

Abstract
Tiny marine animals represent an untapped reservoir for undiscovered, bioactive natural products. However, their small size and extreme chemical variability preclude traditional chemical approaches to discovering new bioactive compounds. Here, we use a metagenomic method to directly discover and rapidly access cyanobactin class natural products from these variable samples, providing proof-of-concept for genome based discovery and supply of marine natural products. We also address practical optimization of complex, multistep ribosomal peptide pathways in heterologous hosts, which is still very challenging. The resulting methods and concepts will be applicable to ribosomal peptide and other biosynthetic pathways.
Keywords: marine natural products, symbiosis, nonribosomal, ribosomal peptide, drug discovery
Introduction
The future of natural products as a discipline depends upon providing new compounds for pharmaceutical and other practical applications. A major limitation of current methods is that the vast majority of marine natural products exist in samples that are far too small for conventional chemical and pharmaceutical methods. For example, the limit of current chemical techniques for compound discovery is ~10 μg.[1] However, this amount of material does not approach the necessary requirement for drug discovery or biological studies.
Marine invertebrate secondary metabolites vary both between and within species, forming large groups of bioactive natural products (Figure 1).[2, 3] This extreme variation has been both a boon and a curse to marine natural products research. It has allowed the identification of many new, potent chemicals from similar species, but it has also meant that resupply is difficult, since there is no guarantee that a compound will be found in the same animal in the same place from year to year. For example, cyanobactin natural products are highly variable even within seemingly identical ascidians, leading to discovery of many new and potent variants.[4] However, a preclinical anticancer lead cyanobactin, trunkamide, has only been found a few times in ascidians, necessitating a complex total synthesis and an expensive supply of the compound.[5-7] Mollamides are anticancer lead cyanobactins that were isolated as milligrams from 10 kilograms of ascidians.[8] As a chemically unrelated example, patellazoles are very potent anticancer polyketides from ascidians, but they cannot be developed because they are rarely found, even in the same location from year to year.[9-11] This variability promises an almost unlimited source of new bioactive marine natural products. Most marine invertebrates are too small to allow isolation of sufficient compound for drug discovery, and yet these tiny animals contain many previously undiscovered metabolites. Therefore, development of techniques to both discover and supply compounds for biological assay is required in order to fully exploit the true promise of marine animal natural products.
Figure 1. Compound variation in tiny ascidian colonies.

Small, adjacent ascidian colonies such as those shown above can contain different and highly variable natural products. These products represent an amazing untapped chemical diversity for further discovery. In this study, we report the first direct metagenomic discovery and supply of marine natural products, using the exact sample shown above, which is much smaller than a US quarter. (Wild ascidian photo: Chris Ireland, University of Utah).
In the course of our studies on cyanobactin ribosomal peptide biosynthesis, we noticed that even neighboring, otherwise identical colonies of ascidian animals contain very diverse natural products, including new compounds that were discovered by solely genetic methods.[12] We previously showed that cyanobactin natural products are made by symbiotic bacteria and not by the animal hosts, and we were able to express cyanobactin genes to known natural products functionally in E. coli.[12-15] Although we could discover new compounds using this method, it was very difficult to supply these compounds for development, leaving a significant gap in the use of metagenomics for marine natural products discovery. Our previous work only provided <100 μg per liter of culture in individual experiments. Much more importantly, these yields were variable, and quite commonly expression would completely fail in individual runs. This problem was worse for engineered natural products, which suffered from even further diminished yields. For example, we synthesized a non-natural product, eptidemnamide, using heterologous expression in E. coli. It required more than 100 individual fermentation experiments and very precise analytical techniques to reproducibly detect this compound in culture, even at a very minimal level. Because of low and variable yields, all expression experiments also required purified standard compounds in order to validate heterologous production. Clearly, although this method provided an excellent means for understanding the science of natural products, it was not ideal for direct discovery and practical supply of compounds and was substantially more difficult than traditional chemical methods.
