

Abstract
Calcium channel blockers (CCBs) are important in the management of hypertension and limit restenosis. Although CCB efficacy could derive from decreased blood pressure, other mechanisms independent of CCB activity also can contribute to antiproliferative action. To understand mechanisms of CCB-mediated antiproliferation, we studied two structurally dissimilar CCBs, diltiazem and verapamil, in cultured rat vascular smooth muscle cells (VSMC). To elucidate CCB-independent effects, pure stereoisomers of verapamil (R-verapamil, inactive VR; S-verapamil, active, VS) were used. The effects of CCB exposure on cell viability (MTT reduction), cell proliferation (3H-thymidine incorporation), VSMC morphology by light and transmission electron microscopy (TEM) and autophagy (LC3I/II, ATG5) were measured. In general, verapamil, VR or VS treatment alone (80 μM) appreciably enhanced MTT absorbance although higher concentrations (VR or VS) slightly decreased MTT absorbance. Diltiazem (140 μM) markedly decreased MTT absorbance (40%) at 120h. VR or VS treatment inhibited 3H-thymidine incorporation (24h) and induced cytological alterations (i.e., karyokinesis, enhanced perinuclear MTT deposition, accumulated perinuclear “vacuoles”). TEM revealed perinuclear “vacuoles” to be aggregates of highly laminated and electron-dense vesicles resembling autophagosomes and lysosomes, respectively. Increased autophagosome activity was confirmed by a concentration-dependent increase in LC3-II formation by Western blotting and by increased perinuclear LC3-GFP+ puncta in verapamil-treated VSMC. Verapamil stereoisomers appeared to decrease perinuclear mitochondrial density. These observations indicate that verapamil stereoisomers are antiproliferative due to enhanced mitochondrial damage and upregulated autophagy in VSMC. These effects are independent of CCB activity indicating a distinct mechanism of action that could be targeted for more efficacious anti-atherosclerotic and anti-restenosis therapy.
Keywords: atherosclerosis, autophagosome, calcium channel blockers, LC3 conversion, restenosis
Introduction
Atherosclerosis and restenosis both involve formation of a neointima by migrating and proliferating vascular smooth muscle cells (VSMC). Studies of experimental vascular injury models and human clinical trials collectively indicate that calcium channel blocker (CCB) treatment prevents VSMC migration and proliferation (Schmitz et al., 1991; Waters and Lesperance, 1994). However, CCB-mediated atheroprotection appears limited because CCB pretreatment in atherosclerosis (e.g., cholesterol-fed rabbits) and carotid injury (e.g., carotid denudation) models substantially retards neointimal formation while CCB treatment after initiation of injury is much less effective (Tulenko et al., 1997). These experimental studies and several clinical trials, e.g., INTACT, REGRESS, indicate that atheroprotection of CCBs is restricted to the early stage of lesion growth, and thus, CCBs act upstream of VSMC proliferative transformation (Armstrong et al., 1991; Schroeder and Gao, 1995; Atkinson, 1997; Jukema and van der Hoorn, 2004). However, newer generation CCBs have been developed and more recent clinical trial data indicate that CCBs exert pleiotropic actions independent of CCB activity, and thus, there may be additional mechanisms of atheroprotection (Jukema and van der Hoorn, 2004).
Multiple mechanisms independent of a blocking action on L-type voltage-gated Ca++ channels are proposed to account for the anti-atherosclerotic action of the three major classes of CCB (e.g., benzothiazepines, dihydropyridines and phenylakylamines) in humans. CCBs act as antioxidants and anti-inflammatory agents, stimulate cholesteryl ester hydrolase, increase cAMP levels, inhibit platelet glycoprotein IIb/IIIa receptor, alter cell membrane cholesterol and membrane thickness, inhibit carbonic anhydrase I, decrease extracellular matrix formation, decrease early response gene transcription, stimulate NF-κB and NF-IL6 transcription factors, block K+ channels, and have “thapsigargin-like” properties, among others (Henry, 1991; Ko et al., 1993; Kauder and Watts, 1996; Eickelberg et al., 1999; Marche and Stepien, 2000; Puscas et al., 2000; Zhang et al., 2000). Some of these effects are independent of calcium channel blockade, and thus, could contribute, in part, to CCB-mediated inhibition of VSMC migration and proliferation (Henry, 1990; Zernig, 1990; Schmitz et al., 1991; Schachter, 1997). As well as having actions independent of calcium channel blockade, CCBs also exert effects that are stereo-independent. In cultured VSMC that express L-type Ca++ channels, lercanidipine stereoisomers inhibit agonist-induced VSMC migration and proliferation independent of calcium channel binding or alterations in intracellular [Ca++](Corsini et al., 1996). Similarly, antiproliferative effects of CCBs occur in cells that do not express L-type voltage-gated Ca++ channels, including lymphocytes, macrophages and platelets (Timar et al., 1992; Weir et al., 1992; Weir et al., 1993a). Thus, several classes of CCBs are antiproliferative, but the specific mechanism(s) of action in VSMC is unknown (Lai et al., 2002).
One possible mechanism for CCB-mediated anti-atherosclerotic effect is by stimulating apoptosis of proliferative VSMC in a calcium channel-independent manner. Nifedipine treatment induces significant carotid artery lesion regression via selective reduction of neointimal VSMC number in association with increased apoptosis independent of antihypertensive effects in WKY rats (Lemay et al., 2001). In addition, verapamil treatment in vivo stimulates thymic cell apoptosis in rat independent of verapamil stereoselectivity or antihypertensive action (Balakumaran et al., 1996). While CCBs may be pro-apoptotic, either directly or indirectly, in VSMC in vivo, there is little evidence for pro-apoptotic effects of CCBs in cultured VSMC. In fact, racemic verapamil (1–80 μM) inhibits 25-hydroxycholesterol- or simvastatin-induced apoptosis but not apoptosis induced by ethanol in cultured VSMC (Ares et al., 1997; Cheng et al., 2003; Li et al., 2004). Thus, there is a need to study the stereo-independent effects of CCBs in cultured VSMC to better understand the antiproliferative action of CCBs.
In the present study, our objective was to investigate the antiproliferative actions of two structurally distinct CCBs, diltiazem (a benzothiazepine) and verapamil (a phenyalkylamine), in cultured VSMC and to use verapamil stereoisomers to define calcium channel blocker independent effects of this drug. Collectively, our results show that two CCBs are antiproliferative in cultured VSMC. Verapamil acts via a stereo-independent mechanism involving decreased 3H-thymidine incorporation, cytological alterations in organelles including mitochondrial damage, and autophagy. Identification of an autophagic mechanism of action stimulated by verapamil and other CCBs in VSMC could potentially improve the therapeutic efficacy of treatments in the management of atherosclerosis and restenosis.
