

SUMMARY
Background
Acute myeloid leukemia (AML) remains a major therapeutic challenge in pediatric oncology even with intensified cytarabine (ara-C)-based chemotherapy. Therefore, new therapies are urgently needed to improve treatment outcome of this deadly disease. In this study, we evaluated antileukemic interactions between clofarabine (a second-generation purine nucleoside analog) and valproic acid (VPA, a FDA-approved agent for treating epilepsy in both children and adult and a histone deacetylase inhibitor), in pediatric AML.
Methodology
In vitro clofarabine and VPA cytotoxicities of the pediatric AML cell lines and diagnostic blasts were measured by using MTT assays. The effects of clofarabine and VPA on apoptosis and DNA double strand breaks (DSBs) were determined by flow cytometry analysis and Western blotting, respectively. Active form of Bax was measured by Western blotting post immunoprecipitation.
Results
We demonstrated synergistic antileukemic activities between clofarabine and VPA in both pediatric AML cell lines and diagnostic blasts sensitive to VPA. In contrast, antagonism between the two agents could be detected in AML cells resistant to VPA. Clofarabine and VPA cooperate in inducing DNA DSBs, accompanied by Bax activation and apoptosis in pediatric AML cells.
Conclusion
Our results document synergistic antileukemic activities of combined VPA and clofarabine in pediatric AML and suggest that this combination could be an alternative treatment option for the disease.
Keywords: pediatric acute myeloid leukemia, clofarabine, valproic acid, histone deacetylase inhibitor, synergistic antileukemic interaction
INTRODUCTION
Acute myeloid leukemia (AML) is a clinically and genetically heterogeneous disease accounting for 15–20% of childhood acute leukemias [1]. However, it is responsible for more than half of the leukemic deaths in this patient population. In contrast to the tremendous success in the treatment of acute lymphoblastic leukemia (ALL) over the last three decades, improvements in AML therapy have been modest, as 20–40% of patients do not respond to initial chemotherapy, and approximately 50% of responding patients will eventually relapse [2,3]. Therefore, new therapies for children with AML, especially those with primary refractory or relapsed disease, need to be developed. Among the newer agents that have been investigated in high-risk adult AML, clofarabine and histone deacetylase (HDAC) inhibitors (HDACIs) are particularly notable [4–7].
Clofarabine (2-chloro-9-[2′-deoxy-2′-fluoro-β-D-arabinofuranosyl]adenine; Cl-F-ara-A; CAFdA) is a rationally designed, second generation purine nucleoside analog [8–11]. Intravenous clofarabine showed significant efficacy in pediatric leukemias (specifically, ALL) [12]. Clofarabine was approved in December 2004 by the United States Food and Drug Administration (US FDA) for the treatment of pediatric patients with relapsed or refractory ALL after at least 2 prior regimens [8,11]. In adults, clofarabine has shown significant efficacy in hematologic malignancies including AML and myelodysplastic syndrome alone and in combinations [13–18].
HDACIs possess antitumor activities and have the potential to induce re-expression of genes abnormally suppressed in cancer cells, thus potentially inducing growth arrest, differentiation, and/or apoptotic cell death of transformed cells in vitro and in vivo [6,7,19–21]. Valproic acid (VPA) is a FDA-approved agent used for treating epilepsy in both children and adults, and was recently reported to be a powerful HDACI [22–24]. We previously demonstrated synergistic antileukemic activities of combined cytarabine and VPA in a panel of pediatric AML cell lines and diagnostic blast samples derived from children with de novo AML [25,26]. Cytarabine and VPA cooperatively induced DNA double-strand breaks (DSBs), as reflected in the induction of γH2AX (a biomarker of DNA DSBs), and apoptosis [25]. However, it is not yet established whether this represents a global mechanism that results in the synergistic cytotoxicity between HDACIs and DNA damaging agents in pediatric AML.
We hypothesized that VPA and clofarabine cooperate in inducing DNA damage and subsequent apoptosis in pediatric AML cells since clofarabine is also a DNA damaging agent. We demonstrate synergistic antileukemic activities of the two agents in VPA-sensitive pediatric AML cells. In contrast, antagonism between the two agents could be detected in VPA-resistant cells. In VPA-sensitive Kasumi-1 cells, clofarabine and VPA cooperate in inducing DNA DSBs and apoptosis, consistent with our previous findings with combined cytarabine and VPA in pediatric AML cells. These results suggest that this drug combination may be beneficial to AML cases which are sensitive to VPA.