This problem is not unique to our system; in fact it is a general property of natural products and especially ribosomal peptides, for which very few heterologous expression tools exist. As one of numerous examples, the Kelly lab recently reported the technical difficulties in heterologously expressing the related ribosomal pathway to thiostrepton and the ultimate need for tedious genetic manipulations to allow the production of derivatives even within the native host, much less in heterologous systems.[16] This solution to the problem is not an option in the case of cyanobactins as the native producer is not yet stably cultivable. Therefore, we decided to systematically explore the expression of cyanobactins in order to obtain a robust, reproducible, and technically simple platform for heterologously expressing diverse cyanobactins in culture (Figure 2). Here, we describe development of new pathway optimization methods that can be applied to ribosomal peptides and other compound classes. We then used these new methods to discover and produce a new marine “animal” natural product, minimide. While it is increasingly common to discover biosynthetic genes in marine animals or to detect new compounds in purified bacteria using genomic methods, there are no other examples in which a marine animal natural product is both discovered and supplied using genetic methods. Therefore, this represents a proof-of-concept for a new mode of marine invertebrate natural products discovery, being the first direct genetic route to supplying new compounds (Figure S1 in the Supporting Information).
Figure 2. Substrate permissivity of the tru pathway.
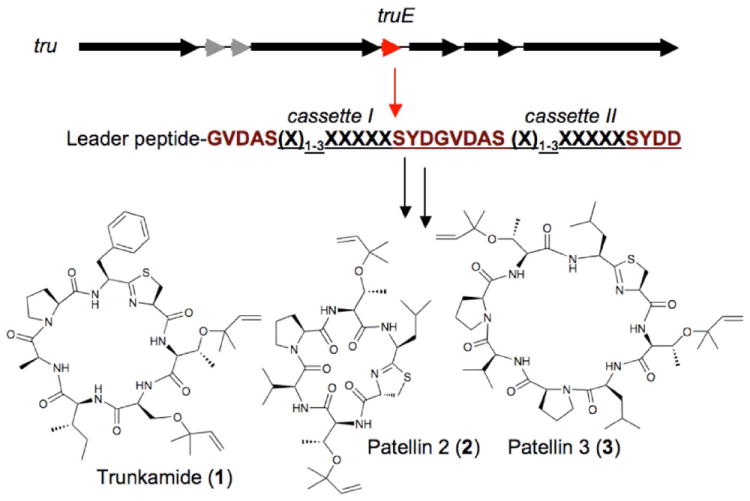
The ~11 kbp tru pathway (top) encodes five probable enzymes, two other proteins, and the TruE precursor peptide (gene in red, sequence shown below). In the TruE peptide, enzyme recognition sequences (brown) flank degenerate sequences of 6-8 amino acids (underlined) that directly encode natural products, such as 1-3 at bottom (Figure S2 in the Supporting Information). By manipulating these sequences, new and known natural products can be produced.
The model for these studies was Didemnum molle, a ubiquitous ascidian that forms small colonies dotting diverse tropical marine habitats. These colonies can fuse and divide in the course of days,[17] and yet individual colonies contain different natural products, including potentially different cyanobactins. However, most D. molle colonies are very small with a diameter of 0.5-1 cm (Figure 1), and they are embedded in mucus, making genetic manipulation (or even DNA isolation) difficult. Also, because each colony is an individual with different cyanbactin composition, it is challenging to chemically or genetically define these minuscule samples. We were particularly interested in rare prenylated cyanobactins that are variants of the potently active anticancer metabolite, trunkamide. Trunkamide variants are synthesized by the tru pathway where the precursor peptide, truE, encodes two hypervariable regions (Figures 2 and S2 in the Supporting Information).[14]
Results and Discussion
Discovery of Minimide
We obtained a large collection of D. molle ascidians from across the tropical Pacific over a period of years. We then used PCR based on truE, to screen our D. molle sample collection for new cyanobactins. For these very small samples, we developed an “ascidian colony” PCR strategy using unpurified DNA from ground small colonies. We identified a new precursor peptide from a tiny D. molle sample, which was predicted to encode a new cyanobactin with the primary sequence, TLATIC. This ascidian was extracted with methanol, and a tiny amount of the putative mature peptide product, dubbed “minimide”, was observed by mass spectrometry. To further confirm that this ion corresponded to the predicted compound (4), we employed a method that we previously developed for prenylated cyanobactins.[12] This Fourier transform-ion cyclotron resonance (FT-ICR) MS/MS method provides high-resolution analysis for the molecular ion. In addition, it specifically eliminates reverse prenyl modifications that are found on tru-pathway cyanobactins, providing high-resolution daughter ions that firmly validate structures. In this case, ions corresponding to loss of both 1 and 2 isoprene units were observed at high resolution, confirming the identity of the compound (Figures 3 and S3 in the Supporting Information). Minimide was present in <10 μg amounts in a single ascidian colony, precluding use of such compounds in drug discovery or development.