Material and methods
Cell Lines and Culture
Two independent lines of smooth muscle cells were used to validate that treatment effects were not cell line specific. For this, neonatal rat heart smooth muscle cell (RHSM) and passaged rat aortic vascular smooth muscle (RASM) cell lines were derived from enzymatically-dispersed hearts of neonatal Sprague-Dawley rats by differential plating to separate smooth muscle cells (RHSM) from beating myocytes (Jones et al., 1979) and from aorta of adult male Sprague-Dawley rats (RASM; Harlan; Indianapolis, IN; 200–300 g)(Conklin et al., 1998), respectively. For RASM isolation, 2 or 3 aortas were removed aseptically and minced, and digested for 1h (37 °C) in enzyme solution (0.1% collagenase, 0.05% elastase type III, 2 mg·ml−1 bovine serum albumin, BSA) and 2 mM CaCl2 (Sigma Chemical Co, St. Louis, MO) in Dulbecco's Modified Eagle Medium (DMEM; Gibco/BRL Life Technologies, Grand Island, NY). The first digest was discarded, fresh enzyme solution added, and a second digestion continued for 2h. After complete tissue dispersion, the released smooth muscle cells were concentrated by a brief centrifugation (400xg; 5 min), washed twice with DMEM containing 10% (v/v) heat-inactivated fetal calf serum (FCS; Hyclone, Logan, UT), and plated into 25 cm2 Corning plastic tissue culture flasks (Fisher Scientific, Houston, TX) at concentrations of 0.5 × 106 cells per flask, and grown in DMEM containing 10% heat-inactivated FCS and 0.1% penicillin G/streptomycin sulfate. Confirmation of smooth muscle cell line purity was performed by immuno-histochemistry for smooth muscle alpha-actin (positive) and Factor VIII (negative) using early cell passages (1–3) grown on cover slips.
Viability (MTT) Assay
Cells from the 3–20th passages were seeded (0.3–1x104 cells per well) in flat-bottom, 96-well tissue culture plates (Falcon, VWR) and allowed to attach and grow for 48–72h until 70–80% confluent, and then serum-deprived (0.1% FCS in DMEM with daily changes) for 72h. Medium was removed prior to treatment (4h) and replaced with fresh medium (0.1% or 10% FCS). Cells were exposed to DMEM alone, dimethyl sulfoxide (DMSO) vehicle, DILT (75 or 140 μM), verapamil, VR, or VS at final concentrations of 40, 80, 115, or 150 μM (concentrations correspond to 5, 10, 15, or 20 μl additions to a single well of a 96-well plate containing 100 μl medium) for a total of 24h, 48h or 120h. Each treatment had 4 replicate wells. At 20h, 44h or 116h post-treatment, 50 μg MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl) tetrasodium bromide (Chemicon International Inc., Temecula, CA; Sigma Chem. Co., St. Louis, MO) in 10 μl of PBS was added to each well and no extracellular chemical reduction was observed upon addition of MTT in any treatment. Solutions were removed after 4h, 10 μl 100% DMSO added to each well, followed by a short incubation (2–3 min at 37 °C) and then addition of 90 μl isopropanol (100%) to each well. Plates were incubated for 3–5 min at 37 °C, mixed on a rocker plate for 5–10 min at room temperature and absorbance read at 595 nm with a 96-well plate reader (3550-UV, BioRad, Hercules, CA)(Conklin et al., 1998). VSMC viability was calculated as a percentage of the average MTT absorbance in matched control cells.
3H-Thymidine Incorporation Assay
VSMC were seeded (~105 cells/well) and grown in 6-well plates until 70–80% confluent and then serum-deprived as above. Cells were treated once with DMSO vehicle, 10% FCS (positive control), DILT (75 or 140 μM), VR, or VR+VS (80 or 150 μM) for 20h, and then exposed to 3H-thymidine (~1 μCi/well) for an additional 4h. The medium was aspirated, cells were washed 3X with Hank’s buffered saline solution (HBSS), and ice cold 10% trichloroacetic acid (TCA) was added for 1h (4 °C). The TCA soluble fraction was removed, cells were washed twice with TCA (1 ml × 2), and remaining cells were solubilized in 0.5N NaOH (0.6 ml) overnight (4 °C). Duplicate 150 μl aliquots of the TCA- (measure of thymidine uptake) and NaOH-soluble (measure of thymidine incorporation in DNA) fractions were counted by liquid scintillation (Weir et al., 1993b). Protein was determined in both fractions with the Bio-Rad Protein Dye Concentrate reagent (Bio-Rad, Hercules, CA) using BSA in saline as the standard, and the absorbance read at 595 nm (Bio-Rad, Hercules, CA).
Cell Morphology and MTT Distribution
VSMC were serum-deprived in 96-well plates with daily media changes over 72h before drug exposure. In preliminary experiments, cells were exposed to DILT, racemic verapamil or verapamil stereoisomers over a concentration range of 1–1,000 μM with appropriate vehicle controls. Following 24h or 48h treatment, cells were exposed to MTT for 3h or stained with 0.5% toluidine blue, and then photographed with a Nikon FE2 camera using a Nikon Diaphot inverted microscope at 300x.
Autophagy Measurements
RASMs (passages 2–7) were grown to ~70% confluency, and then serum-starved in DMEM containing 0.1% FBS for 72h. Cells were then treated with the indicated concentrations of verapamil (dissolved in 0.9% saline/0.1% DMSO) for 24h or 36h. In select experiments, bafilomycin A1 (0.2μM) was added during the last 4 h prior to harvesting cells. After treatment, cells were rinsed twice with PBS, lysed in a protein lysis buffer (25mM HEPES, 1mM EDTA, 1mM EGTA, 0.1% SDS, 1% NP-40 and protease inhibitor), and protein was measured by Lowry’s assay.
Western blotting
Cell protein (15 μg) was applied to each lane of a 10.5–14% Bis–Tris gel and electroblotted onto a PVDF membrane. The membrane was incubated for 1h in 5% milk (w/v) / 0.3% TBS-tween solution (RT). After blocking, membranes were incubated overnight at 4°C using appropriate dilutions of primary antibodies. After 3 washes (10 min each), PVDF membranes were incubated for 1h (RT) with HRP-conjugated secondary antibodies. Immunoreactive bands were detected with Enhanced ChemoLuminescent (ECL) reagent (GE Healthcare) using a GE Typhoon 9410 (Amersham Biosciences). Band intensities were measured using Total Lab software.
