

Abstract
The loop diuretics furosemide and bumetanide are commonly used in neonatal intensive care units (NICUs). Furosemide, due to its actions on the ubiquitous NKCC1 co-transporter and its promotion of prostanoid production and release, also has non-diuretic effects on vascular smooth muscle, airways, the ductus arteriosus, and theoretically the gastrointestinal tract. Loop diuretics also affect the central nervous system through the inhibitory neurotransmitter, GABA.
Conclusion
The loop diuretics have a variety of biological effects that are potentially harmful as well as beneficial. Care should be taken with the use of these agents since the range of their effects may be broader than the single action sought by the prescribing physician.
Keywords: GABA-A receptor, Na-K-2Cl cotransporter, furosemide, bumetanide, ductus arteriosus
Introduction
Loop diuretics get their name from the location of their site of action in the thick ascending limb of the loop of Henle (1). These diuretics also share a common mode of action which results from their inhibition of the Na+-K+-2Cl− cotransporter. This cotransporter is an ion channel which mediates the flux of Na+, K+ and Cl− from the tubular lumen across the cell membrane into the epithelial lining cell (Figure 1). Inhibition of this cotransporter results in a brisk diuresis. There are two isoforms of this cotransporter, NKCC1 and NKCC2. The expression of NKCC2 is limited to the kidney while NKCC1 is widely distributed throughout the body, and is found in cell membranes of vascular smooth cells and neurons as well as of epithelial cells (2).
Figure 1.
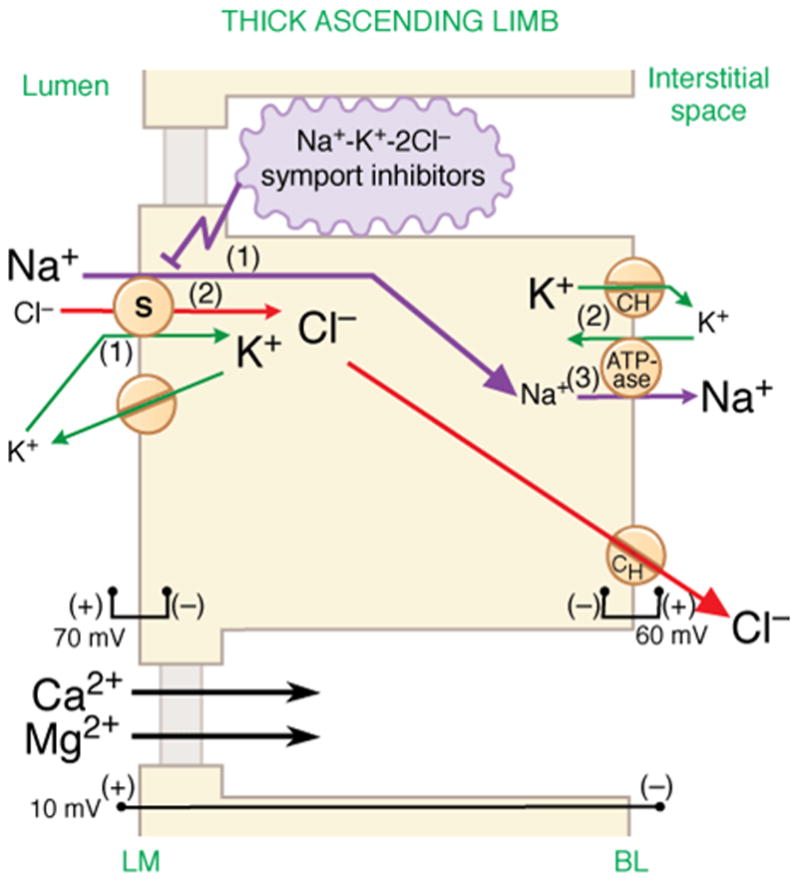
NaCl reabsorption in thick ascending limb and mechanism of diuretic action of Na+-K+-2Cl symport(cotransporter) inhibitors. Numbers in parentheses indicate stoichiometry. Designated voltages are the potential differences across the indicated membrane or cell. The mechanisms illustrated here apply to the medullary, cortical, and postmacular segments of the thick ascending limb. S, symporter; CH, ion channel; BL, basolateral membrane; LM, luminal membrane. From Brunton LL, Chabner BA, Knollmann GC: Goodman & Gilman’s The Pharmacological basis of Therapeutics, 12 Edition: www.accessmedicine.com. With permission.
In addition to furosemide, there are three other loop diuretics available in the United States: bumetanide, ethacrynic acid and torsemide (1). All but ethacrynic acid contain sulfa moieties. The most commonly used loop diuretic is furosemide. Ethacrynic acid is typically used when the patient has a sulfa allergy. Their relative diuretic potencies compared to furosemide are bumetanide (40:1), ethacrynic acid (0.7:1) and torsemide (3:1). Bumetanide, because of its relative potency, is most likely to be used when there is severe oliguria in spite of furosemide treatment. Its use as an antiepileptic agent for the treatment of neonatal seizures has also been considered (see below) (3). Furosemide is one of the most widely used medications in Newborn Intensive Care Units. It accounts for 10% of all medications prescribed in this environment, its use exceeded only by vitamins and antibiotics (4, 5). Its use increases among lower birth weight infants. Over 40% of infants with birth weights less than 1500 g treated in the Vanderbilt Children’s Hospital NICU over the past 4 years were treated with furosemide. It is used in the management of acute respiratory failure in newborn infants, especially those delivered prematurely (6). It is also used in infants with chronic lung disease who have clinical evidence of pulmonary edema accompanied by episodes of apnea and bradycardia and increasing O2 requirement (7). Treatment of other disorders such as congestive heart failure due to symptomatic patent ductus arteriosus often includes the administration of a diuretic, usually furosemide (8). Some neonatologists will order furosemide as part of a red cell transfusion to prevent any adverse effects that may occur due to acute fluid overload (9, 10).
The beneficial effects of furosemide seen in these contexts often are attributed to the diuretic effect of furosemide mediated through its action as an inhibitor of the Na-K+-2Cl− cotransporter located on the luminal surface of the thick ascending limb of the loop of Henle (1, 2). However, this cotransporter is ubiquitous, present in a wide variety of organ systems in addition to the kidney and is involved in multiple biologic actions (2). In addition to inhibiting the Na+-K+-2Cl− cotransporter, furosemide is also an inhibitor of the gamma-amino-butyric acid receptor-A (GABA-A) (11–13). While classically recognized for its action as a mediator of inhibitory neurotransmission in the central nervous system (14) the GABA receptor, like the Na+-K+-2Cl− cotransporter, is widely distributed (15, 16), and may mediate a direct effect on smooth muscle (16) as well as indirect signaling through neural pathways. Given that furosemide is an inhibitor of two different “receptors”, both of which happen to regulate ion flux, and that these transmembrane proteins are distributed throughout multiple organ systems, it is not surprising that that this drug and other Na+-K+-2Cl− cotransporter inhibitors such as bumetanide might have multiple biologic effects, both diuretic and non-diuretic and both beneficial as well as adverse. This review provides a summary of the pulmonary effects of furosemide in newborn premature infants and in infants with chronic lung disease. We then discuss the non-diuretic effects of two of these loop diuretics, furosemide and bumetanide. In some contexts, one or the other may have clinical relevance. In other cases, we describe how the loop diuretics play a key role in experimental systems even though there is no immediate clinical relevance. The overriding aim of this review is to call attention to the myriad biological effects, potentially harmful or beneficial, of certain diuretic agents used in the newborn intensive care unit, when the range of effects is far broader than the single action sought by the prescribing physician.