Figure 3. Discovery of minimide (4).
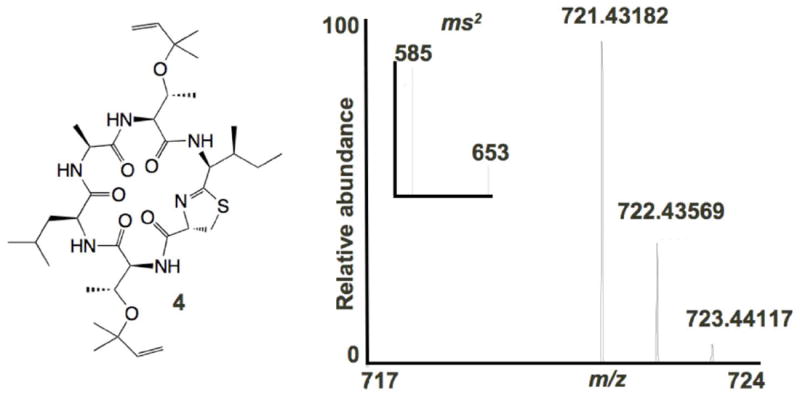
From small, wild D. molle ascidians such as the one shown in Figure 1, a sequence for a new compound minimide (4) was identified. The colony was then extracted. The resulting LC-FT-MS and MS2 spectra match the predicted structure (Figure S3 in the Supporting Information).
Optimization of Expression Conditions
We wished to actually supply this marine invertebrate natural product, not just to observe it once in a tiny animal. Our previous expression method was simply not consistent enough to rapidly supply this compound, so we sought first to optimize the method, rather than struggling for months to express the pathway as in our previous experience.
Previously, we cloned each pat gene into E. coli under the control of a separate T7 promoter.[13] However, yields were low, and more critically yields were quite variable due to accumulating mutations in the expression plasmids. In this work, we extended the same methodology in an attempt to express the tru pathway in E. coli, and we also attempted numerous related methods, all of which led to complete failure to observe heterologous compounds (summarized in Table S1 and Figure S4 in the Supporting information). Previously, we reported production of trunkamide using the lac promoter, although this was unstable and highly variable.[14] Therefore, we attempted to optimize this expression system to achieve greater stability and to understand production conditions.
We required a method to quickly examine the expression levels of the tru operon. We used yeast recombination to generate a green fluorescent protein (gfp) C-terminal fusion to truG, the last gene in the operon (Figure 4 and Figure S5 in the Supporting information). As a control, we also generated a gfp construct directly downstream of the lac promoter in the same vector backbone. In tens of experiments in solid and liquid media, we examined effects of media, cell line, genetic background, and additives on the operon expression via fluorescence. Using this method, we also noted that following transformation about 1% of the colonies appear to be highly fluorescent, while the rest are fluorescent to a lesser extent. Closer analysis of the high-fluorescence clones showed that they result from plasmid rearrangements. Based upon this result and further chemical analysis, we now either analyze individual colonies prior to expression or pick 5-10 individual E. coli colonies to seed each individual expression experiment.
Figure 4. Optimization of expression using gfp fusion.