Adenovirus transfection
To visualize autophagy, RASM were grown in 6-well plates, serum-starved for 48h and then infected with replication-deficient adenovirus carrying GFP linked to LC3 (AdGFP-LC3; MOI=100). AdGFP-LC3 transfected cells were grown for 24h and then treated with vehicle or verapamil (40 μM) for 24h. Cells were incubated with Hoechst stain for nucleus visualization with an AMG EVOS fluorescence microscope.
Transmission Electron Microscopy (TEM)
For TEM, RHSM cells were seeded on Thermanox coverslips in 24-well plates (3x104 cells/well) and treated as described for MTT viability assay. After 24h treatment, cells were fixed in 2% glutaraldehyde, routinely processed, embedded, sectioned, and stained on copper grids with lead citrate and uranyl acetate. Sections were viewed with a Philips EM40 electron microscope at 80 kV at 24,475X.
Stereological Analysis
Electron micrographs (8x10 print; mag. 24,475X) of the perinuclear region were analyzed using a modified Weibel grid method (Weibel, 1969). Micrographs were scanned into Adobe Illustrator and a 6x6 square grid was used as a stereological tool. The grid was placed over the image such that one or two intersecting points of the grid’s outer edge (a 6x6 grid has 49 intersecting points) overlaid the nuclear membrane edge; the remaining portion of the grid stretched across the perinuclear region covering an area of approximately 30.17 μm2. For each image, the total number of points that overlaid the nucleus (TN; i.e., 1 or 2 grid points contacted nucleus), mitochondria (TM), or laminated vesicles (LV; TLV) was counted. Mitochondria were identified by the presence of two or more distinct cristae. An LV was defined as spherical structures containing one or more internal concentric rings with irregular outer borders. The TM or TLV value was transformed as a percentage of the possible intersecting points where alternately TM or TLV was accounted for (i.e., two structures cannot occupy same point on a grid; Eq. 1ab).
| Eq. 1 |
Thus, a structure’s frequency of occurrence was calculated independent of the numerical influence of the other structure in question.
Chemicals
All chemicals were purchased from Sigma Chemical Co. unless stated otherwise. Verapamil R- (VR, inactive) and S-stereoisomers (VS, active) were kindly provided by Dr. Cindi Romerdahl of BASF Bioscience (Worcester, MA). Racemic verapamil (VR+VS) and pure stereoisomers were dissolved in 10% DMSO and then diluted in DMEM. Diltiazem (DILT) was dissolved in saline. All stock solutions were sterile filtered before use (0.2 μm). Rabbit polyclonal anti-LC3 antibody, HRP conjugated anti-rabbit and anti-mouse IgG secondary antibodies were purchased (Cell Signaling Technologies). Mouse monoclonal anti-ATG5 and mouse monoclonal anti-tubulin-α antibodies and bafilomycin A1 were obtained from Sigma-Aldrich. Non-fat dry milk was obtained from BioRad. Antibodies to mitochondrial cytochrome c oxidase (MTCO1), succinate dehydrogenase [ubiquinone] iron-sulfur subunit (SDHB) and NADH dehydrogenase (ubiquinone) 1 beta (NDUFB) were obtained from Abcam. ECL reagent was obtained from GE Healthcare. Ad-GFP-LC3 vector was obtained from Vector Biolabs.
Statistics
Values are reported as means ± standard error of the mean (SEM). For comparison between two groups, we used unpaired t-test (SigmaStat, SPSS Inc., Chicago, IL). For multiple group analysis, we used one-way ANOVA with Bonferroni post-test or one-way ANOVA on Ranks with Dunn’s post-test where appropriate (SigmaStat, SPSS Inc., Chicago, IL). Significance was assumed at P<0.05.
RESULTS
Antiproliferative effects of CCBs
MTT Assay Results
Two-day exposure of serum-starved RHSM to either VR or VS at 80 μM significantly enhanced MTT absorbance (110–115% of control) compared with vehicle control (i.e., same volume of DMSO as in high [VR] treatment; Fig. 1A). In addition, VR or VS (150 μM) treatments appeared to slightly elevate MTT absorbance at 48h, although values were not statistically significant from controls (Fig. 1B). However, 5-day treatments of RHSM with DILT, VR or VS at a higher concentration (150 μM) produced the lowest MTT absorbance values, yet only the DILT–treated MTT values were significantly lower than all other treatments (cytotoxic). Diltiazem, VR or VS treatment induced cytotoxicity, which was observed as increased number of dead, floating cells after 5 days of exposure whereas no treatment increased cytotoxicity or increased proliferation after 48h exposure.
Figure 1.

Treatment with calcium channel blockers altered MTT absorbance in a concentration- and time-dependent manner in rat heart smooth muscle cells (RHSM). RHSM cells (passages 6–14; n = 4–5) were seeded (10,000 cells/well) in 96-well plates, serum-deprived in 0.1% FCS DMEM for 72h, then treated with DMEM (Control), diltiazem (DILT; 75 or 140 μM), DMSO vehicle, or pure verapamil stereoisomer (inactive, VR; active VS; 80 or 150 μM) for 2 (A) or 5 days (B). Four hours before removing solutions, MTT (10 μg in 10 μl) was added to each well. Values = means±SE for a minimum of 4 wells/treatment/96-well plate. * = significant difference (p<0.05) between verapamil treatment (80 μM) and DMSO vehicle treatment at 2 days. † = significant difference between DILT (140 μM) treatment and 0.1% FCS control treatment.
3H-Thymidine Results
To probe the mechanism of increase in MTT absorbance, we measured 3H-thymidine incorporation in RHSM following 24h of CCB treatment. As in the 48h treatment, MTT absorbance was significantly enhanced in cells treated with VR, VS or 10% FCS (positive control) compared with DMSO at 24h (Fig. 2A). Yet 3H-thymidine incorporation was significantly enhanced only by 10% FCS stimulation whereas both VR and VS treatments significantly depressed 3H-thymidine incorporation (Fig. 2A). To demonstrate that this was not specific to RHSM cells, we measured 3H-thymidine incorporation in RASM cells at 24h after verapamil or 10% FCS treatment. Verapamil decreased and 10% FCS treatment increased 3H-thymidine incorporation, respectively, in RASM (data not shown). To determine if verapamil altered either uptake or incorporation of 3H-thymidine, 3H-thymidine levels in NaOH- and TCA- soluble fractions were measured. The VR and verapamil treatments decreased while the 10% FCS treatment increased 3H-thymidine incorporation, respectively (Fig. 2B). When cells were treated with combined verapamil and 10% FCS, 3H-thymidine DNA incorporation (i.e., NaOH fraction) was lower than in cells treated only with 10% FCS without an inhibition of thymidine uptake (i.e., TCA fraction; Fig. 2B). These results indicate specific inhibition of FCS-stimulated DNA synthesis by verapamil.