MATERIALS AND METHODS
Clinical Samples
Diagnostic bone marrow samples (n=9) from children with de novo AML were obtained from the Children’s Hospital of Michigan leukemia cell bank. Patient characteristics and blast percentages in each patient are summarized in Table I. Mononuclear cells were purified by standard Ficoll-Hypaque density centrifugation. Written consent was provided according to the Declaration of Helsinki. The Human Investigation Committee of the Wayne State University School of Medicine approved this study and the procedures involved.
Table I.
Patient characteristics of pediatric AMLs used in the present study.
| Patient ID | FAB | Age (year) | Sex | Cytogenetics | Blasts (%) |
|---|---|---|---|---|---|
| A30307 | AML M6 | 10 | Female | 46, XX | 29 |
| A30308 | AML M4/5 | 14 | Male | 46, XY, inv(16) | 11 |
| A30309 | AML M4 | 0.8 | Male | 46, XY, inv(16) | 30 |
| A30310 | AML M2 | 9 | Male | 46, XY | 57 |
| A30311 | AML M4 | 14 | Male | 46, XY, t(3;5) | 24 |
| A30110 | AML M5a | 0.9 | Male | 46, XY, +9 | 73 |
| A30207 | AML M2 | 4.6 | Male | 46, XY, t(8;21) | 82 |
| A30224 | AML M2 | 14.2 | Female | 46, XX, t(8;21) | 48 |
| A30230 | AML M2 | 6.3 | Female | 46, XX, t(8;21) | 39 |
Drugs
VPA was purchased from Sigma Chemical Company (St Louis, MO). Clofarabine was purchased from Tocris Bioscience (Ellisville, MO).
Cell Culture
The THP-1 (AML M5), Kasumi-1 (AML M2), and MV4-11 (AML M5) pediatric AML cell lines were purchased from the American Type Culture Collection (Manassas, VA). The CMS (AML M7 or AMkL) pediatric AML cell line was a gift from Dr. A Fuse from the National Institute of Infectious Diseases, Tokyo, Japan. The above cell lines were cultured in RPMI 1640 with 10–20% fetal bovine serum (FBS, Hyclone, Logan, UT) and 2 mM L-glutamine, plus 100 U/ml penicillin and 100 μg/ml streptomycin, in a 37°C humidified atmosphere containing 5% CO2/95% air.
In Vitro Cytotoxicity Assays
In vitro clofarabine and VPA cytotoxicities of the pediatric AML cell lines and diagnostic blasts were measured by using MTT (3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium-bromide, Sigma, St Louis, MO) assays, as previously described [25,26]. Briefly, the pediatric AML cell lines and diagnostic AML blasts were cultured in 100 μl of RPMI 1640/10–20% FBS in 96-well plates. For individual drug treatments, pediatric AML cells were treated with variable concentrations of clofarabine (0–1 μM, clinically relevant concentrations [27]) or VPA (0– 8 μM); while for combined treatments, the cells were treated with variable concentrations of clofarabine (0–1 μM) in combination with VPA (0–1 mM, clinically achievable concentrations [28]). After 72 hours, MTT was added to a final concentration of 1 mM. After 4.5 hours, formazan crystals were dissolved by the addition of 100 μl of 10% SDS in 10 mM HCl. Optical densities were measured with a visible microplate reader at 590 nm. IC50 values were calculated as drug concentrations necessary to inhibit 50% proliferation compared to untreated control cells. The extent and direction of clofarabine and VPA interactions were evaluated by standard isobologram analysis as described previously [25,29,30], and by using the CompuSyn software (ComboSyn, Inc., Paramus, NJ). Briefly, synergism, additivity, or antagonism was quantified by determining the combination index (CI), where CI<1, CI=1, and CI>1 indicate synergistic, additive, and antagonistic effects, respectively.
Growth Curve Analysis
Kasumi-1 cells were treated with clofarabine (5 nM) or VPA (0.5 mM) alone or combined for up to 96 hours. Viable cells were counted every 24 hours with trypan blue staining. Data was presented as means ± standard errors of triplicate determinations.
Assessment of Baseline and Drug-Induced Apoptosis
Kasumi-1 cells were treated with variable concentrations of VPA (0.25, 0.5, and 1.0 mM) and 5 nM clofarabine or variable concentrations of clofarabine (5, 10, and 20 nM) and 0.5 mM VPA alone, or in combination for 72 hours. The cells were harvested, vigorously pipetted and triplicate samples taken to determine baseline and drug-induced apoptosis using the Apoptosis Annexin-V fluorescein isothiocyanate (FITC)/propidium iodide (PI) Kit (Beckman Coulter; Brea, CA), as previously described [25,26,31]. Apoptotic events were recorded as a combination of Annexin-V+/PI− (early apoptotic) and Annexin-V+/PI+ (late apoptotic/dead) events and results were expressed as percent of Annexin-V+ cells.