Diuretics in acute neonatal lung disease
There is a well-founded rationale why diuretic therapy might be beneficial in newborn infants with lung disease such as hyaline membrane disease (HMD). Early descriptions of HMD point out the prominence of lung congestion in this disorder (17), consistent with the laboratory findings of increased lung water and lung blood volume in newborn premature lambs with HMD (18). Clinical findings of HMD include peripheral edema, rales and radiographic findings of pulmonary edema (19). Peritoneal dialysis has been reported to reduce mortality in HMD (20), a finding that may have resulted from the removal of water as well as toxic oxidant products as speculated by the authors. In ventilator-dependent infants with HMD, there is a diuresis beginning 24 to 36 hours after birth which typically precedes a significant improvement in aADO2 and the need for ventilator support (21). Magnitude of fluid intake over the first several days after birth has been shown to increase the risk of symptomatic PDA (22) and the risk of BPD (23).
Clinical studies of the effect of furosemide in acute neonatal lung disease have given a variety of results. One study documented enhanced urine output following furosemide administration resulting in a decrease in required mean airway pressure and facilitation of endotracheal tube extubation (24). Other studies showed either improvement (25, 26) or no (27–29) significant improvement in gas exchange and one study reported adverse effects in the form of volume depletion and need for vasopressor treatment (27). A systematic review of the pooled results of 6 studies revealed a transient improvement in pulmonary function without any long-term benefits (30). In spite of the rationale, there is no evidence that furosemide treatment of patients with acute neonatal lung disease will reduce the risk of symptomatic patent ductus arteriosus or chronic lung disease. However, it should be kept in mind that most of these studies were carried out before the routine use of surfactant replacement therapy
Diuretics in chronic neonatal lung disease
As in the premature infant with early neonatal lung disease, there is also a compelling rationale for the use of diuretic therapy in premature infants with chronic neonatal lung disease, also referred to as bronchopulmonary dysplasia (BPD). There are physical findings of pulmonary rales and peripheral edema in association with radiographic evidence of pulmonary edema, a clinical conclusion corroborated by magnetic resonance imaging (31). Infants with chronic lung disease have increased plasma vasopressin levels with hyponatremia, hypotonic plasma, reduced urine output and reduced free water clearance (32).
Furosemide administration to infants with chronic lung disease results in improved lung mechanical properties, decreased PCO2 and improved oxygenation (33–35). However, these effects do not occur with aldactazide, a non-loop diuretic, even though it produces a comparable diuresis (7). In addition, changes in gas exchange and lung mechanical properties do not always correlate in time with the diuresis following furosemide (34). These observations indicate that the beneficial effect of furosemide on lung function in BPD may not result from its diuretic effect per se, but instead from some non-diuretic effect peculiar to the action of this drug as an inhibitor of Na+-K+-2Cl− cotransporter. Improvement in gas exchange and lung function may also be the result of renal production and release of prostaglandins from the kidney or vascular endothelium (36, 37).
Vascular effects of loop diuretics
It has been known for years that the action of furosemide in the treatment of pulmonary congestion in adults is not related to the diuretic properties of the drug. A frequently cited report in 1973 (38) involved patients with left ventricular failure who were treated with intravenous furosemide. Within 5 – 15 minutes left ventricular filling pressure fell significantly and mean calf venous capacitance increased significantly. Over the same period there was no change in urine output, heart rate, systemic blood pressure or cardiac output.
This case is an example of a non-diuretic vascular effect of furosemide in which venous relaxation occurs resulting in a favorable shift of fluid out of alveoli and into the venous compartment. This is the same phenomenon that may also account in part for the improvement in gas exchange and lung function sometimes seen following furosemide treatment of NICU patients with pulmonary edema accompanying hyaline membrane disease or BPD (24, 33). The non-diuretic vasorelaxant effects of the loop-diuretics furosemide and bumetanide are present in humans as well as a variety of experimental animals (39). These effects have been described in both arterial and venous vascular beds including the pulmonary artery and vein (40). The effects are greater in veins than in arteries, a finding attributed to a greater density of the Na+-K+-2Cl− cotransporter in veins than in arteries (40).
There are at least two mechanisms by which furosemide treatment results in non-diuretic vascular relaxation. Furosemide and bumetanide are both inhibitors of NKCC1, one of two isoforms of the Na+-K+-2Cl− cotransporter (2, 41). Inhibition of this isoform, which is located in vascular endothelium and smooth muscle (39), results in hyperpolarization and relaxation of the targeted smooth muscle (39). Another mechanism is vascular relaxation that is secondary to the action of dilating prostanoids formed and released from the kidney (36) and vascular endothelium (37) in response to furosemide (42). For reasons given below, it is likely that the vasorelaxation due to prostanoids is considerably greater than that due to the direct effect of the diuretic on vascular smooth muscle via inhibition of NKCC1.
Vascular relaxation due to treatment with a loop diuretic has been documented both in vivo and in vitro. Using in vitro microvascular myography of resistance arteries from rat and guinea pig mesentery and from human subcutaneous fat, both furosemide and bumetanide produced a concentration-dependent relaxation that was 10X greater with bumetanide than with furosemide and was greater in the guinea pig and rat than in the human (43). Incubation with indomethacin or mechanical removal of the endothelium did not inhibit the loop diuretic-induced relaxation, indicating that neither prostaglandins nor nitric oxide were required for these actions, allowing the conclusion that the furosemide effect may have resulted from a direct smooth muscle inhibitory effect on the Na+-K+-2Cl− cotransporter. In another in vitro study using isolated pulmonary venous vascular rings (40), furosemide was found to cause venous relaxation independent of prostaglandins or nitric oxide, findings consistent with a direct inhibitory effect on the Na+-K+-2Cl− cotransporter.
In vivo venous occlusion plethysmography has been used to assess the vasodilatory effect of furosemide on veins and arteries of hypertensive patients and healthy controls (44). Furosemide induced venodilation in both the control and the hypertensive patients, but no changes were detected in the arterial bed. The venous effects were shown to be nitric oxide-dependent in that the response was blocked by the nitric oxide synthase inhibitor L-NMMA and restored by L-arginine. Other in vivo studies have given similar results (45, 46).
Whether or not the vasorelaxation induced by a loop diuretic is due to renal release of dilating prostaglandins or due to a direct effect on vascular smooth muscle depends on the dose of the diuretic. The high furosemide concentrations needed for a direct effect on Na+-K+-2Cl− cotransporter activity are reached in the renal tubule but not in the cardiovascular system. To achieve sufficiently high vascular levels of a loop diuretic needed to cause a direct vascular action would result in tubular levels that far exceed the threshold for renal injury. For example, in one in vivo study (47) designed to investigate the vascular effects of loop diuretics in the rat, the investigators used 10–80 mg/kg IV of furosemide to show direct drug-induced vasorelaxation. This dose is 10 to 80X the dose used to treat premature infants in the newborn intensive care unit.
Effect of furosemide on the ductus arteriosus
Persistent patent ductus arteriosus (PDA) is one vascular effect of furosemide that has received considerable attention in the literature pertaining to neonatal disorders. The rationale linking furosemide with persistent PDA is the increased production and release of prostanoids from the kidney and endothelium that occurs in response to treatment with this drug (36, 37). Consistent with this rationale, Friedman et al (48) showed that urinary excretion of prostaglandin E increased following furosemide in 5 sick low-birth weight infants (Figure 2A).
Figure 2.
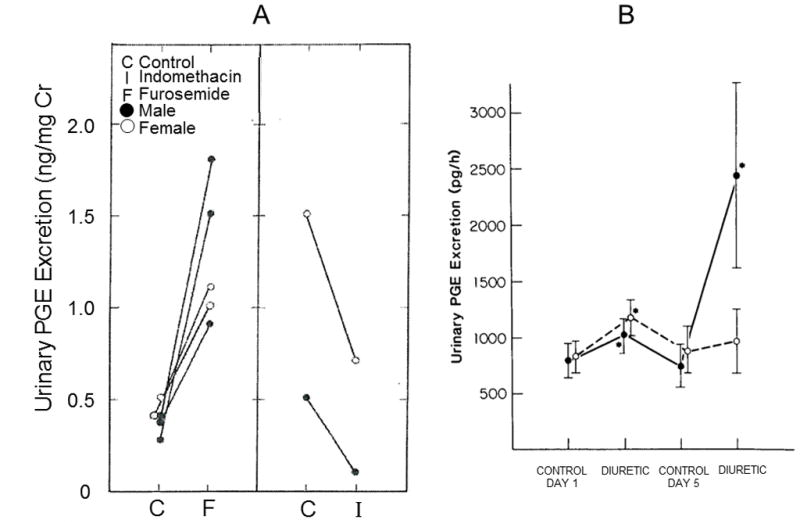
Figure 2A. Changes of urinary excretion of prostaglandin E in five sick low-birth-weight infants before (C) and after (F) the administration of furosemide, and in two sick low-birth-weight infants before (C) and after (I) administration of indomethacin. From Friedman Z, Demers LM, Marks KH, Uhrmann S, Maisels MJ. J Pediatr 1978;93:512–5. With permission.