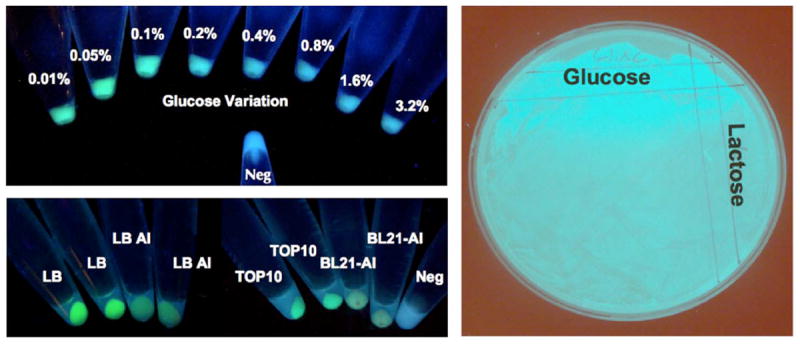
gfp was fused to the C-terminus of truG, the last gene in the pathway. Various media conditions and cell lines in solid and liquid media are shown. This method provided a rapid and conveniently simple readout of pathway expression.
To examine whether the operon is under the tight control of the lac promoter, the gfp fusion construct was introduced in different lac-repressing conditions. Both genetic and chemical repressors were shown to abolish tru expression. For example, BL21-DE3 cell lines contain a chromosomal copy of the lac repressor (lacI), which reduces tru operon expression. Both plasmid-encoded lacI and chemical repressors, such as high concentrations of glucose, effectively repress the operon. Surprisingly, at lower concentrations (0.05%), glucose seems to increase the operon expression (Figure 4). Although lactose should induce lac promotion, it did not affect tru operon expression. We next examined the effect of different E. coli cell lines, using strains TOP10, DH5a, BL21-DE3 and BL21-AI. TOP10 gave the greatest observable fluorescence. Finally, we tried different media for the expressions in solid or liquid state, including marine agar, ISP2, R2A, LB, and autoinducing media. No clear difference in fluorescence was observed except when comparing autoinducing media to LB in liquid culture where LB gave much more fluorescence. Most of these experiments were repeated using the non-fusion operon and analyzed for cyanobactin production. Generally, the fluorescence results were consistent with the chemical results, further validating this tool.
As an outcome of these experiments, we greatly improved the stability of this system, with reproducible, similar expression levels from run to run. We used the lessons learned to guide further experiments.
Pathway Engineering
We next turned our attention to developing convenient engineering strategies for this robust operon system. Although yeast recombination is an efficient technology for engineering large pieces of DNA, it has much lower throughput than regular cloning methods. We designed a double-plasmid system, containing the tru or pat operon on one plasmid and a second “external” copy of the precursor peptide in the pRSFDuet vector (Table S1 and Figure S4 in the Supporting information). This system provides higher expression levels of the precursor peptide, it keeps the operon constant in all experiments, and it is easy to manipulate. Moreover, it provides consistent production of known cyanobactins that serve as internal positive controls.
After a series of failed experiments in BL21-based cell lines using arabinose-T7 promoters, and relying on our fluorescence results in E. coli, we decided to use TOP10 as the host for this double-plasmid system. We inserted a lac promoter and deleted the lacI gene in pRSF-truE, allowing both the operon copy and the external copy of the precursor peptide to be controlled by the lac promoter. In hundreds of E. coli-based expression experiments, compounds encoded on the internal (operon) and external (pRSF) copies of the precursor peptide could be detected by mass spectrometry. This represented a significant advance over previous systems, where even from run to run we observed production changes.
Finally, this robust and reproducible system was subjected to media optimization. Four different rich media were compared for cyanobactin production (Figure S6 in the Supporting information), leading to selection of 2XYT as the production medium.
To determine the yield and scope of this system, we designed vectors wherein known compounds patellin 3 (3) and 2 (2) were encoded on the operon-copy of truE while patellin 3 (3) and trunkamide (1) were encoded on the external copy (Figure 5A). As expected, the three compounds were readily detectable in the E. coli extract (Figure 5C). It is notable that in this experiment identical genes produce three prenylated cyanobactins of different sizes and sequences (hexapeptide, heptapeptide and octapeptide), indicating the broad substrate specificity of the processing enzymes. This experiment was repeated 8 times using different culture volumes (50-10,000 mL). In all cases, we detected the three cyanobactins in very similar yields and ratios, representing a significant improvement over our previous process.
