against ubiquitinated proteins, proteasomes, p62 protein, and Helicobacter pylori VacA, urease and outer membrane proteins.
Results
Many extensively disseminated particulate cytoplasmic structures, accounting for 22.78±5.57% (mean ± standard deviation) of the total cytoplasm, were found in blood neutrophils from mutated Shwachman–Diamond syndrome patients. The particulate cytoplasmic structures showed immunoreactivity for polyubiquitinated proteins and proteasomes, but no reactivity for Helicobacter pylori products, which are present in particulate cytoplasmic structures of Helicobacter pylori-positive gastritis. Neutrophils from patients with Shwachman–Diamond syndrome frequently showed p62-positive autophagic vacuoles and apoptotic changes in 5% of cells. No particulate cytoplasmic structures were observed in most control neutrophils; however, in a few cells from two cases we noted focal development of minute particulate cytoplasmic structures, accounting for 0.74±0.56% of the total cytoplasm (P<0.001 versus particulate cytoplasmic structures from mutated Shwachman–Diamond syndrome patients). Neutrophils from non-mutated Shwachman–Diamond-syndrome-like patients resembled controls in two cases, and a third case showed particulate cytoplasmic structure patterns intermediate between those in controls and those in mutated Shwachman–Diamond syndrome patients.
Conclusions
Particulate cytoplasmic structures are a prominent feature of neutrophils from patients with Shwachman–Diamond syndrome. They may help us to understand the mechanism of granulocyte dysfunction and the neoplastic risk of the disease.
Keywords: ubiquitin, proteasome, particulate cytoplasmic structure, Shwachman-Diamond syndrome, autophagy, apoptosis
Introduction
Shwachman–Diamond syndrome (SDS; OMIM 260400) is an autosomal recessive disorder characterized by exocrine pancreatic insufficiency, skeletal abnormalities and bone marrow dysfunction, resulting in variable degrees of neutropenia, thrombocytopenia and anemia, and an increased risk of developing myelodysplastic syndrome and/or acute myeloid leukemia (MDS/AML). It is a rare disorder with an estimated incidence of 1/76,000.1 Bone marrow failure is associated with reduced numbers and abnormally accelerated apoptosis of hematopoietic progenitor cells.2
Approximately 90% of the patients have a mutation in the SBDS gene,3 coding for a multifunctional protein implicated in ribosome biogenesis, as well as in DNA metabolism and repair.4,5 Of special interest is the observation that the SBDS protein plays a role in cellular stress responses. Indeed, SBDS depletion causes cellular hypersensitivity to a variety of stress conditions, including endoplasmic reticulum stress and DNA damage, which may help explain the patients’ predisposition to MDS/AML.5 In fact, SBDS protein deficiency leads to markedly increased intracellular reactive oxygen species (ROS) in epithelial HeLa and myeloid TF1 cells, with accelerated apoptosis and reduced cell growth, both of which are rescued by antioxidants.6 ROS are known to cause protein damage, with intracellular accumulation of misfolded and partially denatured proteins, which must be rapidly eliminated through hyperfunction of the ubiquitin–proteasome system (UPS) to preserve cell homeostasis and function.7,8 Failure of the UPS leads to accumulation, aggregation and precipitation of ubiquitinated proteins to form aggresome-like induced structures (ALIS; p62 protein-reactive sequestosomes), autophagy activation and increased sensitivity to apoptosis.9–12 Thus, investigation of SDS neutrophils for any sign of UPS change, inclusion body formation and autophagy or apoptosis might contribute to explaining the pathogenesis of the disorder.
During ultrastructural investigations of endoscopic biopsies from patients with Helicobacter pylori chronic gastritis, in human foveolar cells, we found well-defined particulate cytoplasmic structures (PaCS) filled with barrel-like particles, 13 nm thick and 15–40 nm long, which were selectively reactive for polyubiquitinated proteins and proteasomes, as well as for bacterial virulence products.13 H. pylori infection is known to cause markedly increased intracellular ROS,14 which may enhance formation and ubiquitination of misfolded or partially degraded proteins, leading to proteasome stimulation, especially at sites of bacterial toxin concentration.13 In preliminary investigations, PaCS were also found in some neutrophilic granulocytes of H. pylori-infected mucosa (VN and ES, unpublished data). This is of particular interest because bacterial virulence factors such as lipopolysaccharide (LPS) are known to induce ALIS/sequestosome formation with autophagy activation in various cell lines,11,12 and to trigger oxygen-dependent apoptosis in human neutrophils.15
We, therefore, decided to investigate blood neutrophils from SDS patients with known SBDS gene status for the occurrence of ubiquitin–proteasome reactive PaCS, p62-reactive sequestosomes and autophagic and apoptotic changes.