Figure 2B. Rates of urinary prostaglandin excretion (PGE) in furosemide-treated (solid circles) and chlorothiazide-treated (open circles) subjects. Asterisks denote p<0.01 compared with the control value. From Green TP, Thompson TR, Johnson DE, Lock JE. N Engl J Med 1983;308:743–8. With permission
In vivo dilatation of the ductus arteriosus has been shown to be induced by furosemide in the rat (49). This study is important because it demonstrates a causal relationship between furosemide and ductus dilatation under simulated clinical conditions with a clinical dose of the loop diuretic (Figure 3). The response of the ductus to furosemide was partly attenuated when given with indomethacin, implying the involvement of dilating prostanoids. However, the incomplete effect of indomethacin on furosemide-induced ductus dilatation raises the possibility that other factors may be involved as well.
Figure 3.

The neonatal thorax cut along the frontal plane, at the level of the ductus arteriosus (DA). A, The constricted DA in a 60-min-old rat (control). B, The constricted, thick-walled DA in a 120-min-old rat (control). C, The dilated DA in a 120-min-old rat that was s.c. injected with furosemide (1 mg/kg) 60 min after birth. AoA, aortic arch; DA, ductus arteriosus; LPA, left pulmonary artery; LSVC, left superior vena cava; RPA, right pulmonary artery. From Toyoshima K, Momma K, Nakanishi T. Pediatr Res 2010;67:173–6. With permission.
This effect of furosemide on the ductus arteriosus has also been documented in human premature infants. In a randomized clinical trial (42), Green et al found convincing evidence that furosemide increases the risk of persistent PDA in premature infants with the respiratory distress syndrome, probably through a prostaglandin-mediated process. In this study, urinary excretion of PGE was increased following furosemide, but not chlorothiazide, a non-loop diuretic (Figure 2B). However furosemide does not prevent ductus closure in patients given indomethacin (50–54). It appears that the constrictive effect of indomethacin overrides the dilating effect of furosemide in premature infants with a persistent PDA.
The loop diuretic furosemide, but not bumetanide, dilates the ductus arteriosus ex vivo
The studies above summarize clinical and experimental findings that are consistent with the conclusion that furosemide, a loop diuretic, contributes to ductus relaxation. Preliminary data shown in Figure 4 have now been obtained from in vitro and ex vivo studies that confirm the association of furosemide with ductus relaxation and implicate the involvement GABA signaling in this process (55).
Figure 4.
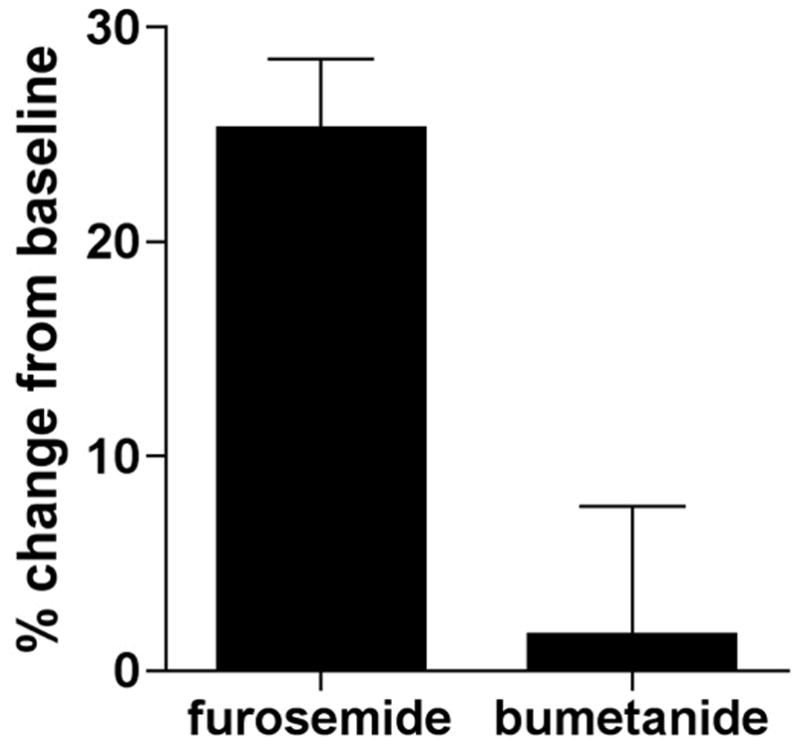
Dilation of the ex vivo ductus arteriosus by loop diuretics. The surgically isolated ductus arteriosus of term gestation fetal mice was mounted on 150 μm cannulae and studied by pressure myography {Reese, 2009 1224 /id}. Exposure to increasing doses of furosemide produced concentration-dependent relaxation of the ductus arteriosus (not shown), whereas exposure to bumetanide had little effect on ductus tone. At the highest concentration (10−3 M), furosemide, which inhibits the GABA-A chloride channel and both of the NKCC isoforms, induced approximately 25% increase in ductus diameter compared to resting baseline tone (p<0.01) and significantly greater than bumetanide (p<0.01). Bumetanide, an NKCC1-specific inhibitor, had no significant effect on ductus diameter compared to baseline.
Effect of loop diuretics on airways
Earlier in this article it was noted that premature infants with chronic lung disease showed an improvement in lung function following intravenous furosemide treatment. This response was thought not to be related to an effect of diuresis since the change in lung function occurred prior to the diuresis. In addition, when a diuresis occurred following aldactazide, a non-loop diuretic, there was no improvement in lung function.
The site and mechanism of the non-diuretic effect of furosemide on lung function is not totally understood. There is clinical and experimental evidence that this response is a direct relaxant effect on airway smooth muscle mediated by the inhibitory effect of furosemide on the Na+-K+-2Cl− cotransporter located in airway smooth muscle. This possibility is consistent with clinical studies showing beneficial effects of furosemide on abnormal lung function when the drug is administered into the airway by nebulization. Rastogi (56) showed significantly improved lung compliance, pulmonary resistance and tidal volume following administration of nebulized furosemide to 8 infants with BPD requiring mechanical ventilation. The beneficial response started as early as 30 minutes after treatment and was not associated with diuresis. Prabhu et al (57) showed significant improvement in tidal volume and compliance without diuresis in 13 ventilator dependent premature infants treated with nebulized furosemide.
While the results of these clinical studies are consistent with a direct effect of furosemide on airway smooth muscle, they do not rule out the involvement of dilating prostanoids released from the kidneys or endothelium, or an action on airway epithelium. To address these possibilities, Stevens et al (58) measured the response of airway (trachea, main stem bronchi) ring segments to furosemide in fetal, newborn and adult guinea pig in vitro. Following preconstriction, furosemide caused significant relaxation of airway smooth muscle at all ages, a response that was enhanced only slightly by removal of the epithelium. These experiments ruled out renal production of dilating prostaglandins as a source of airway smooth muscle relaxation and suggested that an epithelial source of such agents is unlikely.
Additional evidence in support of the hypothesis that airway relaxation to furosemide is mediated via the Na+-K+-2Cl− cotransporter is provided by Lavallee et al (59). These investigators assessed the effect of furosemide on Na+-K+-2Cl− activity by measuring rubidium-86 uptake and tracheal ring response to extracellular [Cl−] in epithelial-denuded tracheal rings from newborn guinea pig. The results of this study indicate that, like in adult guinea pig large airways (60), there is a functional, nonepithelial-dependent, furosemide-sensitive Na-K-2Cl cotransporter in newborn guinea pig trachea.