Figure 2.
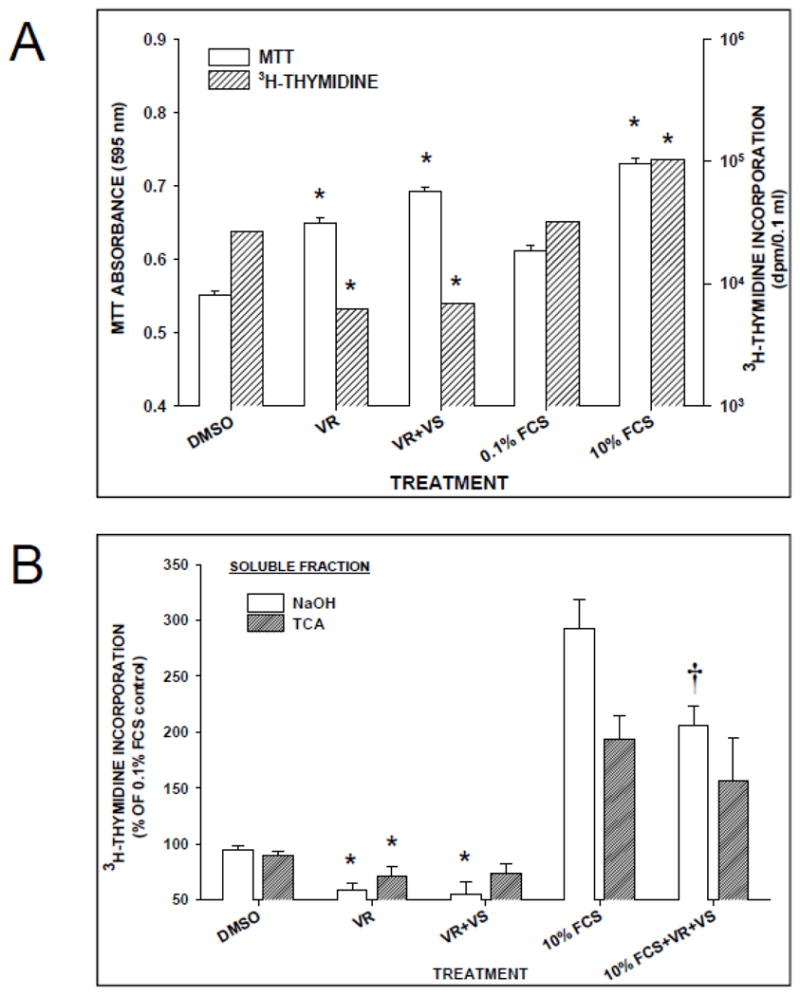
Verapamil treatment for 24h was antiproliferative in cultured VSMC. Verapamil treatment (80 μM) enhanced MTT assay (A) but decreased 3H-thymidine incorporation (B) in serum-deprived rat smooth muscle cells following 24h exposure to R-verapamil (VR; 80 μM) or racemic verapamil (VR+VS; 80 μM). Control cells were exposed to the DMSO vehicle. Cells exposed to 10% FCS served as the positive control group. Effects of verapamil treatments on 3H-thymidine incorporation is expressed as a % of control 3H-thymidine recovered in soluble fractions extracted first with TCA (range = 2,500–25,000 dpm/0.1 ml) and then with NaOH (range = 50,000–450,000 dpm/0.1 ml). * = significant difference (p<0.05) between verapamil treatments with symbol and DMSO control. † = significant difference (p<0.05) between verapamil treatment with symbol and 10% FCS control. Values = means±SE of 2–4 wells/treatment (3H-thymidine) and a minimum of 12 wells/treatment (MTT absorbance).
Verapamil induces stereoindependent changes in cell morphology and ultrastructure
Changes in MTT absorbance and thymidine incorporation were accompanied by visible changes in cytoplasmic structures. Treatment with either diltiazem, racemic verapamil, VR or VS dramatically increased perinuclear accumulation of both “vacuoles” and MTT crystals (3h after MTT addition) following 24h exposure compared with controls (Fig. 3; Suppl. Fig. 1). Concomitantly, CCB treatments appeared to increase karyokinesis (i.e., nuclear division) while halting cytokinesis based on observations of individual VSMC containing 2 or 3 fused nuclei at 2–3-times the frequency observed in control or DMSO treatments (see arrows in Suppl. Fig. 1B,D,E). These changes persisted in cells throughout the treatment duration (up to 120h; data not shown).
Figure 3.
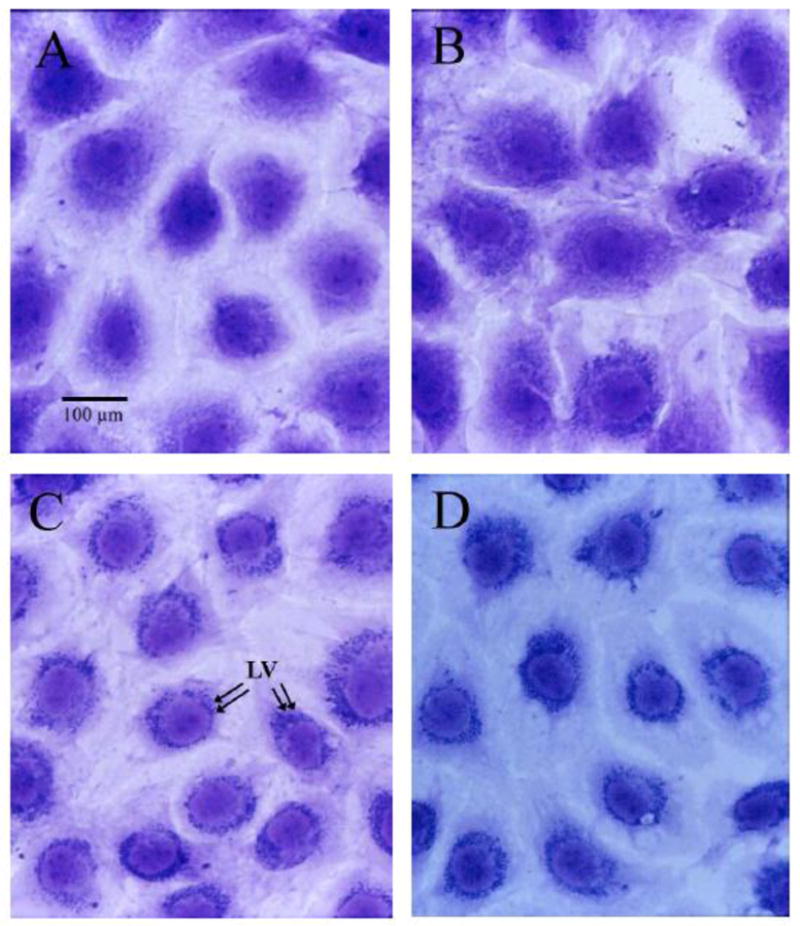
Light micrographs of representative serum-deprived rat aortic smooth muscle cells (RASM) grown for 72h in 96-well plates and then treated for 24h with 0.1% FCS (A, Control), DMSO vehicle (B, equivalent volume to verapamil treatments), R-verapamil (C, 80 μM), or racemic verapamil (D, 80 μM). Note the perinuclear accumulation of vacuoles in C and D (arrows in C), which differed from the random distribution of these structures in control (A) and DMSO (B) treated cells. Cells were stained with 0.5% toluidine blue. Magnification, 300x.