Design and Methods
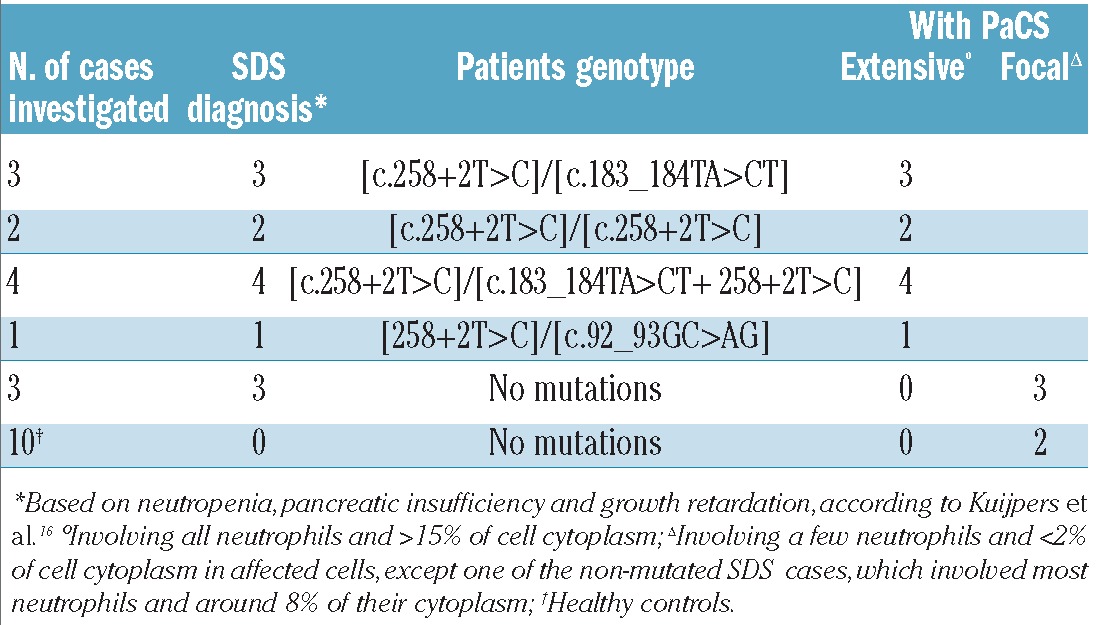
We investigated 13 patients – ten children (3–13 years old) and three young adults (18–38 years old) – in whom a diagnosis of SDS was suspected on the basis of clinical symptoms of pancreatic insufficiency, growth retardation and neutropenia, as suggested by Kuijpers et al.;16 in addition, ten age-matched healthy controls were studied (Table 1). Mutations of the SBDS gene were analyzed as previously described.17 The presence of two of the common mutations ([c.258+2T>C], [c.183_184TA>CT], [c.183_184TA>CT+ 258+2T>C]) was considered diagnostic for SDS; when no or only one of the common mutations was found, sequencing was extended to all exons of the gene. The study was approved by the Ethics Committee of the University of Pavia and written informed consent was obtained from each patient.
Table 1.
Clinical, molecular and cellular findings in patients and controls.
Blood samples were centrifuged at 75 × g for 5 min, and then the upper layer, enriched in white blood cells, was transferred to a new tube, washed twice in 0.9% NaCl buffer and re-centrifuged to obtain a pellet. White blood cell pellets were fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in cacodylate buffer for 4 h at 4°C, post-fixed in 1% osmium tetroxide for 1 h at room temperature, washed in cacodylate buffer for 10 min, and embedded in Epon–Araldite resin mixture. For light microscopy, semi-thin sections (0.5–1 μm) were stained with toluidine blue, and uranyl acetate–lead citrate contrasted ultrathin sections (60 nm) were prepared for transmission electron microscopy (TEM). Biopsies from H. pylori-infected gastric mucosa available from a previous study13 served as controls.
For ultrastructural immunolocalization, we used the colloidal gold technique, as previously reported.13,18 Ultrathin sections collected on 300 mesh nickel grids were pretreated with a saturated water solution of sodium metaperiodate for 10 min, washed with buffer A (0.45 M NaCl, 1% Triton X-100, and 0.05 M Tris–HCl, pH 7.4), and incubated in non-immune goat serum at room temperature for 1 h, to prevent non-specific binding of immunoglobulins. The sections were then incubated at 4°C overnight with antibodies directed against polyubiquitinated proteins (mouse monoclonal, FK1 clone; Enzo Life Sciences International, Plymouth Meeting, PA, USA); 20 S proteasome, αβ or β5i (immunoproteasome) subunits, and 19S proteasome, S2 subunit (all rabbit polyclonal; Calbiochem, La Jolla, CA, USA); p62 protein (rabbit polyclonal H-290; Santa Cruz Biotechnology, Santa Cruz, CA, USA); and H. pylori urease (UreA subunit; mouse monoclonal; Austral Biological, San Ramon, CA, USA), VacA (rabbit polyclonal; Austral Biological) or outer membrane proteins18 (rabbit polyclonal; Biomeda, Forster City, CA, USA) diluted in buffer B (0.45 M NaCl, 1% bovine serum albumin, 0.5% sodium azide, and 0.05 M Tris–HCl, pH 7.4). After washing in buffer B, primary immunoglobulin binding was revealed by appropriate gold-labeled anti-mouse or anti-rabbit immunoglobulins (EM GAMM 15; British Biocell International, Cardiff, UK) diluted in buffer B. The sections were stained with uranyl and lead and then analyzed by TEM using a Jeol 1200 EX II microscope.13,18 Morphometric observations were carried out with the ITEM soft imaging system (Olympus Soft Imaging Solutions GmbH, Münster, Germany). Numerical data were expressed as means ± standard deviation; the statistical significance of the differences was evaluated by Student’s t test, or ANOVA followed by the Newman–Keuls’ Q test, or linear regression analysis.