Iwamoto et al (61) studied isolated human fetal airway and newborn mouse airways to test the hypothesis that furosemide and bumetanide, both loop diuretics, would cause direct relaxation of human fetal airway. They found that human airway relaxed following furosemide after preconstriction with either acetylcholine or the inflammatory mediator leukotriene D-4. Preconsricted newborn mouse airway relaxed following either furosemide or bumetanide. The response was similar whether the diuretics were applied to the luminal (epithelial) surface or abluminal (adventitial) surface. These results provide additional evidence that there is a direct, nonepithelial-dependent effect of furosemide on airway smooth muscle tone and are consistent with the possibility that the Na+-K+-2Cl− cotransporter is present in the human airway smooth muscle cell and that it plays a role in regulating airway tone (61).
Not only does furosemide inhibit the Na+-K+-2Cl− cotransporter, it also inhibits the GABA-A receptor (13, 62) which is known to act as an inhibitory neurotransmitter throughout the central and peripheral nervous systems. The GABA-A receptor may have a role in regulating airway muscle tone. Mizuta et al have reported that mRNA and GABA-A protein are found in human and guinea pig airway smooth muscle and in cultured human airway smooth muscle cells (63). They have also reported that the GABA-A agonist muscimol relaxes the pharmacologic contraction of guinea pig tracheal rings, effects that are inhibited by the GABA-A receptor antagonist gabazine. There is also in vivo evidence of direct GABA-A effect on airway smooth muscle (16). After elimination of neural contributions to airway tone in guinea pigs receiving mechanical ventilation, the GABA-A agonist muscimol was shown to attenuate the increases in airway pressure that occur as a result of induced increases in airway constriction. Even though the pathway is present for GABA-A to mediate changes in airway smooth muscle tone as a result of a furosemide-induced direct inhibition of this receptor, there is little, if any evidence that such a mechanism occurs, but if it did, it would put furosemide in the role of bronchoconstrictor, which is at odds with clinical findings.
Possible effects of loop diuretics on the gastrointestinal tract
Gastroesophageal reflux is one of the most nettlesome clinical problems encountered in the newborn intensive care unit, especially among extremely low birth weight infants with chronic lung disease. The morbidity among these patients is severe, ranging from apnea and nutritional challenges to right heart failure and persistent pulmonary hypertension. Given the widespread involvement of GABA-ergic neural activity in the enteric nervous system, it is tempting to speculate that the loop diuretic furosemide, known to be a potent GABA-A receptor inhibitor (13, 62), might have an effect, beneficial or adverse, on gastrointestinal mobility. In an in vivo study of dogs (64), the GABA-A agonist muscimol produced a dose-dependent inhibition of transient lower esophageal sphincter relaxations, which are regarded as the event causing GER. Using mouse whole stomach in vitro (15), GABA was shown to induce gastric relaxation, an action which was antagonized by the GABA-A receptor antagonist bicuculline. In addition, muscimol, a GABA-A receptor agonist, mimicked the GABA effect (relaxation) which was reduced by bicuculline, a GABA-A receptor antagonist. In another study (65), GABA induced an excitatory effect in the longitudinal muscle of the mouse duodenum. This effect consisted of an increase in basal tone, which was antagonized by bicuculline.
These studies illustrate that activation of the GABA-A receptors can lead to variable effects depending on the part of the gut or the animal species studied (65). In addition, activation of the GABA-A receptors may stimulate the enteric cholinergic excitatory and the non-adrenergic, non-cholinergic (NANC) inhibitory motor neurons, leading to either contraction or relaxation of the intestinal smooth muscle (65). For these reasons, it would be hazardous to predict where in the gastrointestinal tract a loop diuretic such as furosemide might exert an effect on motility and what that effect might be – inhibitory or excitatory.
There is little, if any, clinical evidence that furosemide has an effect on gastrointestinal motility. Several case-control retrospective studies (66–68) have looked for associations between GER and medication use, including caffeine, surfactant, opioid, dopamine, prenatal and postnatal corticosteroids and bronchodilators. It would appear that even though furosemide is a drug with manifold pharmacologic actions, it has not been critically scrutinized as a risk factor for GER.
Effects of loop diuretics on GABA-mediated events in the central nervous system
GABA is well known for its action as an inhibitory neurotransmitter which acts throughout the central and peripheral nervous system. Three receptor subtypes mediate this action: GABA-A, GABA-B and GABA-C. The most prominent GABA-receptor subtype, the GABA-A receptor, is a ligand-gated Cl− ion channel. (14). The GABA-A receptor is inhibited by the loop diuretic, furosemide (13, 62), but not by bumetanide.
Although GABA is best known as an inhibitory neurotransmitter, it actually can go both ways, either inhibitory or excitatory, depending on intraneuronal [Cl−] (69, 70). Intraneuronal [Cl−] is modulated by two isoforms of the cation-Cl− cotransporter family. These two isoforms are NKCC1, an isoform of the Na+-K+-2Cl− cotransporter, and KCC2, an isoform of the K+-Cl− cotransporter. NKCC1 transports Cl− (along with Na+ and K+) into the neuron which increases [Cl−] leading to depolarization (excitatory state) upon the opening of Cl− channels, such as GABA-A receptors. KCC2 transports Cl− (along with K+) out of the neuron which decreases [Cl−] leading to hyperpolarization (inhibitory state) with Cl− channel opening, as illustrated in Figure 5.
Figure 5.
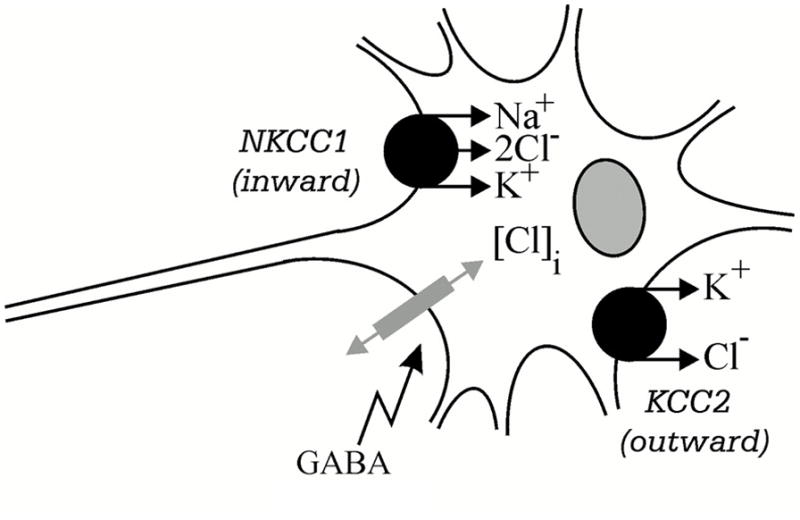
Two secondary active cation-Cl− cotransporters expressed in GABA-responsive neurons. NKCC1 is an isoform of the Na+-K+-2Cl− cotransporter and KCC2 is an isoform of the K+-Cl− cotransporter; [Cl−]i is intracellular Cl− concentration. From Delpire E (2000). Cation-Chloride Cotransporters in Neuronal Communication. News Physiol Sci 15, 309–312, with permission.
Whether a neuron responds to GABA activation of the Cl− channel in an excitatory or inhibitory fashion depends on the relative expression of KCC2 versus NKCC1. A high expression of KCC2 along with a low expression of NKCC1 would result in an inhibitory response to GABA, while a high expression of NKCC1 coupled with a low expression of KCC2 would result in an excitatory response to GABA (70, 71).