To qualify and quantify the nature of these perinuclear structures observed at the light microscope level, we performed TEM. The apparent vacuoles appeared as highly laminated, aggregates of vesicles in TEM (Fig. 4). In VR- or VS-treated cells, many of the laminated vesicles were part of larger, membrane-bound aggregates (>1.5 μm) composed of 4–8 smaller laminated vesicles (<0.5 μm) with a minority of vesicles containing electron-dense inclusions, which were not characteristic of the laminated vesicles observed in control and DMSO-treated cells (Fig. 4). Notably, DMSO-, VR-, or VS-treated cells had slightly increased frequency of perinuclear vesicles (Suppl. Fig. 2). Moreover, no obvious morphological indices of apoptosis were observed in any cells treated at any concentration of DMSO, DILT, VR, VS or racemic verapamil by light microscopy or TEM.
Figure 4.

Transmission electron micrographs of representative rat heart smooth muscle cell (RHSM) ultrastructure following 72h of serum-deprivation and then 24h treatment with 0.1% FCS (A, Control), DMSO vehicle (B, equal volume of verapamil treatments), R-verapamil (C; VR, 150 μM) or S-verapamil (D; VS, 150 μM). Normal appearing mitochondria and few laminated vesicles (LV) were evident in the perinuclear area in control (A) and DMSO (B) treated cells. In contrast, there were many perinuclear highly laminated vesicles (see arrows) and a notable absence of mitochondria (M) in VR- and VS-treated cells (C and D). N=nucleus. Magnification, 24,500x.
Perinuclear mitochondria of control- and DMSO-treated cells had distinct outer and inner membranes and cristae and were of comparable size and frequency of occurrence. However, mitochondria of VR- and VS-treated (150 μM) VSMC were severely affected in quality (distorted and swelled)(Fig. 4). Moreover, mitochondrial frequency in the perinuclear region was significantly decreased with the VR (150 μM) treatment compared with control (Suppl. Fig. 2; q=4.548, p=0.016) or DMSO treatment (Suppl. Fig. 2; q=4.556, p=0.008). Similarly, the VS (150 μM) treatment appeared to reduce perinuclear mitochondrial frequency compared with control (q=3.179, p=0.079) or with DMSO (p=0.034) treatments. The lower dose VR or VS treatment (80 μM) had minimal effect on perinuclear mitochondrial frequency due to reduced sample sizes (n=2,3, respectively; Suppl. Fig. 2).
At 24h or 36h post-treatment with racemic verapamil (10–160 μM), autophagy was strongly stimulated in smooth muscle cells as evinced by both upregulated conversion of LC3-I to LC3-II (i.e., ratio LC3-II/LC3-I) and ATG5 induction (Fig. 5; Suppl. Fig. 3). Increased autophagy was visualized in cells infected with ad-LC3-GFP and treated with verapamil (40 μM; 24h). Distinct GFP-positive puncta were seen as perinuclear GFP+ bodies indicating increased autophagosome formation (Fig. 5C-E). Because lipidation of LC3-I with phosphatidyl ethanolamine forms LC3-II (or LC3-PE) and is necessary for circularization of the autophagosome, the LC3-II/LC3-I ratio also indicates autophagy. Additionally, the Atg5 protein is part of an Atg12-Atg5-Atg16L1 complex that localizes to autophagosome precursors and plays an essential role in autophagosome formation (Mizushima et al., 2003). Similarly, GFP+ puncta formation indicates GFP-LC3 is present in autophagosomes. Bafilomycin A1 (0.2 μM; inhibitor of autolysosome formation)(Klionsky, 2012) increased LC3-II accumulation as expected (Fig. 5F) yet co-treatment with verapamil led to a significantly greater abundance of LC3-II than either treatment alone (Fig. 5G) indicating racemic verapamil stimulates autophagy.
Figure 5.

Verapamil induces autophagy in smooth muscle cells. A) Western blot of verapamil induced (10–40 μM; 36h) expression of LC3-I, LC3-II and ATG5 (and α-tubulin as loading control). B) Quantification of ratio of LC3-II to LC3-I shows enhanced autophagy by verapamil. C-E) Perinuclear GFP-positive puncta formation in verapamil-treated (E; white arrows) but not in vehicle-treated (C, nothing; D, DMSO) LC3-GFP-transfected RASM indicates autophagosome formation. F) Western blot of LC3-II protein in Bafilomycin A1-, verapamil- and co-treated RASM. Bafilomycin A1 (0.2 μM) was added 4h before harvest. G) Quantification of LC3-II protein (n=3; *p<0.05).
Discussion
A prominent observation in this study is the consistent intensity and incidence of effects induced by the pure stereoisomers of verapamil, R- and S- (e.g., MTT viability assay, 3H-thymidine incorporation, light microscopy, and TEM), under all the varying experimental conditions (i.e., concentrations, durations, and in 2 different VSMC types). Based on these collective data, we conclude that these effects stimulated by the pure stereoisomers of verapamil are independent of L-type calcium channel blockade. In addition, diltiazem, a structurally distinct CCB benzothiazepine, also induced similar changes in cell morphology and MTT absorbance over similar concentrations and exposure durations. These observations indicate that a common mechanism could be shared by verapamil and diltiazem that is independent of calcium channel blockade.