For confocal microscopy, 0.5-μm sections from aldehyde–osmium-fixed Epon–Araldite blocks were processed for immunofluorescence with antibodies directed against 20S proteasome (rabbit polyclonal PW 8155: Enzo Life Sciences International), mono- and polyubiquitinated (FK2 clone) or polyubiquitinated only (FK1 clone) proteins (both mouse monoclonal; Enzo Life Sciences International), and p62 protein (Santa Cruz Biotechnology), followed by Alexa488-labeled anti-mouse or anti-rabbit antibodies as appropriate. A TCS SP2 confocal laser scanning microscope (Leica, Heidelberg, Germany) equipped with a 63× oil-immersion objective was used. For correlative confocal microscopy and TEM13,19 the two faces of an ultrathin resin section collected on a 200-mesh gilder finder grid (Electron Microscopy Sciences, Hatfield, PA, USA) were processed separately. First, one face was immunostained and viewed by confocal microscopy, as above, then the reverse face of the section was processed for immunogold labeling and observed by TEM after uranyl-lead staining. The resulting confocal and TEM images of the same area were then overlapped using Adobe Photoshop.
For sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting, peripheral blood collected in a heparinized syringe was centrifuged on a Ficoll–Hypaque gradient at 400 × g for 35 min. A granulocyte pellet was recovered, resuspended in 90% NH4Cl in phosphate-buffered saline (PBS), and incubated on ice for 15 min to obtain complete lysis of residual red blood cells. Cells were then rinsed with PBS and recovered at 690 g for 10 min.20 Granulocyte purity >98% was obtained with this protocol. The granulocyte pellet was then lysed with lysis buffer (1.5 M Tris–HCl, pH 6.8, 8% SDS, and 40% glycerol) supplemented with 20% 2-mercaptoethanol. Volumes equivalent to a total of 50,000 cells for healthy controls or SDS patients were subjected to SDS-PAGE in 4–20% polyacrylamide gel. Proteins were then blotted onto nitrocellulose membranes (Bio-Rad Laboratories, Richmond, CA, USA) and the immunological analyses were performed using antibodies directed against 20S proteasome (rabbit polyclonal PW 8155; Enzo Life Sciences International), polyubiquitinated proteins (mouse monoclonal FK1 clone; Enzo Life Sciences International), p62 protein (SQSTM1, rabbit polyclonal; Santa Cruz Biotechnology), and β-actin (clone AC-74, mouse monoclonal; Sigma–Aldrich, St Louis, MO, USA), followed by secondary anti-mouse or anti-rabbit horseradish-peroxidase-labeled antibodies as appropriate.
For in vitro investigation of the role of ROS, purified human granulocytes (5×105 cells/mL) were incubated at 37°C for 5 h in RPMI cell culture medium, not supplemented with fetal calf serum, in the absence or presence of a ROS generating system, as described previously21 (i.e. 50 mU/mL xanthine oxidase plus 1 mM xanthine; both from Sigma–Aldrich). After incubation, cells were processed for TEM or SDS-PAGE as described above.
Results
SBDS gene analysis was carried out in 13 patients with neutropenia, pancreatic insufficiency and growth retardation, the characteristic clinical signs of SDS.16 Ten patients showed the common mutations of the disease, one of whom also showed a rare mutation [c.92_93GC>AG], as detailed in Table 1.
PaCS filled with barrel-like particles, 13 nm thick and 15–40 nm long, were found by TEM to be extensively distributed throughout the cytoplasm in all blood neutrophils taken from the 10 SDS patients with SBDS gene mutations (Figure 1A). High-resolution analysis of the PaCS particles showed a regularly arranged, aligned, punctuate substructure like that found in H. pylori-infected gastric epithelium.13 In 30 representative cells (three from each case), the PaCS had a mean diameter of 373±116 nm and accounted for 22.78±5.57% (range, 16.04±1.54 to 27.12±2.05%) of the total cytoplasmic area. No PaCS were seen in erythrocytes, lymphocytes, monocytes, eosinophils or basophils.