Immunoprecipitation (IP) of Bax
To detect the active form of Bax, Kasumi-1 cells treated with clofarabine or VPA alone, or in combination, for 72 hours were lysed in Tris buffer (10 mM, pH 8.0) containing protease inhibitors (Roche, Indianapolis, IN). After determination of protein concentrations, cell lysates were diluted with the above Tris buffer to 1.33 μg/μl. After pre-clearing 400 μl of the sample with 20 μl Dynabeads® Protein G (Invitrogen, Carlsbad, CA) at 4°C for 1 hour, immunoprecipitation was performed by incubating 400 μl of the lysates with 4 μg of anti-Bax monoclonal antibody (clone 6A7, BD-Pharmingen, San Diego, CA; or B9, Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. After extensive washing with PBS containing protease inhibitors, Dynabeads-Ig-Antigen complex was heated at 70 °C for 10 min in 40 μl loading buffer, and 20 μl of the eluted proteins were analyzed by Western blotting.
Western Blot Analysis
Soluble protein extracts or immunoprecipitated proteins were subjected to SDS-polyacrylamide gel electrophoresis. Separated proteins were electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Thermo Fisher Inc., Rockford, IL) and immunoblotted with anti-Bax, -PARP, -cleaved-caspase 3, -γH2AX (Cell Signaling Technology, Danvers, MA), or -β-actin (Sigma, St Louis, MO) antibody, as described previously [32,33]. Immunoreactive proteins were visualized using the Odyssey Infrared Imaging System (Li-Cor, Lincoln, NE), as described by the manufacturer.
Statistical Analysis
Differences in clofarabine IC50s (drug concentrations necessary to inhibit 50% proliferation compared to untreated control cells) between VPA-treated and -untreated AML cells and differences in cell apoptosis between clofarabine- and VPA-treated (individually or combined) and untreated cells were compared using the paired t-test. The relationship between apoptosis and levels of γH2AX was determined by the Pearson test. Statistical analyses were performed with GraphPad Prism 4.0.
RESULTS
VPA Synergistically enhanced clofarabine cytotoxicity in Kasumi-1 cells
To test our hypothesis that clofarabine and VPA synergize in their antileukemic acitivities against pediatric AML cells, we first evaluated the antileukemic activities of clofarabine in combination with VPA in Kasumi-1 cell line using MTT assays. When simultaneously administered with clofarabine, VPA at 0.15, 0.3 and 0.5 mM, significantly enhanced clofarabine sensitivities (reflected by the decreased IC50s) by 1.4-, 2.1- and 4.2-fold, respectively (Figure 1A). The combined effects of clofarabine with VPA on cell proliferation were clearly synergistic, as determined by standard isobologram analysis (Figure 1C) and by calculating combination index (CI) values by using the CompuSyn software (Table II).
Figure 1. Synergistic antileukemic interactions between VPA and clofarabine in Kasumi-1 cells.

Panel A: Kasumi-1 cells were cultured at 37 °C for 72 h in complete medium with dialyzed fetal bovine serum in 96-well plates at a density of 1.2 × 104 cells/well, with a range of concentrations of clofarabine and/or VPA, and viable cell numbers were determined using the MTT reagent and a visible microplate reader. The IC50 values were calculated as the concentrations of drug necessary to inhibit 50% proliferation compared to control cells cultured in the absence of drug. Clofarabine IC50s of Kasumi-1 cells were determined in the absence or presence of VPA treated simultaneously. * indicates statistically significant difference (p<0.05). The data are presented as means ± standard errors from at least 3 independent experiments. Panel B: Kasumi-1 cells were treated with clofarabine (5 nM) or VPA (0.5 mM) alone or combined for up to 96 h. Viable cells were counted every 24 h with trypan blue staining. Data are presented as means ± standard errors of triplicate determinations. C, clofarabine; V, VPA. Panel C: Standard isobologram was used to analyze the antileukemic interactions between VPA and clofarabine in Kasumi-1 cells. The IC50 values of each drug are plotted on the axes; the solid line represents the additive effect, while the points represent the concentrations of each drug resulting in 50% inhibition of proliferation. Points falling below the line indicate synergism between drug combinations whereas those above the line indicate antagonism. → indicates pre-treatment with the first drug for the indicated times shown in parentheses. C, clofarabine; V, VPA.
Table II.