The so-called “GABA switch” is a developmental phenomenon whereby GABA-A receptor (a Cl− channel) activation excites immature neurons, unlike in mature neurons, where GABA-A receptor activation is inhibitory. GABA acts as an excitatory neurotransmitter in immature neurons, a process enabled by the developmental upregulation of NKCC1 (a Cl− importer) and late onset expression of KCC2 (Cl− exporter) (72). As neurons mature the process is reversed, so that KCC2 increases and NKCC1 decreases, resulting in GABA taking on the classic role of an inhibitory neurotransmitter where upon ligand-gated channel opening, Cl− flows down its concentration gradient into the cell (See Figure 6).
Figure 6.
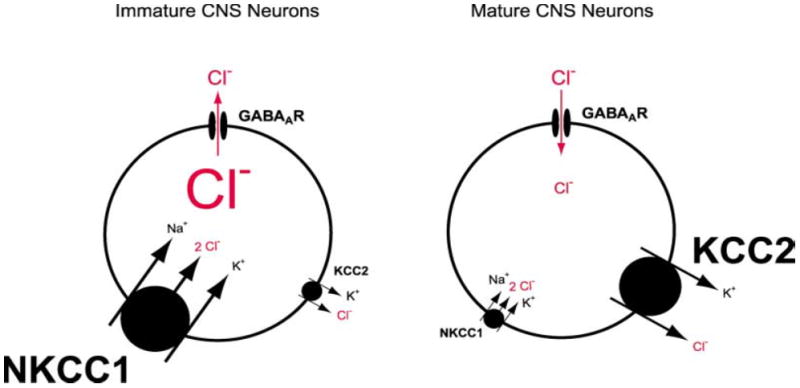
Developmental regulation of chloride homeostasis in neurons. Immature neurons and DRGs express primarily the Na+-K+-2Cl− cotransporter NKCC1 and to a lesser extent the K+-Cl− cotransporter KCC2. This results in a high intracellular concentration of Cl− so that GABA-A receptor activation causes Cl− efflux and a depolarization. In mature CNS neurons, the expression of NKCC1 decreases and the expression of KCC1 increases. This results in a low intracellular Cl− concentration so that GABA-A receptor activation causes a Cl− influx and a hyperpolarization. The font size of the lettering for the transporters depicts their relative importance, and the relative font size for Cl− depicts the gradient across the membrane. From Stein V & Nicoll RA (2003). GABA generates excitement. Neuron 37, 375–378, with permission.
One effect of the GABA switch is that the excitatory state thereby imposed on the immature developing brain is thought to increase the risk of neonatal seizures as a result of an upregulated expression of NKCC1 (70). If so, these results would provide evidence supporting the idea that the NKCC1 inhibitor bumetanide should be useful in the treatment of neonatal seizures.
There are several lines of evidence in support of this idea. The NKCC1-specific blocker bumetanide has been shown to suppress epileptiform activity in immature brain slices in vitro and to attenuate electrographic seizures in neonatal rats in vivo (70). Changes in Cl− transporter expression have been observed to contribute to epileptiform activity in humans (73). In post partum d 6–17 mice, bumetanide has been shown to inhibit the generation of inter-ictal seizures and prevent their transformation to ictal seizures, indicating the role of excitatory GABA in epilepsies (74). Alteration of Cl− transport by bumetanide has been reported to augment the anticonvulsant effect of phenobarbital in adult rats in vitro. (75). Continuous electroencephalography of a neonate with intractable multifocal seizures showed significant reductions in seizure duration and frequency after treatment with bumetanide, indicating that this drug may exert antiepileptic effects in human neonates (3).
While this is a novel and refreshing idea about the pathophysiology and potential treatment of neonatal seizures, there are troublesome issues that arise related to switching GABA from an excitatory to inhibitory neurotransmitter in the developing brain. In a comprehensive array of in vitro and in vivo studies in mice ranging from embryonic to postnatal ages, Wang et al (76) demonstrated that blocking NKCC1 with bumetanide during cortical development revealed a crucial period for the development of certain synapses and that disruption of GABA signaling during this window resulted in permanent decreases in excitatory synaptic transmission and sensorimotor deficits, a common feature found in schizophrenia. In addition, these investigators showed that bumetanide also disrupts the morphology of cortical neurons and causes significant behavioral and developmental abnormalities.
Like bumetanide, furosemide also inhibits NKCC1 in the brain (77) and has anticonvulsant action (78). However this action is thought to be associated with interference of neuronal excitability unrelated to NKCC1 inhibition (78). Not only does furosemide inhibit NKCC1 in the brain (77), this loop diuretic, but not bumetanide, has also been shown to modulate uniquely a GABA-A receptor subtype found in the cerebellum (11, 12). These known actions of furosemide, GABA-A inhibition, NKCC1 inhibition, and interference with neuronal excitability have the potential to add up to a triple threat to the brain, especially in the case of a developing newborn infant.
What oxytocin and bumetanide have in common in regard to asphyxial brain injury during the perinatal period
In studies using fetal and neonatal rat hippocampal slices, Tyzio et al (79) showed that the GABA switch phenomenon may be an important mechanism that serves to protect the brain from asphyxial injury during the intrapartum period. The transition from fetal to neonatal conditions during this period is associated with an increased risk of asphyxia. Since increased metabolic demand and neuronal activity characterize the GABA excitatory state (80), temporary relief from these energy demands would allow the fetus to tolerate an asphyxial insult that otherwise would cause significant brain damage.
These investigators made the pertinent observation that GABA activity in fact does switch temporarily from excitatory to inhibitory in rats during the intrapartum period, about 1–2 hours before birth. This finding supports their hypothesis that an excitatory to inhibitory switch in GABA signaling reduces neuronal activity and metabolic demand, thus helping to protect fetal neurons from hypoxic insults.
Since the observed GABA switch was associated temporally with parturition, it was thought that maternal hormones released during labor and delivery might be the trigger for the switch. Suspecting that oxytocin might be such a hormone, these investigators looked for and found abundant oxytocin receptor immunoreactivity in the hippocampus and neocortex during the perinatal period. Next, exposure of brain slices to oxytocin was observed to suppress GABA-mediated excitation, an effect completely prevented by a selective oxytocin receptor antagonist. Using an electrophysiological marker of neuronal death, control rat fetuses were shown to undergo neuronal death at 55.6 minutes after perfusion of an anoxic-aglycemic solution. In vivo treatment of the mother or the fetus with oxytocin receptor antagonists decreased the time of neuronal survival in vitro to 44.1 minutes, an effect that was blocked by bumetanide.
Since the loop diuretic bumetanide restores survival time of neurons that have been denied the respite of GABA inhibitory signaling, it raises the possibility that pharmacologic inhibition of the Na+-K+-2Cl− cotransporter might be considered a therapeutic strategy when birth of a fetus occurs without the protection of a maternal oxytocin surge. The irony here is that bumetanide, when given as an antiepileptic agent, is associated with neurobehavioral abnormalities (76), but in this experimental context, it can protect against neuronal damage, Additional in vitro and in vivo studies are necessary to explore these mechanisms.
Summary
In addition to enhancing urine output, the loop diuretics furosemide and bumetanide have a variety of non-diuretic effects. This review brings together reports showing how these agents might affect vascular beds, airways, the ductus arteriosus, the brain, and possibly the gastrointestinal tract. These effects and their mediators are shown in the Table.
Table.
A Representative Reference to Each Study Showing a Non-diuretic Effect of a Loop Diuretic and the Mediator Involved (except prostanoids).
| Effects | Furosemide | Bumetanide | NKCC1 | GABA |
|---|---|---|---|---|
| BPD | Engelhardt (33) | |||
| Vascular | Dormans (39) | Pickkers (43) | Dormans (39) | |
| PDA | Green (42) | Suarez (55) | ||
| Airway | Iwamoto (61) | Iwamoto (61) | Lavallee (59) | Gleason (16) |
| GI | Rotondo (15) | |||
| Anti-Epileptic | Dzhala (70) | Dzhala (70) | ||
| Anti-Epileptic | Gutschmidt (78) | |||
| Other CNS | Wang (76) | Wang (76) | Wang (76) | |
| Other CNS | Korpi (12) | Korpi (12) | ||
| Other CNS | Payne (77) | Payne (77) | Payne (77) | |
| Intrapartum | Tyzio (79) | Tyzio (79) | Tyzio (79) |
Acknowledgments
This work was presented in part by Dr. Reese at the biennial meeting of the International Perinatal Collegium held at Amalfi, Italy, June 25–29, 2011.