We also show for the first time that the antiproliferative effects of verapamil in smooth muscle cells are closely associated in both time and concentration with the onset of autophagy. This conclusion is supported by multiple lines of evidence including the striking appearance of distinct LC3-GFP+ puncta in the perinuclear region where highly lamellated vesicles (autophagosomes), electron-dense bodies (lysosomes) and damaged mitochondria accrue. Although verapamil strongly upregulates autophagy (i.e., LC3-I to LC3-II conversion, increased atg5, bafilomycin A1-independent), it did not necessarily promote VSMC cell death yet appears to suspend cell division resulting in an antiproliferative state. An earlier report indicated that verapamil (or propranol) treatment triggered autophagy in cardiac myocytes in vivo and the authors concluded this process was due to cardiodepressant action (Bahro and Pfeifer, 1987). It is not known if verapamil induces cardiomyocyte or VSMC autophagy in vivo independent of CCB activity but will be an important question of future studies.
Because CCBs are known to be antiproliferative in VSMC (Waters and Lesperance, 1994), we are surprised that verapamil treatment (<80 μM) increases MTT absorbance in VSMC at 24–48h after exposure. MTT absorbance is used as a marker of cell viability and/or cell proliferation because MTT absorbance is positively and linearly correlated with cell number (Conklin et al., 1998). In our current study, verapamil stimulates an increase in MTT absorbance that corresponds with verapamil-induced inhibition of 3H-thymidine incorporation in VSMC. Thus, the enhanced MTT absorbance in verapamil-treated VSMC is not due to increased cell number, but due to a change in the level of MTT reduction. Although the specific mechanism for the enhanced MTT absorbance observed with verapamil treatment is unknown, it is associated with significant cytologic changes in nuclear and perinuclear organelle morphology (i.e., increased karyokinesis, increased autophagosomes, altered deposition of MTT formazan crystals). Enhanced MTT absorbance in the absence of increased cell number is seen in other cell types after treatment with dicumarol or H2O2 regardless of the tetrazolium salt used, which is converted by reduction to soluble or insoluble formazan crystals, e.g., XTT (Mitsuyoshi et al., 1999; Collier and Pritsos, 2003). Although increased MTT (formazan) absorbance (independent of increased cell number) could be due to increased ROS production and direct chemical reduction of MTT (or XTT) in media/solution (Bernas and Dobrucki, 2000), we reject the latter explanation because we did not observe a MTT color change (yellow to purple) in the media when CCB and MTT were mixed (an event indicative of direct MTT reduction). Moreover, although we did not measure ROS production, CCBs are considered antioxidants not pro-oxidants. Because increased MTT absorbance is associated with enhanced MTT reduction and we observe increased deposition of formazan crystals in the perinuclear area, we conclude this is due to altered and/or impaired mitochondrial integrity with increased enzyme (e.g., succinate dehydrogenase, SDH) leakage and increased MTT reduction.
The source of the highly laminated vesicles, which superficially resemble autophagosomes and phospholipidosis inclusion bodies (Sawada et al., 2005), is unknown. The lamellated vesicles could be remnants of swollen and injured mitochondria because there is a loss of mitochondrial structural integrity in verapamil-treated VSMC by electron microscopy (see Fig. 4CD) but this is not clear that mitochondria are the sole contents of these structures, and mitochondrial proteins are unaffected after 24h of verapamil (see Suppl. Fig. 4). As mentioned above, mitochondrial injury could contribute to enhanced MTT absorbance due to increased availability of mitochondrial enzymes, e.g., SDH. Mitochondria account for 25–45% of MTT reduction in HepG2 cells as estimated by co-localization of formazan crystals with mitochondrial fluorescent probes (Bernas and Dobrucki, 2002). In our study, MTT crystal deposition in the perinuclear region is coincident with both the decrease in perinuclear mitochondrial frequency and the appearance of laminated vesicles and LC3-GFP+ puncta (i.e., autophagosomes). Thus, these three events are linked both in time and cellular localization.
Although apoptosis has been demonstrated with CCB treatment in a variety of cells both in vitro and in vivo and in cells without L-type Ca++ channels, we did not observe any morphological indices of apoptosis in cultured VSMC, which is important because morphological/cytological change is the “gold standard” to judge apoptosis (Lemay et al., 2001). However, apoptosis in thymic cells following CCB treatment in vivo is prevented by pretreatment with RU486 indicating a role of glucocorticoids as mediators of CCB-induced thymic apoptosis in vivo (Balakumaran et al., 1996). The lack of apoptosis in VSMC in vitro is consistent with findings of other investigators using racemic verapamil (1–80 μM), which show verapamil is more anti- than pro-apoptotic in cultured VSMC (Ares et al., 1997; Cheng et al., 2003). More importantly, we show that verapamil induces autophagy not apoptosis in VSMC in vitro.
In conclusion, we observed antiproliferative and autophagic effects of verapamil stereoisomers in VSMC that are independent of calcium channel blocker activity. These effects occurred at concentrations consistent with other reports of verapamil-induced antiproliferative effects in VSMC and in non-VSMC (Weir et al., 1992; Munro et al., 1994; Patel et al., 1995). Additionally, the antiproliferative action was in vitro within the therapeutic treatment range (i.e., 10–160 μM in vitro; est. instantaneous blood level is [500–1000 μM] in a 60 kg bwt human on 120–240 mg once a day; measured steady state human serum verapamil is [0.2–0.8 μM]) (Weir et al., 1992). Racemic verapamil (Calan®, Covera®, Isoptin®, Verelan®) is prescribed, and thus, the antiproliferative action of the combined stereoisomers could contribute independently and/or in addition to calcium channel blockade in the overall anti-atherosclerotic action of verapamil. Identification of this stereo-independent antiproliferative and autophagic mechanism of action could reveal a novel therapeutic target in proliferating smooth muscle cells as seen for mTOR inhibitors (e.g., Everolimus) that promote stabilization of atherosclerotic plaque (Martinet et al., 2007). This highlights an intriguing possibility of using higher dosages of inactive verapamil stereoisomers without the risk of hypotensive crisis.
Supplementary Material
Highlights.
Calcium channel blockers (CCB) are antiproliferative in vascular smooth muscle cells
Verapamil steroisomers are antiproliferative in VSMC independent of CCB activity
Verapamil steroisomers alter mitochondrial appearance and frequency in VSMC
Verapamil stimulates autophagy in cultured VSMC
Acknowledgments
This work was supported by NIH Grants HL26189 and HL65416 (PJB), NIEHS Center Grant P30ES06676-11 (PJB, MTM), Houston Endowment (MTM), NIEHS Toxicology Training Grant T32ESO7254 (DJC), NIEHS Academic Research Enhancement Award Grant 1 R15-ES011141 (DJC), NIEHS 1P01ES11860, RR024489, HL89380 (DJC), HL89380-S2 (DJC) and the Ronald E. McNair Postbaccalaureate Scholar Program (JCF). We thank C. Romerdahl of BASF for the kind gift of the verapamil stereoisomers. We acknowledge the expert assistance of the Department of Pathology Staff, J. Wen and V. Han. We thank M.B. Trent and Dr. U. Tipnis for assistance with cell culture. Funding agencies had no part in the preparation of this work.