Effects of VPA on clofarabine cytotoxicities in pediatric AML cell lines and diagnostic AML blasts
| Cell line/Patient | VPA IC50 (mM) | Clofarabine IC50 (nM) | p value | |||||
|---|---|---|---|---|---|---|---|---|
| 0 mM VPA | 0.15 mM VPA | 0.3 mM VPA | 0.5 mM VPA | 0.75 mM VPA | 1.0 mM VPA | |||
| MV4-11 | 0.7±0.1 | 29.4±0.6 | 21.5±0.6 (0.9) | 16.0±0.3 (1.0) | 6.9±0.4 (1.0) | ND | ND | <0.008 |
| Kasumi-1 | 0.9±0.1 | 49.0±3.7 | 35.0±3.1 (0.9) | 23.6±5.6 (0.8) | 11.6±1.5 (0.9) | ND | ND | <0.014 |
| CMS | 2.7±0.2 | 14.4±0.4 | ND | ND | 13.1±0.3 (1.0) | 12.5±0.8 (1.0) | 11.0±0.9 (1.0) | NS |
| THP-1 | 3.3±0.1 | 33.2±0.5 | ND | ND | 30.5±1.0 (1.0) | 29.5±0.3 (1.1) | 28.8±1.1 (1.1) | NS |
| A30307 | 1.1 | 18.2 | ND | ND | 1.2 (0.5) | 0.8 (0.6) | ND | NA |
| A30308 | 4.9 | 337.8 | ND | ND | 259.0 (0.9) | 191.1 (0.7) | 148.3 (0.6) | NA |
| A30309 | 2.0 | 129.3 | ND | ND | 78.02 (0.8) | 52.2 (0.9) | 40.0 (0.8) | NA |
| A30310 | 1.7 | 38.9 | ND | ND | 22.2 (0.7) | 12.8 (0.6) | 13.2 (0.7) | NA |
| A30311 | 0.9 | 67.6 | ND | ND | 12.6 (0.8) | 4.4 (0. 9) | ND | NA |
| A30110 | 1.0 | 55.9 | ND | ND | 7.5 (0.6) | 3.2 (0.7) | ND | NA |
| A30207 | 0.7 | 36.8 | ND | ND | 2.0 (0.8) | ND | ND | NA |
| A30224 | 0.7 | 136.8 | 46.6 (0.5) | 12.1 (0.5) | 5.9 (0.7) | ND | ND | NA |
| A30230 | 0.2 | 189.1 | 3.6 (0.9) | ND | ND | ND | ND | NA |
Note: For individual drug treatments, pediatric AML cells were treated with variable concentrations of clofarabine (0–1 μM) or VPA (0– 8 μM) for 72h; for combined treatments, the cells were treated with variable concentrations of clofarabine (0– 1μM) in combination with VPA (0–1 mM) for 72h. Viable cells were measured by using the MTT reagent and a visible microplate reader. Clofarabine and VPA IC50s are presented as mean ± standard errors from at least three independent experiments for the pediatric AML cell lines, while those for the diagnostic AML blasts are presented as mean of duplicates from one experiment. Numbers in parentheses represent the combination index values. NA, not applicable; NS, not significant; ND, not determined.
To determine the effects of treatment time on the antileukemic interactions between the two agents, Kasumi-1 cells were treated with 5 nM clofarabine or 0.5 mM VPA alone, or in combination, for up to 96 hours, and cells were counted every 24 hours with trypan blue staining. As shown in Figure 1B, a minimum 48 hour co-treatment was needed to detect enhanced inhibition on cell proliferation. Interestingly, the synergism between clofarabine and VPA in Kasumi-1 cells was independent of the sequence of drug administration (Figure 1C).
Differential effects of VPA on clofarabine cytotoxicities in other pediatric AML cell lines and diagnostic blasts
To determine whether the synergistic antileukemic activities of VPA with clofarabine were unique to the Kasumi-1 subline, analogous cytotoxicity experiments were performed with MV4-11, THP-1, and CMS cell lines derived from children with different AML subtypes. Surprisingly, additive-to-synergistic (in MV4-11), additive (in CMS), or additive-to-antagonistic (in THP-1) results were obtained in these additional lines (Table II). At 0.5 mM VPA, simultaneous treatment with clofarabine resulted in 4.3-fold decrease in clofarabine IC50 in MV4-11 cells, compared to that from clofarabine alone. In contrast, VPA only had minimal effects on clofarabine IC50s at this concentration in both THP-1 and CMS cells (Table II). Synergistic antileukemic activities (Table II) were detected in diagnostic AML blasts derived from 9 children with diverse subtypes of de novo AML (Table I), when VPA (0.15 to 1 mM) was administered simultaneously with clofarabine. Analogous to our previous study with cytarabine and VPA [25], diagnostic blasts from t(8;21) AML cases (n=3, patients 7–9) showed significantly increased sensitivities to clofarabine (range 18.4–52.5 fold) compared to those for non-t(8;21) AML cases (n=6, patients 1–6, range 1.3–15.2 fold) when combined with VPA at doses 0.5 mM or lower (p<0.05, Table II).