Figure 1.

(A) Electron microscopy (4,000x) of blood neutrophils from an SDS patient with SBDS gene mutation showing several PaCS scattered in the cytoplasm. The boxed area is enlarged in a1 (20,000x) to show PaCS-concentrated FK1 immunogold reactivity for polyubiquitinated proteins. In the inset (a2, 200,000x) from the same specimen as a1, the finely punctuate ultrastructure and barrel-like morphology of PaCS are shown at high resolution. Preferential PaCS reactivity of 20S (a3, 25,000x) and 19S (a4, 14,000x) proteasome antibodies is also seen. Black asterisk: PaCS; g: secretory granule. (B) PaCS-free control neutrophil (4,000x) enlarged in b1 (12,000x) to show thin areolae (white arrows) with preferential FK1 immunoreactivity. Scarce 20S proteasome immunoreactivity is seen in another section of the same control cell, with scattered areolae (b2, 12,000x). (C) Control neutrophil with focal development of small PaCS (boxed in C; 3,000x) enlarged in c1 (10,000x) and c2 (40,000x), with PaCS-concentrated FK1 immunogold. White asterisk: ribosomes. (D) One of the few neutrophils from an SDS-like syndrome without SBDS gene mutation showing PaCS. Note focal development of small PaCS, boxed in D (6,000x) and enlarged in d1 (35,000x) to recognize their particulate substructure and FK1 immunoreactivity. (E, 3,000x) A representative neutrophil from another SDS-like case shows on the left large, polarized PaCS, one of which is enlarged in e1 (30,000x) to illustrate its barrel-like particles and FK1 immunoreactivity; note PaCS-free cytoplasm on the right.
In healthy control neutrophils, minute, clear “areolae” (112±39 nm in diameter, P<0.001 versus PaCS of neutrophils from mutated SDS cases) with poorly defined contents were seen scattered in the cytoplasm (Figure 1B). No particle-rich PaCS were detected in 125 of 127 neutrophils analyzed (10–15 for each case) from the ten healthy controls during systematic TEM analysis. However, in two neutrophils from two separate control cases, focal development of small PaCS with their usual particulate content was observed (Figure 1C). Further analysis of an additional 40 neutrophils from the same two cases yielded three more such cells, giving a total of five out of 64 (8%) investigated from the two patients. Their PaCS had a mean diameter of 159.35±77.66 nm and accounted for 0.74±0.56% of the total cytoplasmic area (P<0.001 versus mutated SDS PaCS).
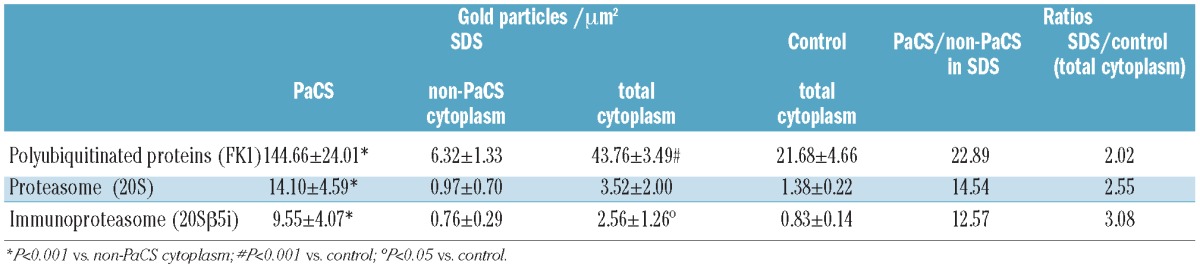
Cytochemical tests for polyubiquitinated proteins using the FK1 antibody showed a high concentration of immunogold particles inside the PaCS in mutated SDS neutrophils, with only a sparse distribution in non-PaCS cytoplasm; intermediate concentrations were seen in the PaCS-free cytoplasm of control neutrophils (Table 2 and Figure 1A and B). Mitochondria, endoplasmic reticulum cisternae and secretory granules remained unreactive. The total, PaCS-inclusive, cytoplasmic concentration of FK1 reactivity in SDS neutrophils increased by about 100% compared to that in the cytoplasm of control cells. In mutated SDS neutrophils, we also found a selective PaCS concentration of 20S, 20S β5i and 19S proteasome immunoreactivities, although less intense than that of ubiquitinated proteins. Proteasome reactivities were scarce (<1 gold particle/μm2 cytoplasm) and scattered in the PaCS-free cytoplasm of control neutrophils (Figure 1A and B), whereas their total cytoplasm concentration increased substantially in SDS neutrophils (by 150–200%) (Table 2). Immunoblotting analyses on whole cell lysates confirmed an increase in both 20S proteasome and FK1 reactive polyubiquitinated proteins in SDS neutrophils from patients with SBDS gene mutations, compared to control cells (Online Supplementary Figure S1).