Abbreviations
- CCB
calcium channel blocker
- DILT
diltiazem
- GFP
green fluorescent protein
- LC3
microtubule-associated light chain I protein 3ß
- MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl) tetrasodium bromide
- RHSM
neonatal rat heart smooth muscle cells
- RASM
rat aortic vascular smooth muscle
- TEM
transmission electron microscopy
- VR
verapamil R-isomer
- VS
verapamil S-isomer
- VSMC
vascular smooth muscle cell
Footnotes
Conflict of Interest Statement
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ares MP, Porn-Ares MI, Thyberg J, Juntti-Berggren L, Berggren PO, Diczfalusy U, Kallin B, Bjorkhem I, Orrenius S, Nilsson J. Ca2+ channel blockers verapamil and nifedipine inhibit apoptosis induced by 25-hydroxycholesterol in human aortic smooth muscle cells. J Lipid Res. 1997;38:2049–2061. [PubMed] [Google Scholar]
- Armstrong ML, Heistad DD, Lopez JA. Regression of atherosclerosis. A role for calcium antagonists. Am J Hypertens. 1991;4:503S–511S. doi: 10.1093/ajh/4.7.503s. [DOI] [PubMed] [Google Scholar]
- Atkinson JB. Accelerated arteriosclerosis after transplantation: the possible role of calcium channel blockers. Int J Cardiol. 1997;62(Suppl 2):S125–134. doi: 10.1016/s0167-5273(97)00250-7. [DOI] [PubMed] [Google Scholar]
- Bahro M, Pfeifer U. Short-term stimulation by propranolol and verapamil of cardiac cellular autophagy. J Mol Cell Cardiol. 1987;19:1169–1178. doi: 10.1016/s0022-2828(87)80527-8. [DOI] [PubMed] [Google Scholar]
- Balakumaran A, Campbell GA, Moslen MT. Calcium channel blockers induce thymic apoptosis in vivo in rats. Toxicol Appl Pharmacol. 1996;139:122–127. doi: 10.1006/taap.1996.0150. [DOI] [PubMed] [Google Scholar]
- Bernas T, Dobrucki J. Mitochondrial and nonmitochondrial reduction of MTT: interaction of MTT with TMRE, JC-1, and NAO mitochondrial fluorescent probes. Cytometry. 2002;47:236–242. doi: 10.1002/cyto.10080. [DOI] [PubMed] [Google Scholar]
- Bernas T, Dobrucki JW. The role of plasma membrane in bioreduction of two tetrazolium salts, MTT, and CTC. Arch Biochem Biophys. 2000;380:108–116. doi: 10.1006/abbi.2000.1907. [DOI] [PubMed] [Google Scholar]
- Cheng G, Shan J, Xu G, Huang J, Ma J, Ying S, Zhu L. Apoptosis induced by simvastatin in rat vascular smooth muscle cell through Ca2+-calpain and caspase-3 dependent pathway. Pharmacol Res. 2003;48:571–578. doi: 10.1016/s1043-6618(03)00245-7. [DOI] [PubMed] [Google Scholar]
- Collier AC, Pritsos CA. The mitochondrial uncoupler dicumarol disrupts the MTT assay. Biochem Pharmacol. 2003;66:281–287. doi: 10.1016/s0006-2952(03)00240-5. [DOI] [PubMed] [Google Scholar]
- Conklin DJ, Langford SD, Boor PJ. Contribution of serum and cellular semicarbazide-sensitive amine oxidase to amine metabolism and cardiovascular toxicity. Toxicol Sci. 1998;46:386–392. doi: 10.1006/toxs.1998.2528. [DOI] [PubMed] [Google Scholar]
- Corsini A, Bonfatti M, Quarato P, Accomazzo MR, Raiteri M, Sartani A, Testa R, Nicosia S, Paoletti R, Fumagalli R. Effect of the new calcium antagonist lercanidipine and its enantiomers on the migration and proliferation of arterial myocytes. J Cardiovasc Pharmacol. 1996;28:687–694. doi: 10.1097/00005344-199611000-00012. [DOI] [PubMed] [Google Scholar]
- Eickelberg O, Roth M, Mussmann R, Rudiger JJ, Tamm M, Perruchoud AP, Block LH. Calcium channel blockers activate the interleukin-6 gene via the transcription factors NF-IL6 and NF-kappaB in primary human vascular smooth muscle cells. Circulation. 1999;99:2276–2282. doi: 10.1161/01.cir.99.17.2276. [DOI] [PubMed] [Google Scholar]
- Henry PD. Atherogenesis, calcium and calcium antagonists. Am J Cardiol. 1990;66:3I–6I. doi: 10.1016/0002-9149(90)91256-6. [DOI] [PubMed] [Google Scholar]
- Henry PD. Antiperoxidative actions of calcium antagonists and atherogenesis. J Cardiovasc Pharmacol. 1991;18(Suppl 1):S6–10. [PubMed] [Google Scholar]
- Jones PA, Scott-Burden T, Gevers W. Glycoprotein, elastin, and collagen secretion by rat smooth muscle cells. Proc Natl Acad Sci. 1979;76:353–357. doi: 10.1073/pnas.76.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jukema JW, van der Hoorn JW. Amlodipine and atorvastatin in atherosclerosis: a review of the potential of combination therapy. Expert Opin Pharmacother. 2004;5:459–468. doi: 10.1517/14656566.5.2.459. [DOI] [PubMed] [Google Scholar]
- Kauder WF, Watts JA. Antioxidant properties of dihydropyridines in isolated rat hearts. Biochem Pharmacol. 1996;51:811–819. doi: 10.1016/0006-2952(95)02404-2. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ. Protocols, Toolboxes and Resource papers. Autophagy. 2012:8. doi: 10.4161/auto.19460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko Y, Totzke G, Graack GH, Heidgen FJ, Meyer zu Brickwedde MK, Dusing R, Vetter H, Sachinidis A. Action of dihydropyridine calcium antagonists on early growth response gene expression and cell growth in vascular smooth muscle cells. J Hypertens. 1993;11:1171–1178. [PubMed] [Google Scholar]
- Lai YM, Fukuda N, Su JZ, Suzuki R, Ikeda Y, Takagi H, Tahira Y, Kanmatsuse K. Novel mechanisms of the antiproliferative effects of amlodipine in vascular smooth muscle cells from spontaneously hypertensive rats. Hypertens Res. 2002;25:109–115. doi: 10.1291/hypres.25.109. [DOI] [PubMed] [Google Scholar]
- Lemay J, Tea BS, Hamet P, deBlois D. Regression of neointimal lesions in the carotid artery of nifedipine-treated SHR and WKY rats: possible role of apoptosis. J Vasc Res. 2001;38:462–470. doi: 10.1159/000051079. [DOI] [PubMed] [Google Scholar]