We then divided the AML cell lines and diagnostic blast samples into VPA-sensitive (IC50 ≤1.0 mM) and VPA-resistant (IC50 > 1.0 mM) groups by using the median VPA IC50 (1.0 mM) as the cut off. Interestingly, the VPA-sensitive AML cell lines and diagnostic blast samples showed 4.2- to 52.5-fold decreased clofarabine IC50s when combined with VPA at doses 0.5 mM or lower. This is in great contrast to VPA-resistant cells which showed 1.1- to 15.2-fold decreased clofarabine IC50s under the same experimental conditions (p=0.014, Figure 2). These results demonstrate that there are two groups of pediatric AMLs that show differential responses to the combination of clofarabine and VPA.
Figure 2. Differential responses of pediatric AML cells to the combination of clofarabine and VPA.
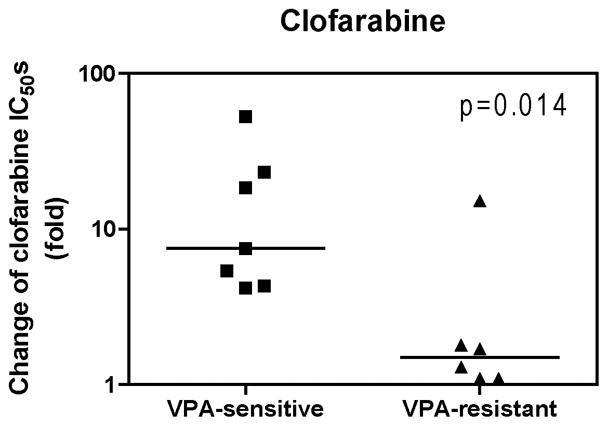
The pediatric AML cell lines and diagnostic AML blast samples were divided into two groups based the median VPA IC50 (1.0 mM) measured by MTT assays. Cells with VPA IC50s ≤ 1.0 mM were defined as VPA-sensitive, while cells with VPA IC50 > 1.0 mM were defined as VPA-resistant. Fold decrease of clofarabine IC50s for pediatric AML cell lines and diagnostic AML blasts measured by MTT assays in the presence of 0.5 mM or lower VPA was compared with that from clofarabine alone. The horizontal lines indicate the median fold change in each group of AML cell lines and patient samples. The p value was determined by the nonparametric Mann-Whitney U test.
Cooperative induction of DNA DSBs and apoptosis by clofarabine and VPA in Kasumi-1 cells
Efforts were then undertaken to determine the molecular mechanisms that underlie the synergistic antileukemic interactions between the two agents. Our previous study demonstrated that VPA and cytarabine cooperatively caused DNA DSBs in Kasumi-1 cells [25]. It is conceivable that clofarabine and VPA may also cooperate in inducing DNA DSBs which subsequently trigger apoptosis since clofarabine is also a DNA-damaging agent. To test these possibilities, Kasumi-1 cells were treated with variable concentrations of clofarabine or VPA, alone or combined for 72 hours, and protein lysates were subjected to Western blotting to detect γH2AX. Consistent with our hypothesis, co-treatment with VPA and clofarabine resulted in cooperative induction of γH2AX, which was both clofarabine and VPA concentration-dependent (Figure 3). This was accompanied by cooperative induction of apoptosis determined by flow cytometry analysis and cleavage of caspase-3 and PARP, and activation of Bax in the cells (Figures 4A–E). Further, the levels of γH2AX significantly correlated with apoptosis (r= 0.93 and p <0.0001, Figure 4F). These results strongly suggest that VPA augments clofarabine-induced DNA DSBs which trigger apoptosis in Kasumi-1 cells.
Figure 3. Cooperative induction of DNA DSBs by clofarabine and VPA in Kasumi-1 cells.

Kasumi-1 cells were treated with variable concentrations of VPA and fixed concentration of clofarabine (Panel A) or variable concentrations of clofarabine and fixed concentration of VPA (Panel B) alone or in combination for 72 h. Whole cell lysates were extracted and subjected to Western blotting probed by anti-γH2AX or -actin antibody. C, clofarabine; V, VPA.