Table 2.
Cellular distribution of polyubiquitinated proteins, the proteasome and immunoproteasome in five SDS patients with SBDS gene mutations, and five age-matched controls.
In two of the three SDS-like cases lacking mutations, blood neutrophils resembled those of healthy controls in showing either no or only small focal PaCS development (Figure 1D). In the third case, larger and polarized PaCS were observed in most cells (Figure 1E), accounting for 7.97±3.29% of total cell cytoplasm.
Under confocal microscopy, many intensely fluorescent cytoplasmic spots were detected in granulocytes from patients with mutated SBDS tested with FK2, FK1 or 20S proteasome antibodies (Figure 2A). Correlative, confocal immunofluorescence/immunogold electron microscopy, allowed us to overlap and compare directly light and electron microscopy findings on the same cell section using the same or different antibodies, thus confirming the PaCS localization of most of such immunoreactivity in mutated SDS neutrophils (Figure 2A). We did not identify membrane-free p62-reactive fibrillary/amorphous cytoplasmic bodies (sequestosomes, ALIS) of the type induced in various cell lines by stressful conditions (e.g. oxidative stress, LPS, or puromycin).10–12
Figure 2.

Correlative confocal and electron microscopy of PaCS in SDS neutrophils. (A, 3,000x) Confocal microscopy of an SDS neutrophil immunostained with FK2 antibody against ubiquitinated proteins. Note intensely immunofluorescent areas which, when projected on a TEM micrograph at the same enlargement (A1, 3,000x), show substantial overlapping with PaCS (A2, 3,000x) as confirmed by the intense FK1 immunogold reactivity at higher enlargement (a3, 20,000x). (A4, 3,000x) 20S proteasome immunofluorescence of cytoplasmic areas in another neutrophil from the same SDS patient. (B and C) Autophagic vacuoles and PaCS in SDS neutrophils. Note in B (10,000x) the FK1 reactivity of PaCS, one of which (arrowhead) is apparently discharging its content into the vacuole, and in C (3,000x; enlarged in c1, 20,000x), the p62 reactivity of the vacuole and cytoplasm with, however, no reactivity of PaCS. (D–F, 3,000x) Three apoptotic neutrophils with typical chromatin condensation and segregation (target-type in E and crescentic in F) from SDS patients with SBDS gene mutations. In D, enlarged in d1 (10,000x), cytoplasmic homogenization with disappearance of organelles structure can be seen, whereas in E, enlarged in e1 (10,000x), secretory granules and some FK1 immunoreactivity are still recognizable. Note cytoplasmic vacuoles (of autophagic origin?) in all apoptotic cells. (G, 2,500x) Intraepithelial neutrophil in a gastric biopsy specimen from a patient with H. pylori gastritis, enlarged in g1 (36,000x) to show several PaCS intensely reactive for H. pylori urease. ep: epithelial cell; bm: basal membrane; sg: secretory granules; arrowheads: ribosomes.
In SDS neutrophils from mutated patients, we frequently observed membrane-delimited p62-reactive vesicles, some of which were formed by a double membrane enclosing degenerating cytoplasmic organelles and lysosome-like dense bodies, thus closely resembling autophagic vesicles (Figure 2B and C). Immunoblotting analysis of whole cell lysates showed increased p62 protein in mutated SDS compared to control granulocytes (Online Supplementary Figure S1). As a rule, the autophagic vesicles remained physically separated from the PaCS, which were p62-unreactive; direct contact between the two structures, occasionally with apparent discharge of PaCS contents into the vesicle, was observed only rarely (Figure 2B).
Light microscopy of toluidine-blue-stained semithin resin sections showed round, dense pyknotic nuclei suggestive of apoptosis in 3–8% of neutrophils from mutated SDS cases, and in 0–1% of those from control or non-mutated SDS-like cases. In accordance with the available literature,22,23 TEM of consecutive ultrathin sections allowed us to confirm the apoptotic nature of most of such cells from seven patients with mutated SDS from whom enough material remained for extensive ultrastructural investigation. The more prominent and distinctive features included chromatin compaction, often forming dense crescents abutting the inner nuclear membrane, or round, target-like bodies surrounded by residual karyoplasm, with or without nuclear membrane retention (Figure 2D–F). Membrane-delimited cytoplasmic vesicles, a possible remnant of autophagic vesicles, were frequently found in such cells, whose cytoplasm was usually dense and homogeneous, often in the absence of recognizable organelles or PaCS, and with sharply reduced UPS immunoreactivity. Intermediate patterns showing a frankly apoptotic nucleus coexisting with partly preserved cytoplasmic structures and immunoreactivities were also seen in some mutated SDS neutrophils (Figure 2D d1). When blood neutrophils from healthy subjects were incubated with the ROS-generating xanthine/xanthine oxidase system, we found a large increase of apoptotic features compared with those incubated with culture medium only (Online Supplementary Figure S2). However, neither PaCS nor sequestosomes were a consistent finding in such ROS-exposed cells.