Figure 5. Optimized expression system.
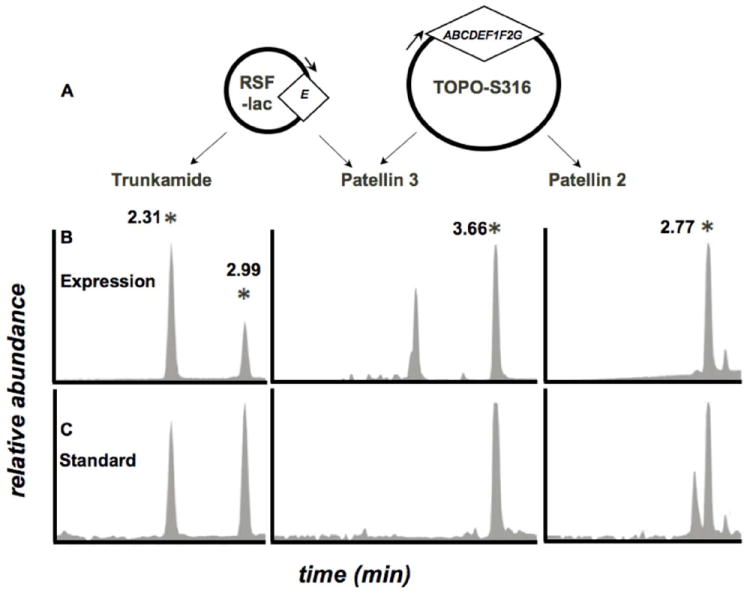
Expression of compounds 1-3 was established and quantified by LC-MS. A. The dual expression system, with trunkamide and patellin 3 encoded on a modified pRSF vector and patellins 2 and 3 encoded on the operon. B. Filtered LC-MS traces showing authentic standards of 1-3. The numbers indicate time, in minutes, of the elution of each compound. C. Filtered traces of an E. coli expression producing compounds 1-3. Trunkamide is present as 2 peaks because it undergoes racemization at Phe.
In representative experiments, the consistent yield of recombinant trunkamide was measured as 9.9 ± 1.0 μg L-1. By assuming that the similar compounds 2 and 3 had similar ionization efficiencies, a total of 13.7 μg of 2 and 10.3 μg of 3 was estimated, for a total of ~34 μg L-1 of cyanobactins. In occasional experiments, the yield was >1 mg L-1. By contrast, in previous work our occasional best level produced ~10-fold less material. However, the major advantage was in the ability to now produce new derivatives in a controlled environment. Previously, expression experiments sometimes worked and sometimes did not, making it impossible for us to tell whether heterologous experiments truly failed or if there was variation in production in the individual run.
Expression of minimide
With the dual vector system, we met the two main criteria for an efficient heterologous expression system: reproducible and robust yield and an easily manipulable system. We then functionally expressed the new ascidian natural product, minimide, in this system and confirmed that the heterologous E. coli host did indeed produce the new product (Figure 6). Importantly, this positive expression result was obtained on the first try and repeated to confirm its reproducibilty. This is much different than our previous experience with engineering this system where hundreds of experiments had to be performed to confirm the expression of the new compounds, further validating that the system had been indeed greatly optimized. Moreover, we had not previously produced any newly discovered ascidian natural products in E. coli.
Figure 6. MS analysis of minimide production.