Figure 4. Cooperative induction of apoptosis by clofarabine and VPA in Kasumi-1 cells.

Panels A–D: Kasumi-1 cells were treated with variable concentrations of VPA and fixed concentration of clofarabine (Panels A&B) or variable concentrations of clofarabine and fixed concentration of VPA (Panels C&D) alone or in combination for 72 h. Early and late apoptotic events in the cells post drug treatments were determined by annexin V/PI staining and flow cytometry analysis (Panels A&C). Whole cell lysates were extracted and subjected to Western blotting probed by anti-cleaved caspase-3 (CF casp-3), -PARP, or -actin antibody (Panels B&D). Panel E: Whole cell lysates from Kasumi-1 cells treated with clofarabine and VPA, alone or combined for 72 h, were subjected to immunoprecipitation with the Bax 6A7 (active Bax) or B9 (total Bax) antibody, as described in the Materials and Methods. The immunoprecipitated proteins were subjected to Western blotting probed by anti-Bax or -actin antibody. Panel F: The relationship between the levels for γH2AX and apoptosis in Kasumi-1 cells treated with clofarabine and VPA, alone or combined, were determined by the Pearson test. CF, cleaved form; Clof, clofarabine; ** indicates p<0.005.
DISCUSSION
In our previous study, we demonstrated global synergistic antileukemic activities of combined VPA/cytarabine in pediatric AMLs and suggested that VPA could be an attractive agent for combination therapies of this deadly disease [25]. In the present study, we investigated whether combining the novel nucleoside analog clofarabine with VPA could be an effective treatment option for children with AML.
Our initial results demonstrated synergistic antileukemic activities of combined VPA and clofarabine in a clinically-relevant pediatric AML cell line, Kasumi-1. Interestingly, this drug synergy was independent of the sequence of drug administration. We then expanded our study to include additional pediatric AML cell lines and diagnostic blasts from children with de novo AML. Consistent with our previous study [25], global synergistic results with clofarabine and VPA were obtained in the diagnostic blasts. In the additional cell lines (MV4-11, CMS, and THP-1), synergistic-to-additive and additive effects were observed in the MV4-11 and CMS cells, respectively, while additive-to-antagonistic effects were detected in the THP-1 cells (Table II). These results are different from our previous study which showed synergistic effects of combined VPA and cytarabine in THP-1 cells. The molecular mechanism underlying this difference is unknown. Nonetheless, it may reflect the different mechanisms of action between clofarabine and cytarabine in pediatric AML cells.
Of particular interest, responses of pediatric AML cells to the combination of clofarabine and VPA seemed to reflect the sensitivities of the cells to VPA (Figure 2). In addition, VPA-sensitive pediatric AML cells also showed significantly greater median fold change of cytarabine IC50s than that of VPA-resistant cells when cytarabine was combined with VPA at 0.5 mM or lower concentrations (16.7- and 1.8-fold, respectively, p=0.008) [25]. Clinically, it would be difficult to estimate which pediatric AMLs are sensitive or resistant to the combination of VPA and clofarabine or cytarabine. Our results suggest, however, that sensitivities to VPA could be biomarkers for predicting responses of pediatric AML to this drug combination. In relation to this, we found that t(8;21) AML cases were uniformly sensitive to VPA, and so to the combination of VPA and clofarabine or cytarabine. This was not unexpected, given that several fusion proteins, such as AML1-ETO, recruit nuclear corepressor complexes, which contain HDACs, [34]. Thus, AML cases harboring these fusion genes might be preferentially susceptible to VPA and its combinations with clofarabine or cytarabine. Additional studies are warranted to determine the molecular basis underlying the differential responses of pediatric AML cells to VPA.
Our laboratory has previously showed that induction of apoptosis along with DNA DSBs was a major mechanism responsible for the anti-leukemic synergism between cytarabine and VPA [25]. Interestingly, this also seems the major molecular mechanism underlying the synergistic antileukemic interactions between clofarabine and VPA in pediatric AML cells. However, the molecular mechanisms by which VPA cooperates with clofarabine/cytarabine in inducing DNA DSBs and apoptosis remain unknown. Recent studies with solid tumor cell lines suggested that HDACIs can suppress the expression of DNA DSB repair genes, such as RAD51 and BRCA1 [7]. This could be the molecular basis underlying the synergy between VPA and clofarabine or cytarabine. Studies are underway investigating this possibility and results will be reported elsewhere.