In a parallel investigation of H. pylori-infected gastric mucosa, extensive PaCS were found in neutrophilic granulocytes, but not in lymphocytes, macrophages or fibroblasts of the inflamed lamina propria (Figure 2G). Similarly to PaCS in epithelial cells of the same biopsies, PaCS of gastric mucosal neutrophils showed prominent reactivity for H. pylori products such as VacA, urease (Figure 2G g1) and outer membrane proteins, which, on the contrary, lacked reactivity on PaCS of blood SDS neutrophils. Interestingly, in both neutrophils and epithelial cells, PaCSs of H. pylori-infected gastric mucosa, the concentration of proteasome immunogold (63.23±25.30 particles for 20S in epithelial PaCSs) was of the same order as that of FK1-reactive polyubiquitinated proteins (56.46±10.27 in consecutive sections of the same PaCS). This can be compared with a much lower (P<0.001) FK1/20S or FK1/20Sβ1 ratio in PaCS of SDS blood neutrophils (Table 2), even when both gastric biopsies and blood neutrophils were tested simultaneously using the same procedures and reagents.
Discussion
In blood neutrophils from SDS patients with SBDS gene mutations, we showed the presence of extensive PaCS, that is, cytoplasmic structures characterized by barrel-like particles resembling, both in size and in high-resolution ultrastructure, those of the proteasome unit.13 In SDS neutrophils, PaCS were found to resemble closely the morphology and UPS reactivity of those first observed in gastric epithelium, and here described in neutrophils from H. pylori-infected gastric mucosa. H. pylori-dependent PaCS were believed to result from focal UPS overstimulation at sites of accumulation of bacterial products, either as an attempt to degrade them or in response to infection-elicited cellular stress. Indeed, H. pylori virulence factors such as VacA, urease and outer membrane proteins were found to be selectively concentrated inside PaCS of H. pylori-infected gastric mucosa, both in epithelial cells13 and in neutrophils (this investigation). However, no evidence of H. pylori infection was seen in blood SDS neutrophils and no H. pylori products were detected in their PaCS, which also differed from gastric mucosa PaCS with regard to their lower concentration of proteasomes compared to polyubiquitinated proteins. Proteasomes and polyubiquitinated proteins were both increased quantitatively in SDS compared to in control neutrophils, and were selectively concentrated in the PaCS. This has already been seen in both H. pylori-infected mucosa cells and in a variety of, mostly H. pylori-unrelated, epithelial neoplasms.13,19
SBDS gene mutations of the type commonly reported in patients with SDS3,17 were found in ten of the 13 cases investigated in the present study. Such mutations are known to result in severe loss of the corresponding protein,24 whereas SBDS protein deficiency in epithelial and myeloid cell lines has been shown to increase ROS levels, leading to decreased cell growth and accelerated apoptosis, which are rescued by antioxidants.6 Thus, it seems likely that mutation-induced SBDS protein deprivation causes neutropenia in SDS patients, possibly through oxidative-stress-induced apoptosis.2,5,6 Indeed, more apoptotic figures were observed in mutated SDS neutrophils and in control neutrophils treated with the ROS-generating xanthine/xanthine oxidase system than in untreated control neutrophils. In addition, the high concentration of polyubiquitinated proteins that we found in PaCS of SDS neutrophils, in apparent excess of their proteasome content, suggests a relative insufficiency of proteasome degradative function as a probable cause of disproportionate accumulation of polyubiquitinated proteins which, in turn, may activate autophagy and apoptosis.8,9,11 The known role of SBDS protein in ribosome biogenesis and translation activation,4,25 coupled with the free ribosomal origin of proteasomal proteins13 and the translational burst usually associated with the formation of ubiquitinated protein aggregates,10 may also support the hypothesis of a ribosome-dependent insufficient proteasome response in SBDS-deprived SDS neutrophils.