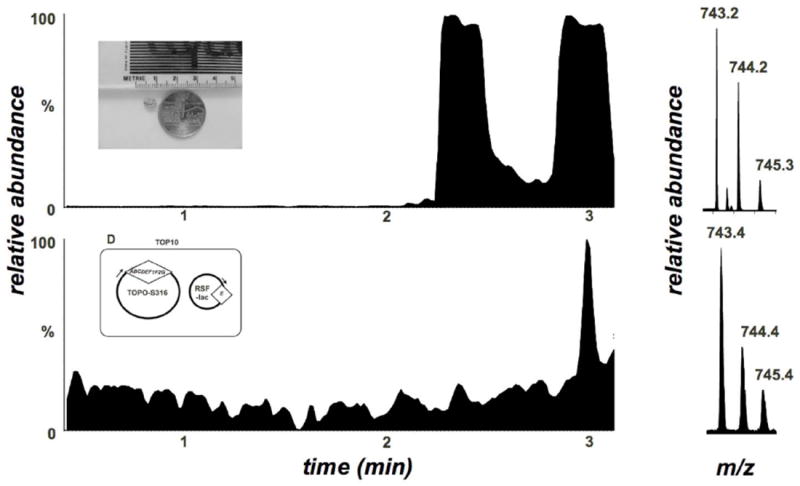
Filtered HPLC-ESIMS spectra of minimide expression in E. coli vs. the natural products. Two isomers are found in the natural product due to slow epimerization adjacent to the thiazoline residue, as found in natural trunkamide (Figure 5). Only a single isomer is isolated from E. coli expression, since the expression takes place over days instead of months or years, as found in the ascidian.
Conclusion
Marine animals contain an enormous diversity of undiscovered natural products. Because most of these likely exist in animals that are too small to be compatible with modern drug discovery, direct genomic tools to compound discovery and supply are desirable. Here, we report the first application of genomic technology for this purpose. Moreover, it is the first application of metagenomics to the discovery and supply of new natural products that have actually been shown to exist in their natural environment. Therefore, this represents a proof-of-concept for a new discovery method that bypasses traditional grind-and-find.
The methods described here will be directly applicable to expression and rapid engineering of ribosomal peptide pathways, which are ubiquitous bioactive components of bacteria and are important drug leads.[18] Recently, ~50 derivatives of the more complex ribosomal peptides such as thiocillin have been made in the native host, building on a decade of elegant work that includes hundreds of derivatives in the simpler lantibiotic groups.[19-21] By contrast, heterologous expression has been difficult for complex, multistep pathways.[16] Indeed, even for the one- or two-step lantibiotic pathways, optimization in E. coli was not reported until relatively recently despite more than a decade of study.[22] Therefore, tools for rapid optimization of ribosomal peptide biosynthesis are urgently needed to enable progress in this growing field. Here, we report a rapid method that is directly applied in E. coli, representing the first route to optimize a complex, multistep ribosomal peptide pathway in a heterologous host. Although the average yield reported here is modest, achievement of robust consistency allows standard metabolic engineering methods to be easily applied, whereas beforehand such methods would be extremely laborious.
A few research groups have identified complete biosynthetic gene clusters from symbionts of marine invertebrates; however, pathway expression has been problematic except in the cyanobactins case.[23, 24] We hope that the experiments described here will represent failure and success examples that will help to accelerate this process for more complex pathways. The engineering described here is especially applicable to ribosomal peptide pathways, but it also highlights the main issues likely to face any natural product optimization, and key steps will be useful for many different pathway types. In our view, the main difference between the ribosomal peptide groups and other pathways lies not in the difficulty of expression and optimization, but rather in the relative difficulty in genetic manipulation of the larger polyketide and nonribosomal peptide genes. It required numerous gene manipulations and more than 1000 individual experiments to optimize the cyanobactin pathways, which previously would not be practical for the larger polyketide and nonribosomal pathways. However, as these manipulations become simpler and cheaper, the optimization strategy described here will become more broadly practical.