Collectively, our results demonstrate synergistic antileukemic interactions between clofarabine and VPA in VPA-sensitive pediatric AML cells. Thus, children with VPA-sensitive AML may benefit from combination therapies involving VPA and clofarabine.
Acknowledgments
This study was supported by a Start-up Fund from the Barbra Ann Karmanos Cancer Institute, grants from the St. Baldrick’s Foundation, American Cancer Society (IRG #11-053-01-IRG), the Herrick Foundation, Children’s Research Center of Michigan, Leukemia Research Life, National Cancer Institute (CA120772), Elana Fund, Justin’s Gift Charity, the Buric Family, Sehn Family Foundation, and the Ring Screw Textron Endowed Chair for Pediatric Cancer Research.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors have no conflict of interest to disclose.
References
- 1.Manola KN. Cytogenetics of pediatric acute myeloid leukemia. European Journal of Haematology. 2009;83(5):391–405. doi: 10.1111/j.1600-0609.2009.01308.x. [DOI] [PubMed] [Google Scholar]
- 2.Meshinchi S, Arceci RJ. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist. 2007;12(3):341–355. doi: 10.1634/theoncologist.12-3-341. [DOI] [PubMed] [Google Scholar]
- 3.Kaspers GJL, Zwaan CN. Pediatric acute myeloid leukemia: towards high-quality cure of all patients. Haematologica-the Hematology Journal. 2007;92(11):1519–1532. doi: 10.3324/haematol.11203. [DOI] [PubMed] [Google Scholar]
- 4.Jeha S, Gandhi V, Chan KW, et al. Clofarabine, a novel nucleoside analog, is active in pediatric patients with advanced leukemia. Blood. 2004;103(3):784–789. doi: 10.1182/blood-2003-06-2122. [DOI] [PubMed] [Google Scholar]
- 5.Abujamra AL, dos Santos MP, Roesler R, et al. Histone deacetylase inhibitors: A new perspective for the treatment of leukemia. Leukemia Research. 2010;34(6):687–695. doi: 10.1016/j.leukres.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 6.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nature Reviews Drug Discovery. 2006;5(9):769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 7.Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert opinion on investigational drugs. 2010;19(9):1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonate PL, Arthaud L, Cantrell WR, Jr, et al. Discovery and development of clofarabine: a nucleoside analogue for treating cancer. Nat Rev Drug Discov. 2006;5(10):855–863. doi: 10.1038/nrd2055. [DOI] [PubMed] [Google Scholar]
- 9.Faderl S, Gandhi V, Kantarjian HM. Potential role of novel nucleoside analogs in the treatment of acute myeloid leukemia. Curr Opin Hematol. 2008;15(2):101–107. doi: 10.1097/MOH.0b013e3282f46e94. [DOI] [PubMed] [Google Scholar]
- 10.Faderl S, Gandhi V, Keating MJ, et al. The role of clofarabine in hematologic and solid malignancies--development of a next-generation nucleoside analog. Cancer. 2005;103(10):1985–1995. doi: 10.1002/cncr.21005. [DOI] [PubMed] [Google Scholar]
- 11.Kantarjian HM, Jeha S, Gandhi V, et al. Clofarabine: past, present, and future. Leuk Lymphoma. 2007;48(10):1922–1930. doi: 10.1080/10428190701545644. [DOI] [PubMed] [Google Scholar]
- 12.Jeha S, Gaynon PS, Razzouk BI, et al. Phase II study of clofarabine in pediatric patients with refractory or relapsed acute lymphoblastic leukemia. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24(12):1917–1923. doi: 10.1200/JCO.2005.03.8554. [DOI] [PubMed] [Google Scholar]
- 13.Faderl S, Gandhi V, O’Brien S, et al. Results of a phase 1–2 study of clofarabine in combination with cytarabine (ara-C) in relapsed and refractory acute leukemias. Blood. 2005;105(3):940–947. doi: 10.1182/blood-2004-05-1933. [DOI] [PubMed] [Google Scholar]
- 14.Faderl S, Ravandi F, Huang X, et al. A randomized study of clofarabine versus clofarabine plus low-dose cytarabine as front-line therapy for patients aged 60 years and older with acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood. 2008;112(5):1638–1645. doi: 10.1182/blood-2007-11-124602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faderl S, Verstovsek S, Cortes J, et al. Clofarabine and cytarabine combination as induction therapy for acute myeloid leukemia (AML) in patients 50 years of age or older. Blood. 2006;108(1):45–51. doi: 10.1182/blood-2005-08-3294. [DOI] [PubMed] [Google Scholar]
- 16.Kantarjian H, Gandhi V, Cortes J, et al. Phase 2 clinical and pharmacologic study of clofarabine in patients with refractory or relapsed acute leukemia. Blood. 2003;102(7):2379–2386. doi: 10.1182/blood-2003-03-0925. [DOI] [PubMed] [Google Scholar]