The authors are grateful for the analysis by Matt Marshall, Pharm.D, of furosemide usage in the Vanderbilt Children’s Hospital NICU. Dr. Marshall is an Informatics Pharmacist at Vanderbilt Children’s Hospital.
This review was supported by NIH HL77395, HL96967, and HL109199.
Abbreviations
- NICUs
Neonatal intensive care units
- Na+-K+-2Cl−
Sodium potassium chloride cotransporter
- NKCC1/2
Sodium potassium chloride cotransporter isoform 1/2
- K+-Cl−
Potassium chloride cotransporter
- KCC2
Potassium chloride cotransporter isoform
- [Cl−]
Chloride ion concentration
- GABA
Gamma-amino-butyric acid
- GABA-A/B/C
gamma-amino-butyric acid receptor-A/B/C
- HMD
hyaline membrane disease
- PDA
Patent ductus arteriosus
- BPD
bronchopulmonary dysplasia
- aADO2
Alveolar/Arterial difference in oxygen tension
- PCO2
Partial pressure of carbon dioxide
- L-NMMA
L-NG monomethyl Arginine
- GERD
Gastroesophageal reflux disease
Footnotes
Key Notes
In addition to enhancing urine output, the loop diuretics furosemide and bumetanide have a variety of non-diuretic effects. This review brings together reports showing how these agents might affect vascular beds, airways, the ductus arteriosus, the brain, and possibly the gastrointestinal tract.
References
- 1.Reilly RF, Jackson EK. Regulation of Renal Function and Vascular Volume. In: Brunton LL, Chabner BA, Knollmann BC, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12. Chapter 25. New York: McGraw-Hill; 2011. [Accessed March 18, 2012]. http://www.accessmedicine.com/content.aspx?aID=16666605. [Google Scholar]
- 2.Haas M, Forbush B., III The Na-K-Cl cotransporters. J Bioenerg Biomembr. 1998;30:161–72. doi: 10.1023/a:1020521308985. [DOI] [PubMed] [Google Scholar]
- 3.Kahle KT, Barnett SM, Sassower KC, Staley KJ. Decreased seizure activity in a human neonate treated with bumetanide, an inhibitor of the Na(+)-K(+)-2Cl(−) cotransporter NKCC1. J Child Neurol. 2009;24:572–6. doi: 10.1177/0883073809333526. [DOI] [PubMed] [Google Scholar]
- 4.Aranda JV, Cohen S, Neims AH. Drug utilization in a newborn intensive care unit. J Pediatr. 1976;89:315–7. doi: 10.1016/s0022-3476(76)80478-7. [DOI] [PubMed] [Google Scholar]
- 5.Clark RH, Bloom BT, Spitzer AR, Gerstmann DR. Reported medication use in the neonatal intensive care unit: data from a large national data set. Pediatrics. 2006;117:1979–87. doi: 10.1542/peds.2005-1707. [DOI] [PubMed] [Google Scholar]
- 6.Guignard JP, Dubourg L, Gouyon JB. [Diuretics in the neonatal period] Rev Med Suisse Romande. 1995;115:583–90. [PubMed] [Google Scholar]
- 7.Engelhardt B, Blalock WA, DonLevy S, Rush M, Hazinski TA. Effect of spironolactone-hydrochlorothiazide on lung function in infants with chronic bronchopulmonary dysplasia. J Pediatr. 1989;114:619–24. doi: 10.1016/s0022-3476(89)80708-5. [DOI] [PubMed] [Google Scholar]
- 8.Cotton RB, Stahlman MT, Kovar I, Catterton WZ. Medical management of small preterm infants with symptomatic patent ductus arteriosus. Journal of Pediatrics. 1978;92:467–473. doi: 10.1016/s0022-3476(78)80451-x. [DOI] [PubMed] [Google Scholar]
- 9.Betremieux P, Hartnoll G, Modi N. Should frusemide be prescribed after packed red cell transfusions in the newborn? Eur J Pediatr. 1997;156:88–9. doi: 10.1007/s004310050560. [DOI] [PubMed] [Google Scholar]
- 10.Stefano JL, Bhutani VK. Role of furosemide therapy after booster-packed erythrocyte transfusions in infants with bronchopulmonary dysplasia. J Pediatr. 1990;117:965–8. doi: 10.1016/s0022-3476(05)80146-5. [DOI] [PubMed] [Google Scholar]
- 11.Korpi ER, Luddens H. Furosemide interactions with brain GABA-A receptors. Br J Pharmacol. 1997;120:741–8. doi: 10.1038/sj.bjp.0700922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korpi ER, Kuner T, Seeburg PH, Luddens H. Selective antagonist for the cerebellar granule cell-specific gamma-aminobutyric acid type A receptor. Mol Pharmacol. 1995;47:283–9. [PubMed] [Google Scholar]
- 13.Makela R, Uusi-Oukari M, Oja SS, Alho H, Anghelescu I, Klawe C, et al. Furosemide action on cerebellar GABA(A) receptors in alcohol-sensitive ANT rats. Alcohol. 1999;19:197–205. doi: 10.1016/s0741-8329(99)00040-3. [DOI] [PubMed] [Google Scholar]
- 14.Molinoff PB. Neurotransmission and the Central Nervous System. In: Brunton LL, Chabner BA, Knollmann BC, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 12. Chapter 14. New York: McGraw-Hill; 2011. [Accessed March 19, 2012]. http://www.accessmedicine.com/content.aspx?aID=16662724. [Google Scholar]
- 15.Rotondo A, Serio R, Mule F. Functional evidence for different roles of GABA-A and GABA-B receptors in modulating mouse gastric tone. Neuropharmacology. 2010;58:1033–7. doi: 10.1016/j.neuropharm.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Gleason NR, Gallos G, Zhang Y, Emala CW. The GABA-A agonist muscimol attenuates induced airway constriction in guinea pigs in vivo. J Appl Physiol. 2009;106:1257–63. doi: 10.1152/japplphysiol.91314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avery ME, Taeusch HW. Schaffer’s Diseases of the Newborn. Philadelphia: WB Saunders; 1984. Hyaline membrane disease; pp. 133–47. [Google Scholar]
- 18.Sundell HW, Harris TR, Cannon JR, Lindstrom DP, Green R, Rojas J, et al. Lung water and vascular permeability-surface area in premature newborn lambs with hyaline membrane disease. 60. 1987. pp. 923–32. [DOI] [PubMed] [Google Scholar]
- 19.Cotton RB. Pathophysiology of hyaline membrane disease (excluding surfactant) In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology. 3. Philadelphia: W. B. Saunders; 2004. pp. 926–34. [Google Scholar]
- 20.Boda D, Muranyi L, Belay M, Ebrey P, Eck E. Demonstration of the accumulation of metabolites in hypoxic conditions of premature infants to indicate the gravity of the disease. 1. 1967. pp. 411–2. [Google Scholar]
- 21.Langman CB, Engle WD, Baumgart S, Fox WW, Polin RA. The diuretic phase of respiratory distress syndrome and its relationship to oxygenation. J Pediatr. 1981;98:462–6. doi: 10.1016/s0022-3476(81)80723-8. [DOI] [PubMed] [Google Scholar]
- 22.Bell EF, Warburton D, Stonestreet BS, Oh W. Effect of fluid administration on the development of symptomatic patent ductus arteriosus and congestive heart failure in premature infants. N Engl J Med. 1980;302:598–604. doi: 10.1056/NEJM198003133021103. [DOI] [PubMed] [Google Scholar]
- 23.Van Marter LJ, Leviton A, Allred EN, Pagano M, Kuban KC. Hydration during the first days of life and the risk of bronchopulmonary dysplasia in low birth weight infants. J Pediatr. 1990;116:942–9. doi: 10.1016/s0022-3476(05)80658-4. [DOI] [PubMed] [Google Scholar]