Experimental Section
Vector synthesis
PCR experiments were performed using Taq HiFi DNA Polymerase (Invitrogen). PCR experiments were always run with a negative control (sterile water) and a positive control. Products were viewed by agarose gel electrophoresis and bands needed were cut and gel extracted using the QIAquick Gel Extraction Kit (Qiagen) or TOPO-XL cloning kit (Invitrogen). To obtain genes from D. molle ascidians, individual colonies of D. molle were separated and saved in RNAlater (Amersham). Each colony was gently squeezed to release Prochloron cells. Prochloron cells (10 μl) were added to sterile DMSO (200 μl; sterilized by UV treatment for 15 min). The mixture was then ground in a microcentrifuge tube using a small plastic tissue grinder. Serial dilutions (1/10x and 1/100x) of this original solution were made in sterile water. PCR was performed as above except with the addition of a 1/10 dilution of betaine solution (16 μl of 6.5 M betaine; 0.5 μl DMSO) that was UV-sterilized before use. Purified PCR products were cloned into the desired vector using one of two methods: 1. Digestion with the appropriate restriction enzymes and ligation directly to the digested desired vector; 2. Blunt cloning into the vector pCR2.1-TOPO (Invitrogen) following the manufacturer’s protocol. Transformants were spread on plates with appropriate antibiotics and grown at 30 °C. Viable transformants were screened by restriction digest and sequencing to ensure that mutation-free plasmids were obtained in all cases. Selection at >30 °C usually did not yield any plasmids with intact inserts, probably due to a rearrangement problem. Top 10 or DH5α E. coli cells (Invitrogen) were used for vector propagation. For a list of constructs, see Table S2 in the Supporting Information. To delete lacI, pRSFDuet-truE1 was digested with BssHII and purified on a 1% agarose gel. The fragment lacking lacI was purified and self-ligated to yield the plasmid pRSF-truE1-DlacI.
Yeast recombination to generate gfp fusions
The plasmid TOPO-E1-S316 was described previously and contains the tru pathway in an E. coli-yeast shuttle vector.[14] Linear pieces required for the homologous recombination were generated and transformed into yeast strain BY4741 (Research Genetics). The resulting vector was rescued from the yeast cells into E. coli Top 10 strain (Invitrogen) and validated by restriction digests. Yeast transformation and plasmid rescue were performed following standard procedures.[25] pGFP-UV vector (Clontech) was used as a template for PCR. The resulting fragment contains homologous ends to the region of interest in the plasmid TOPO-E1-S316. Plasmid TOPO-E1-S316 was linearized by digestion with the restriction enzyme NotI. The two linear fragments were transformed to yeast and the resulting constructs were rescued into E. coli. Restriction digests and sequencing reactions confirmed the fusion of gfp to truG, the last gene in the tru operon. A lac-gfp control lacking the operon was synthesized by a similar method.
Heterologous expression
Several E. coli cell lines were used in optimization experiments (Top10, BL21-DE3, BL21-AI and DH5α (Invitrogen)). Top10 was selected for constitutive expression of the operon via the lac promoter and used for the majority of the successful expressions. Cell lines harboring the desired plasmid(s) were constructed by chemical transformation (following the manufacturer protocol) or electroporation. Fresh colonies (from glycerol stock streaks or fresh transformations) were picked and seed cultures were grown overnight at 30 °C in LB with appropriate antibiotics. For shake flasks, seed culture (1 mL) was used to inoculate medium (1 L) with antibiotics in Fernbach baffled flasks. Seed cultures originating from 5-10 different transformed colonies were mixed and maintained for 5 days at 30 °C in a rotary shaker to allow accumulation of products. Controls consisted of identical expression and extraction conditions. Positive controls employed strains known to produce cyanobactins in previous experiments. Negative controls employed strains harboring various other vectors with inserts modified to produce no products. In nearly all cases, identical experiments were repeated more than once for confirmation of the reproducibility of the system. For example, the optimized trunkamide expression was repeated 8 times and analyzed by HPLC-ESIMS using different instruments and columns. Expected products were observed in the replicates and never in negative controls lacking the correct genetic elements. In fermentation experiments, seed culture in LB (50 mL) with the appropriate antibiotics were transferred to media (1 or 10 L) with the same antibiotics. Fermentation was run for ~36 h at 30 °C with agitation at 500 rpm using a BIOFLO 110 fermentor. In the case of experiments with plasmids harboring inducible promoters, cells were grown to OD600 ~ 0.4-0.6 then induced with the relevant inducer. In BL21-DE3 cell lines, IPTG was used for induction of expression at a concentration of 1 mM. In BL21-AI cell lines, arabinose was used for induction of expression at a concentration of 0.2%.