- 17.Kantarjian HM, Gandhi V, Kozuch P, et al. Phase I clinical and pharmacology study of clofarabine in patients with solid and hematologic cancers. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2003;21(6):1167–1173. doi: 10.1200/JCO.2003.04.031. [DOI] [PubMed] [Google Scholar]
- 18.Karp JE, Ricklis RM, Balakrishnan K, et al. A phase 1 clinical-laboratory study of clofarabine followed by cyclophosphamide for adults with refractory acute leukemias. Blood. 2007;110(6):1762–1769. doi: 10.1182/blood-2007-03-081364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond DC, Noble CO, Kirpotin DB, et al. Clinical development of histone deacetylase inhibitors as anticancer agents. Annual Review of Pharmacology and Toxicology. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 20.Ellis L, Pan Y, Smyth GK, et al. Histone deacetylase inhibitor panobinostat induces clinical responses with associated alterations in gene expression profiles in cutaneous T-cell lymphoma. Clinical Cancer Research. 2008;14(14):4500–4510. doi: 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- 21.Kelly WK, O’Connor OA, Krug LM, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. Journal of Clinical Oncology. 2005;23(17):3923–3931. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, et al. Valproic acid as epigenetic cancer drug: Preclinical, clinical and transcriptional effects on solid tumors. Cancer Treatment Reviews. 2008;34(3):206–222. doi: 10.1016/j.ctrv.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 23.Gottlicher M, Minucci S, Zhu P, et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. Embo Journal. 2001;20(24):6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cimino G, Lo-Coco F, Fenu S, et al. Sequential valproic acid/all-trans retinoic acid treatment reprograms differentiation in refractory and high-risk acute myeloid leukemia. Cancer Research. 2006;66(17):8903–8911. doi: 10.1158/0008-5472.CAN-05-2726. [DOI] [PubMed] [Google Scholar]
- 25.Xie C, Edwards H, Xu X, et al. Mechanisms of synergistic antileukemic interactions between valproic acid and cytarabine in pediatric acute myeloid leukemia. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16(22):5499–5510. doi: 10.1158/1078-0432.CCR-10-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu X, Xie C, Edwards H, et al. Inhibition of histone deacetylases 1 and 6 enhances cytarabine-induced apoptosis in pediatric acute myeloid leukemia cells. PloS one. 2011;6(2):e17138. doi: 10.1371/journal.pone.0017138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeha S, Gandhi V, Chan KW, et al. Clofarabine, a novel nucleoside analog, is active in pediatric patients with advanced leukemia. Blood. 2004;103(3):784–789. doi: 10.1182/blood-2003-06-2122. [DOI] [PubMed] [Google Scholar]
- 28.Su JM, Li XN, Thompson P, et al. Phase 1 study of valproic acid in pediatric patients with refractory solid or CNS tumors: a children’s oncology group report. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17(3):589–597. doi: 10.1158/1078-0432.CCR-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tallarida RJ. Drug synergism: its detection and applications. J Pharmacol Exp Ther. 2001;298(3):865–872. [PubMed] [Google Scholar]
- 30.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological Reviews. 2006;58(3):621–681. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
- 31.Edwards H, Xie CZ, LaFiura KM, et al. RUNX1 regulates phosphoinositide 3-kinase/AKT pathway: role in chemotherapy sensitivity in acute megakaryocytic leukemia. Blood. 2009;114(13):2744–2752. doi: 10.1182/blood-2008-09-179812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ge Y, Dombkowski AA, LaFiura KM, et al. Differential gene expression, GATA1 target genes, and the chemotherapy sensitivity of Down syndrome megakaryocytic leukemia. Blood. 2006;107(4):1570–1581. doi: 10.1182/blood-2005-06-2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ge Y, Stout ML, Tatman DA, et al. GATA1, cytidine deaminase, and the high cure rate of Down syndrome children with acute megakaryocytic leukemia. J Natl Cancer Inst. 2005;97(3):226–231. doi: 10.1093/jnci/dji026. [DOI] [PubMed] [Google Scholar]
- 34.Berman JN, Look AT. Targeting transcription factors in acute leukemia in children. Current drug targets. 2007;8(6):727–737. doi: 10.2174/138945007780830818. [DOI] [PubMed] [Google Scholar]