- 24.Yeh TF, Shibli A, Leu ST, Raval D, Pildes RS. Early furosemide therapy in premature infants (less than or equal to 2000 gm) with respiratory distress syndrome: a randomized controlled trial. J Pediatr. 1984;105:603–9. doi: 10.1016/s0022-3476(84)80431-x. [DOI] [PubMed] [Google Scholar]
- 25.Belik J, Spitzer AR, Clark BJ, Gewitz MH, Fox WW. Effect of early furosemide administration in neonates with respiratory distress syndrome. Pediatr Pulmonol. 1987;3:219–25. doi: 10.1002/ppul.1950030405. [DOI] [PubMed] [Google Scholar]
- 26.Green TP, Thompson TR, Johnson DE, Lock JE. Diuresis and pulmonary function in premature infants with respiratory distress syndrome. J Pediatr. 1983;103:618–23. doi: 10.1016/s0022-3476(83)80601-5. [DOI] [PubMed] [Google Scholar]
- 27.Green TP, Johnson DE, Bass JL, Landrum BG, Ferrara TB, Thompson TR. Prophylactic furosemide in severe respiratory distress syndrome: blinded prospective study. J Pediatr. 1988;112:605–12. doi: 10.1016/s0022-3476(88)80182-3. [DOI] [PubMed] [Google Scholar]
- 28.Marks KH, Berman W, Jr, Friedman Z, Whitman V, Lee C, Maisels MJ. Furosemide in hyaline membrane disease. Pediatrics. 1978;62:785–8. [PubMed] [Google Scholar]
- 29.Savage MO, Wilkinson AR, Baum JD, Roberton NR. Frusemide in respiratory distress syndrome. Arch Dis Child. 1975;50:709–13. doi: 10.1136/adc.50.9.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brion LP, Soll RF. Diuretics for respiratory distress syndrome in preterm infants. Cochrane Database Syst Rev. 2008:CD001454. doi: 10.1002/14651858.CD001454.pub2. [DOI] [PubMed] [Google Scholar]
- 31.Adams EW, Harrison MC, Counsell SJ, Allsop JM, Kennea NL, Hajnal JV, et al. Increased lung water and tissue damage in bronchopulmonary dysplasia. J Pediatr. 2004;145:503–7. doi: 10.1016/j.jpeds.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 32.Hazinski TA, Blalock WA, Engelhardt B. Control of water balance in infants with bronchopulmonary dysplasia: role of endogenous vasopressin. Pediatr Res. 1988;23:86–8. doi: 10.1203/00006450-198801000-00019. [DOI] [PubMed] [Google Scholar]
- 33.Engelhardt B, Elliott S, Hazinski TA. Short- and long-term effects of furosemide on lung function in infants with bronchopulmonary dysplasia. J Pediatr. 1986;109:1034–9. doi: 10.1016/s0022-3476(86)80295-5. [DOI] [PubMed] [Google Scholar]
- 34.Najak ZD, Harris EM, Lazzara A, Jr, Pruitt AW. Pulmonary effects of furosemide in preterm infants with lung disease. J Pediatr. 1983;102:758–63. doi: 10.1016/s0022-3476(83)80253-4. [DOI] [PubMed] [Google Scholar]
- 35.Rush MG, Engelhardt B, Parker RA, Hazinski TA. Double-blind, placebo-controlled trial of alternate-day furosemide therapy in infants with chronic bronchopulmonary dysplasia. J Pediatr. 1990;117:112–8. doi: 10.1016/s0022-3476(05)82458-8. [DOI] [PubMed] [Google Scholar]
- 36.Wiemer G, Fink E, Linz W, Hropot M, Scholkens BE, Wohlfart P. Furosemide enhances the release of endothelial kinins, nitric oxide and prostacyclin. J Pharmacol Exp Ther. 1994;271:1611–5. [PubMed] [Google Scholar]
- 37.Patak RV, Fadem SZ, Rosenblatt SG, Lifschitz MD, Stein JH. Diuretic-induced changes in renal blood flow and prostaglandin E excretion in the dog. Am J Physiol. 1979;236:F494–F500. doi: 10.1152/ajprenal.1979.236.5.F494. [DOI] [PubMed] [Google Scholar]
- 38.Dikshit K, Vyden JK, Forrester JS, Chatterjee K, Prakash R, Swan HJ. Renal and extrarenal hemodynamic effects of furosemide in congestive heart failure after acute myocardial infarction. N Engl J Med. 1973;288:1087–90. doi: 10.1056/NEJM197305242882102. [DOI] [PubMed] [Google Scholar]
- 39.Dormans TP, Pickkers P, Russel FG, Smits P. Vascular effects of loop diuretics. Cardiovasc Res. 1996;32:988–97. doi: 10.1016/s0008-6363(96)00134-4. [DOI] [PubMed] [Google Scholar]
- 40.Greenberg S, McGowan C, Xie J, Summer WR. Selective pulmonary and venous smooth muscle relaxation by furosemide: a comparison with morphine. J Pharmacol Exp Ther. 1994;270:1077–85. [PubMed] [Google Scholar]
- 41.Greger R, Wangemann P. Loop diuretics. Ren Physiol. 1987;10:174–83. doi: 10.1159/000173128. [DOI] [PubMed] [Google Scholar]
- 42.Green TP, Thompson TR, Johnson DE, Lock JE. Furosemide promotes patent ductus arteriosus in premature infants with the respiratory-distress syndrome. N Engl J Med. 1983;308:743–8. doi: 10.1056/NEJM198303313081303. [DOI] [PubMed] [Google Scholar]
- 43.Pickkers P, Russel FG, Thien T, Hughes AD, Smits P. Only weak vasorelaxant properties of loop diuretics in isolated resistance arteries from man, rat and guinea pig. Eur J Pharmacol. 2003;466:281–7. doi: 10.1016/s0014-2999(03)01536-x. [DOI] [PubMed] [Google Scholar]
- 44.deBerrazueta, Gonzalez JP, de MI, Poveda JJ, Garcia-Unzueta MT. Vasodilatory action of loop diuretics: a plethysmography study of endothelial function in forearm arteries and dorsal hand veins in hypertensive patients and controls. J Cardiovasc Pharmacol. 2007;49:90–5. doi: 10.1097/FJC.0b013e31802e3c39. [DOI] [PubMed] [Google Scholar]
- 45.Mackay IG, Muir AL, Watson ML. Contribution of prostaglandins to the systemic and renal vascular response to frusemide in normal man. Br J Clin Pharmacol. 1984;17:513–9. doi: 10.1111/j.1365-2125.1984.tb02383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jhund PS, Davie AP, McMurray JJ. Aspirin inhibits the acute venodilator response to furosemide in patients with chronic heart failure. J Am Coll Cardiol. 2001;37:1234–8. doi: 10.1016/s0735-1097(01)01169-x. [DOI] [PubMed] [Google Scholar]
- 47.Barthelmebs M, Stephan D, Fontaine C, Grima M, Imbs JL. Vascular effects of loop diuretics: an in vivo and in vitro study in the rat. Naunyn Schmiedebergs Arch Pharmacol. 1994;349:209–16. doi: 10.1007/BF00169839. [DOI] [PubMed] [Google Scholar]
- 48.Friedman Z, Demers LM, Marks KH, Uhrmann S, Maisels MJ. Urinary excretion of prostaglandin E following the administration of furosemide and indomethacin to sick low-birth-weight infants. J Pediatr. 1978;93:512–5. doi: 10.1016/s0022-3476(78)81182-2. [DOI] [PubMed] [Google Scholar]
- 49.Toyoshima K, Momma K, Nakanishi T. In vivo dilatation of the ductus arteriosus induced by furosemide in the rat. Pediatr Res. 2010;67:173–6. doi: 10.1203/PDR.0b013e3181c2df30. [DOI] [PubMed] [Google Scholar]
- 50.Brion LP, Campbell DE. Furosemide for symptomatic patent ductus arteriosus in indomethacin-treated infants. Cochrane Database Syst Rev. 2001:CD001148. doi: 10.1002/14651858.CD001148. [DOI] [PubMed] [Google Scholar]
- 51.Lee BS, Byun SY, Chung ML, Chang JY, Kim HY, Kim EA, et al. Effect of furosemide on ductal closure and renal function in indomethacin-treated preterm infants during the early neonatal period. Neonatology. 2010;98:191–9. doi: 10.1159/000289206. [DOI] [PubMed] [Google Scholar]