The considerable amount of polyubiquinated proteins found scattered in the cytoplasm of control neutrophils, in the absence of or with very scarce PaCS, sometimes with preferential distribution in minute clear areas (areolae) of amorphous to poorly structured content, is noteworthy. Our ultrastructural findings suggest that the areolae may work as a starting point for PaCS formation when most of the ubiquitinated proteins apparently shift from the cytosol to developing PaCS, where the proteasomes also concentrate and barrel-like particles appear. The origin and functional role of the high content of polyubiquitinated proteins in healthy human circulating neutrophils (a novel finding to the best of our knowledge) remains unexplained. It may be recalled that blood neutrophils are short-lived cells whose programmed death by apoptosis, central to their homeostasis as well as to the resolution of inflammation, is known to involve special ROS-dependent mechanisms.15,26,27
Unequivocal ultrastructural signs of apoptosis21,22 were a prominent finding in a limited fraction of circulating neutrophils from SDS patients, but not in those from healthy controls. Accelerated apoptosis, both Fas-dependent and -independent, has already been documented in bone marrow cells and suggested to be a crucial cause of SDS patients’ neutropenia,2 a major clinical sign of the disease. Autophagy-related programmed cell death has also been proposed to occur in neutrophils,28 which may fit with the p62-positive autophagic vesicles that we observed in well-preserved, PaCS-positive neutrophils, as well as with the frequent membrane-delimited vacuoles that we found in the cytoplasm of otherwise classically apoptotic cells. Despite these associations and the known regulatory role of the UPS on several crucial factors of the apoptotic process,9 at present, there is no direct evidence for a role of PaCS in accelerated SDS neutrophil apoptosis. In particular, no PaCS development was seen in granulocytes in the presence of ROS-generating treatment that caused apoptosis.
Direct ultrastructural and correlative confocal/TEM analysis proved that, in most SDS neutrophils, polyubiquitinated protein spots corresponded essentially to proteasome-positive, p62-negative PaCS. In particular, no p62-reactive, free cytoplasmic bodies resembling ALIS or sequestosomes of the type found in vitro in macrophages or epithelial cells under various stress conditions,11,29,30 have been consistently detected in our preparations. This underlines a distinct cellular biology of PaCS compared to ALIS/sequestosomes, two structures that also differ in their high-resolution ultrastructure and cytochemistry.13,19
The few patients showing SDS-like disease in the absence of SBDS gene mutation form a subgroup whose genetic background remains to be clarified.21,31 The poor or reduced PaCS development that we observed in blood neutrophils from three such patients discloses some differences in cell biology compared to that in their mutated counterparts, and outlines the need for further molecular and cellular investigation to characterize them positively as a distinct clinicopathological entity.
In conclusion, extensive development of UPS-rich PaCS is a prominent, novel finding in neutrophils from SDS patients carrying SBDS gene mutations, which may offer a new opportunity to investigate cellular mechanisms underlying the disease, including its predisposition to neoplasia. In particular, given the widespread occurrence of PaCS in neoplastic and pre-neoplastic lesions,19 a search for their presence in leukemia, especially of myeloid type, is needed.
Footnotes
Funding: this study was supported in part by grants from the Italian Ministry of Health to Fondazione IRCCS Policlinico San Matteo (RF PSM 2006 401345), and from AISS – Associazione Italiana Sindrome di Shwachman, and Regione Lombardia (Progetto SAL-45).
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Rommens JM, Durie PR. Shwachman-Diamond Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993–2008. Jul 17, [Google Scholar]
- 2.Dror Y, Freedman Shwachman-Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood. 2001;97(10):3011–6. doi: 10.1182/blood.v97.10.3011. [DOI] [PubMed] [Google Scholar]
- 3.Boocock GR, Morrison JA, Popovic M, Richards N, Ellis N, Durie PR, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 4.Menne TF, Goyenechea B, Sanchez-Puig N, Wong CC, Tonkin LM, Ancliff PJ, et al. The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39(4):486–95. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 5.Ball HL, Zhang B, Riches JJ, Gandhi R, Li J, Rommens JM, et al. Shwachman-Bodian Diamond syndrome is a multi-functional protein implicated in cellular stress responses. Hum Mol Gen. 2009;18(19):3684–95. doi: 10.1093/hmg/ddp316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambekar C, Das B, Yeger H, Dror Y. SBDS-deficiency results in deregulation of reactive oxygen species leading to increased cell death and decreased cell growth. Pediatr Blood Cancer. 2010;55(6):1138–44. doi: 10.1002/pbc.22700. [DOI] [PubMed] [Google Scholar]