- Li W, Li J, Liu W, Altura BT, Altura BM. Alcohol-induced apoptosis of canine cerebral vascular smooth muscle cells: role of extracellular and intracellular calcium ions. Neurosci Lett. 2004;354:221–224. doi: 10.1016/j.neulet.2003.10.047. [DOI] [PubMed] [Google Scholar]
- Marche P, Stepien O. Calcium antagonists and vascular smooth muscle cell reactivity. Z Kardiol. 2000;89(Suppl 2):140–144. doi: 10.1007/s003920070114. [DOI] [PubMed] [Google Scholar]
- Martinet W, Verheye S, De Meyer GR. Everolimus-induced mTOR inhibition selectively depletes macrophages in atherosclerotic plaques by autophagy. Autophagy. 2007;3:241–244. doi: 10.4161/auto.3711. [DOI] [PubMed] [Google Scholar]
- Mitsuyoshi H, Nakashima T, Sumida Y, Yoh T, Nakajima Y, Ishikawa H, Inaba K, Sakamoto Y, Okanoue T, Kashima K. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem Biophys Res Commun. 1999;263:537–542. doi: 10.1006/bbrc.1999.1403. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116:1679–1688. doi: 10.1242/jcs.00381. [DOI] [PubMed] [Google Scholar]
- Munro E, Patel M, Chan P, Betteridge L, Gallagher K, Schachter M, Wolfe J, Sever P. Effect of calcium channel blockers on the growth of human vascular smooth muscle cells derived from saphenous vein and vascular graft stenoses. J Cardiovasc Pharmacol. 1994;23:779–784. doi: 10.1097/00005344-199405000-00013. [DOI] [PubMed] [Google Scholar]
- Patel MK, Chan P, Betteridge LJ, Schachter M, Sever PS. Inhibition of human vascular smooth muscle cell proliferation by the novel multiple-action antihypertensive agent carvedilol. J Cardiovasc Pharmacol. 1995;25:652–657. doi: 10.1097/00005344-199504000-00020. [DOI] [PubMed] [Google Scholar]
- Puscas L, Gilau L, Coltau M, Pasca R, Domuta G, Baican M, Hecht A. Calcium channel blockers reduce blood pressure in part by inhibiting vascular smooth muscle carbonic anhydrase I. Cardiovasc Drugs Ther. 2000;14:523–528. doi: 10.1023/a:1007893207279. [DOI] [PubMed] [Google Scholar]
- Sawada H, Takami K, Asahi S. A toxicogenomic approach to drug-induced phospholipidosis: analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol Sci. 2005;83:282–292. doi: 10.1093/toxsci/kfh264. [DOI] [PubMed] [Google Scholar]
- Schachter M. Vascular smooth muscle cell migration, atherosclerosis, and calcium channel blockers. Int J Cardiol. 1997;62(Suppl 2):S85–90. doi: 10.1016/s0167-5273(97)00245-3. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Hankowitz J, Kovacs EM. Cellular processes in atherogenesis: potential targets of Ca2+ channel blockers. Atherosclerosis. 1991;88:109–132. doi: 10.1016/0021-9150(91)90074-d. [DOI] [PubMed] [Google Scholar]
- Schroeder JS, Gao SZ. Calcium blockers and atherosclerosis: lessons from the Stanford Transplant Coronary Artery Disease/Diltiazem Trial. Can J Cardiol. 1995;11:710–715. [PubMed] [Google Scholar]
- Timar J, Chopra H, Rong X, Hatfield JS, Fligiel SE, Onoda JM, Taylor JD, Honn KV. Calcium channel blocker treatment of tumor cells induces alterations in the cytoskeleton, mobility of the integrin alpha IIb beta 3 and tumor-cell-induced platelet aggregation. J Cancer Res Clin Oncol. 1992;118:425–434. doi: 10.1007/BF01629425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulenko TN, Laury-Kleintop L, Walter MF, Mason RP. Cholesterol, calcium and atherosclerosis: is there a role for calcium channel blockers in atheroprotection? Int J Cardiol. 1997;62(Suppl 2):S55–66. doi: 10.1016/s0167-5273(97)00242-8. [DOI] [PubMed] [Google Scholar]
- Waters D, Lesperance J. Calcium channel blockers and coronary atherosclerosis: from the rabbit to the real world. Am Heart J. 1994;128:1309–1316. doi: 10.1016/0002-8703(94)90253-4. [DOI] [PubMed] [Google Scholar]
- Weibel ER. Stereological principles for morphometry in electron microscopic cytology. Int Rev Cytol. 1969;26:235–302. doi: 10.1016/s0074-7696(08)61637-x. [DOI] [PubMed] [Google Scholar]
- Weir MR, Gomolka D, Peppler R, Handwerger BS. Mechanisms responsible for inhibition of lymphocyte activation by agents which block membrane calcium or potassium channels. Transplant Proc. 1993a;25:605–609. [PubMed] [Google Scholar]
- Weir MR, Peppler R, Gomolka D, Handwerger BS. Evidence that the antiproliferative effect of verapamil on afferent and efferent immune responses is independent of calcium channel inhibition. Transplantation. 1992;54:681–685. doi: 10.1097/00007890-199210000-00023. [DOI] [PubMed] [Google Scholar]
- Weir MR, Peppler R, Gomolka D, Handwerger BS. Calcium channel blockers inhibit cellular uptake of thymidine, uridine and leucine: the incorporation of these molecules into DNA, RNA and protein in the presence of calcium channel blockers is not a valid measure of lymphocyte activation. Immunopharmacology. 1993b;25:75–82. doi: 10.1016/0162-3109(93)90032-l. [DOI] [PubMed] [Google Scholar]
- Zernig G. Widening potential for Ca2+ antagonists: non-L-type Ca2+ channel interaction. Trends Pharmacol Sci. 1990;11:38–44. doi: 10.1016/0165-6147(90)90040-f. [DOI] [PubMed] [Google Scholar]
- Zhang YZ, Gao PJ, Wang XY, Stepien O, Marche P, Zhang ZL, Zhu DL. The inhibitory mechanisms of amlodipine in human vascular smooth muscle cell proliferation. Hypertens Res. 2000;23:403–406. doi: 10.1291/hypres.23.403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.