- 52.Romagnoli C, Zecca E, Papacci P, De Carolis MP, Giannini R, Gallini F, et al. Furosemide does not prevent indomethacin-induced renal side effects in preterm infants. Clin Pharmacol Ther. 1997;62:181–6. doi: 10.1016/S0009-9236(97)90066-7. [DOI] [PubMed] [Google Scholar]
- 53.Andriessen P, Struis NC, Niemarkt H, Oetomo SB, Tanke RB, Van OB. Furosemide in preterm infants treated with indomethacin for patent ductus arteriosus. Acta Paediatr. 2009;98:797–803. doi: 10.1111/j.1651-2227.2009.01224.x. [DOI] [PubMed] [Google Scholar]
- 54.Yeh TF, Wilks A, Singh J, Betkerur M, Lilien L, Pildes RS. Furosemide prevents the renal side effects of indomethacin therapy in premature infants with patent ductus arteriosus. J Pediatr. 1982;101:433–7. doi: 10.1016/s0022-3476(82)80079-6. [DOI] [PubMed] [Google Scholar]
- 55.Suarez S, Cotton R, Ehinger N, Poole SD, Slaughter J, Stoller J, Reese J. Mechanisms for direct vasodilatory actions of loop diuretics via the NKCC1 and GABA-A receptor chloride channels in the ductus arteriosus. 2011. E-PAS2011:3809.69. [Google Scholar]
- 56.Rastogi A, Luayon M, Ajayi OA, Pildes RS. Nebulized furosemide in infants with bronchopulmonary dysplasia. J Pediatr. 1994;125:976–9. doi: 10.1016/s0022-3476(05)82018-9. [DOI] [PubMed] [Google Scholar]
- 57.Prabhu VG, Keszler M, Dhanireddy R. Dose-dependent evaluation of the effects of nebulized furosemide on pulmonary function in ventilated preterm infants. J Perinatol. 1998;18:357–60. [PubMed] [Google Scholar]
- 58.Stevens EL, Uyehara CF, Southgate WM, Nakamura KT. Furosemide differentially relaxes airway and vascular smooth muscle in fetal, newborn, and adult guinea pigs. Am Rev Respir Dis. 1992;146:1192–7. doi: 10.1164/ajrccm/146.5_Pt_1.1192. [DOI] [PubMed] [Google Scholar]
- 59.Lavallee SL, Iwamoto LM, Claybaugh JR, Dressel MV, Sato AK, Nakamura KT. Furosemide-induced airway relaxation in guinea pigs: relation to Na-K-2Cl cotransporter function. Am J Physiol. 1997;273:L211–L216. doi: 10.1152/ajplung.1997.273.1.L211. [DOI] [PubMed] [Google Scholar]
- 60.Rhoden KJ, Douglas JS. Evidence of Na-K-Cl cotransport in airway smooth muscle. Am J Physiol. 1995;268:L551–L557. doi: 10.1152/ajplung.1995.268.4.L551. [DOI] [PubMed] [Google Scholar]
- 61.Iwamoto LM, Gries DM, Nakamura KT. Loop diuretics and in vitro relaxation of human fetal and newborn mouse airways. Pediatr Res. 2001;50:273–6. doi: 10.1203/00006450-200108000-00018. [DOI] [PubMed] [Google Scholar]
- 62.Minier F, Sigel E. Positioning of the alpha-subunit isoforms confers a functional signature to gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A. 2004;101:7769–74. doi: 10.1073/pnas.0400220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mizuta K, Xu D, Pan Y, Comas G, Sonett JR, Zhang Y, et al. GABA-A receptors are expressed and facilitate relaxation in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1206–L1216. doi: 10.1152/ajplung.00287.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beaumont H, Jonsson-Rylander AC, Carlsson K, Pierrou S, Ahlefelt M, Branden L, et al. The role of GABA(A) receptors in the control of transient lower oesophageal sphincter relaxations in the dog. Br J Pharmacol. 2008;153:1195–202. doi: 10.1038/sj.bjp.0707681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zizzo MG, Mule F, Serio R. Functional evidence for GABA as modulator of the contractility of the longitudinal muscle in mouse duodenum: role of GABA(A) and GABA(C) receptors. Neuropharmacology. 2007;52:1685–90. doi: 10.1016/j.neuropharm.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 66.Mendes TB, Mezzacappa MA, Toro AA, Ribeiro JD. Risk factors for gastroesophageal reflux disease in very low birth weight infants with bronchopulmonary dysplasia. J Pediatr (Rio J) 2008;84:154–9. doi: 10.2223/JPED.1764. [DOI] [PubMed] [Google Scholar]
- 67.Malcolm WF, Gantz M, Martin RJ, Goldstein RF, Goldberg RN, Cotten CM. Use of medications for gastroesophageal reflux at discharge among extremely low birth weight infants. Pediatrics. 2008;121:22–7. doi: 10.1542/peds.2007-0381. [DOI] [PubMed] [Google Scholar]
- 68.Mezzacappa MA, Rosa AC. Clinical predictors of abnormal esophageal pH monitoring in preterm infants. Arq Gastroenterol. 2008;45:234–8. doi: 10.1590/s0004-28032008000300013. [DOI] [PubMed] [Google Scholar]
- 69.Delpire E. Cation-Chloride Cotransporters in Neuronal Communication. News Physiol Sci. 2000;15:309–12. doi: 10.1152/physiologyonline.2000.15.6.309. [DOI] [PubMed] [Google Scholar]
- 70.Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–13. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- 71.Fukuda A. Diuretic soothes seizures in newborns. Nat Med. 2005;11:1153–4. doi: 10.1038/nm1105-1153. [DOI] [PubMed] [Google Scholar]
- 72.Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, et al. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–5. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 73.Huberfeld G, Wittner L, Clemenceau S, Baulac M, Kaila K, Miles R, et al. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci. 2007;27:9866–73. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rheims S, Represa A, Ben-Ari Y, Zilberter Y. Layer-specific generation and propagation of seizures in slices of developing neocortex: role of excitatory GABAergic synapses. J Neurophysiol. 2008;100:620–8. doi: 10.1152/jn.90403.2008. [DOI] [PubMed] [Google Scholar]
- 75.Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–35. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- 76.Wang DD, Kriegstein AR. Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex. 2011;21:574–87. doi: 10.1093/cercor/bhq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- 78.Gutschmidt KU, Stenkamp K, Buchheim K, Heinemann U, Meierkord H. Anticonvulsant actions of furosemide in vitro. Neuroscience. 1999;91:1471–81. doi: 10.1016/s0306-4522(98)00700-3. [DOI] [PubMed] [Google Scholar]
- 79.Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hubner CA, Represa A, et al. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314:1788–92. doi: 10.1126/science.1133212. [DOI] [PubMed] [Google Scholar]
- 80.Dzhala V, Ben-Ari Y, Khazipov R. Seizures accelerate anoxia-induced neuronal death in the neonatal rat hippocampus. Ann Neurol. 2000;48:632–40. [PubMed] [Google Scholar]
- 81.Reese J, O’Mara PW, Poole SD, Brown N, Tolentino C, Eckman DM, et al. Regulation of the fetal mouse ductus arteriosus is dependent on interaction of nitric oxide and COX enzymes in the ductal wall. Prostaglandins Other Lipid Mediat. 2009;88:89–96. doi: 10.1016/j.prostaglandins.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]