- 7.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426(6968):895–9. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 8.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schroter F, et al. Immunoproteasomes preserve protein homeastasis upon interferon-induced oxidative stress. Cell. 2010;142(4):613–24. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 9.Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol. 2002;3(2):112–21. doi: 10.1038/nrm731. [DOI] [PubMed] [Google Scholar]
- 10.Lelouard H, Ferrand V, Marguet D, Bania J, Camosseto V, David A, et al. Dendritic cell aggresome-like induced structures are dedicated areas for ubiquitination and storage of newly synthesized defective proteins. J Cell Biol. 2004;164(5):667–75. doi: 10.1083/jcb.200312073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szeto J, Kanuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, et al. ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy. 2006;2(3):189–99. doi: 10.4161/auto.2731. [DOI] [PubMed] [Google Scholar]
- 12.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 13.Necchi V, Sommi P, Ricci V, Solcia E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS ONE. 2010;5(3):e9716. doi: 10.1371/journal.pone.0009716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawahara T, Kohjima M, Kuwano Y, Mino H, Teshima-Kondo S, Takeya R, et al. Helicobacter pylori lipopolysaccharide activates Rac1 and transcription of NADPH oxidase Nox 1 and its organizer NOXO1 in guinea pig gastric mucosal cells. Am J Physiol Cell Physiol. 2005;288(2):C450–7. doi: 10.1152/ajpcell.00319.2004. [DOI] [PubMed] [Google Scholar]
- 15.Blomgran R, Zheng L, Stendahl O. Uropathogenic Escherichia coli triggers oxygen-dependent apoptosis in human neutrophils through the cooperative effect of type 1 fimbriae and lipopolysaccharide. Infect Immun. 2004;72(8):4570–8. doi: 10.1128/IAI.72.8.4570-4578.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuijpers TW, Alders M, Tool AT, Mellink C, Roos D, Hennekam RC. Hematologic abnormalities in Shwachman Diamond syndrome: lack of genotype-phenotype relationship. Blood. 2005;106(1):356–61. doi: 10.1182/blood-2004-11-4371. [DOI] [PubMed] [Google Scholar]
- 17.Minelli A, Maserati E, Nicolis E, Zecca M, Sainati L, Longoni D, et al. The isochromosome i(7)(q10) carrying c.258+2t>c mutation of the SBDS gene does not promote development of myeloid malignancies in patients with Shwachman syndrome. Leukemia. 2009;23(4):708–11. doi: 10.1038/leu.2008.369. [DOI] [PubMed] [Google Scholar]
- 18.Necchi V, Candusso ME, Tava F, Luinetti O, Ventura U, Fiocca R, et al. Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterology. 2007;132(3):1009–23. doi: 10.1053/j.gastro.2007.01.049. [DOI] [PubMed] [Google Scholar]
- 19.Necchi V, Sommi P, Vanoli A, Manca R, Ricci V, Solcia E. Proteasome particle-rich structures are widely present in human epithelial neoplasms. Correlative light, confocal and electron microscopy study. PLoS ONE. 2011;6(6):e21317. doi: 10.1371/journal.pone.0021317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pecci A, Canobbio I, Balduini A, Stefanini L, Cisterna B, Marseglia C, Noris P, Savoia A, Balduini CL, Torti M. Pathogenetic mechanisms of hematological abnormalities of patients with MYH9 mutations. Hum Mol Genet. 2005;14(21):3169–78. doi: 10.1093/hmg/ddi344. [DOI] [PubMed] [Google Scholar]
- 21.Hiraishi H, Terano A, Ota S, Mutoh H, Razandi M, Sugimoto T, Ivey KJ. Role for iron in reactive oxygen species-mediated cytotoxicity to cultured rat gastric mucosal cells. Am J Physiol. 1991;260(4 Pt 1):G556–63. doi: 10.1152/ajpgi.1991.260.4.G556. [DOI] [PubMed] [Google Scholar]
- 22.Lambertenghi Deliliers G, Caneva L. Images in clinical medicine. Apoptosis in myelodysplastic syndromes. N Engl J Med. 1999;340(21):1639. doi: 10.1056/NEJM199905273402105. [DOI] [PubMed] [Google Scholar]
- 23.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14(7):1237–43. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 24.Woloszyneck JR, Rothbaum RJ, Rawls AS, Minx PJ, Wilson RK, Mason PJ, et al. Mutations of the SBDS gene are present in most patients with Shwachman-Diamond syndrome. Blood. 2004;104(12):3588–90. doi: 10.1182/blood-2004-04-1516. [DOI] [PubMed] [Google Scholar]
- 25.Ganapathi KA, Austin KM, Lee CS, Dias A, Malsch MM, Reed R, et al. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007;110(5):1458–65. doi: 10.1182/blood-2007-02-075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK, et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood. 2004;104(8):2557–64. doi: 10.1182/blood-2004-01-0191. [DOI] [PubMed] [Google Scholar]
- 27.Blomgran R, Zheng L, Stendahl O. Cathepsin-cleaved bid promotes apoptosis in human neutrophils via oxidative stress-induced lysosomal membrane permeabilization. J Leuk Biol. 2007;81(5):1213–22. doi: 10.1189/jlb.0506359. [DOI] [PubMed] [Google Scholar]
- 28.van Gunten S, Simon HU. Autophagic-like cell death in neutrophils induced by autoantibodies. Autophagy. 2007;3(1):67–8. doi: 10.4161/auto.3436. [DOI] [PubMed] [Google Scholar]
- 29.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujita K, Maeda D, Xiao Q, Srinivasula S. Nrf2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci USA. 2011;108(4):1427–32. doi: 10.1073/pnas.1014156108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Austin KM, Leary RJ, Shimamura A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood. 2005;106(4):1253–8. doi: 10.1182/blood-2005-02-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]