Chemical analysis
Cultures were centrifuged at 5000 rpm for 10 min and the broth and pellet were separated and treated independently. The broth was subjected to chemical extraction by shaking for about 30 min with diaion HP20 or HP20SS resins (SUPELCO). The resin was then washed with water (500 ml) and eluted with 100% methanol (250 ml). The methanol fraction was dried by rotary evaporation and the remaining residue was dissolved in water (30 ml). The water fraction was then extracted three times with equal volumes of ethyl acetate. The organic extracts were combined, dried by rotary evaporation and fractionated by reverse phase C18 chromatography using a 3-step gradient (100% water, 25% acetonitrile in water and 100% acetonitrile). Fractions of 100% water and 25 % acetonitrile in water were discarded while the 100% acetonitrile fraction was dried and used further for MS. The procedure was scaled up proportionally for larger fermentation experiments. To extract cells, pellets were sonicated 3 times (1 h each) in 100-200 ml HPLC methanol (Fischer) depending on the pellet size. The combined extracts were then filtered through a porcelain funnel (150 ml × 60F, Kimax). The methanol filtrate was then treated as described for the methanol broth extracts. For further analysis, samples were dissolved in 1:1 water : acetonitrile and injected on a Waters Alliance 2795 HPLC, using an analytical C18 column (Phenomenex) and a gradient of acetonitrile (aqueous, 1% formic acid) was applied. For high-resolution HPLC mass spectrometry (HPLC-HRESIMS), a Micromass Q-TOF mass spectrometer was used with a LockSpray running concurrently to ensure higher mass accuracy. For low-resolution HPLC mass spectrometry (HPLC-LRESIMS), a Quattro II mass spectrometer was used. All analyses were done in positive ion mode. In some experiments, HPLC-HRESIMS was performed using a UPLC system (Waters) connected to an ESI-TOF Micromass mass spectrometer. All known compounds were identified by comparison with available authentic standards, while new compounds were validated by FT-ICR MS/MS.
Whole ascidian animal sample 06-017 was collected at Faila Island, Solomon Islands (S 9° 3.03’ E 159° 15.03’) and frozen until further analysis. Two single colonies were extracted twice with methanol. The dried extracts were combined, desalted on a C18 column, dissolved in 1:1 water : acetonitrile solvent mixture and analyzed by MS using the same methods as for E. coli extracts.
Minimide (4) FTMS: m/z = 721.43182 [M+H]+; predicted 721.43166 for C36H60N6O8S (Δ = 0.2 ppm). IRMPD of m/z = 721: -1 × isoprene: m/z = 653.36928; predicted 653.36907 for C31H52N6O7S (Δ = 0.2 ppm); -2 × isoprene: m/z = 585.30625; predicted 585.30647 for C26H44N6O7S (Δ = -0.4 ppm).
Quantification of cyanobactin production
An authentic standard of trunkamide was isolated from tropical marine ascidians as previously described and characterized by spectroscopy, including NMR and MS.[14] Clones expressing the dual operon system were grown in 1-10L. Batches were removed and pelleted by centrifugation. Broth and pellet were separately purified both with and without addition of fixed amounts of trunkamide standard prior to purification. Production of cyanobactins was measured on a Micromass ZQ single quad by HPLC-ESIMS.
Supplementary Material
Acknowledgments
We thank J. Muller, K. Parsawar, and C. Nelson (U. Utah) for help with mass spectrometric analysis. We thank C. Ireland (U. Utah) and the government of Solomon Islands for the sample reported herein, which was obtained with appropriate permits and intellectual property agreements. This work was funded by NIH GM071425 and GM071425-S1.
Footnotes
Conflict of interest statement. D.E.R. and E.W.S. are officers in and co-owners of Symbion Discovery, aimed at synthesizing and testing derivatives of cyanobactins.
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Molinski TF. Curr Opin Biotechnol. 2010;21:819–826. doi: 10.1016/j.copbio.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molinski TF, Dalisay DS, Lievens SL, Saludes JP. Nat Rev Drug Discov. 2009;8:69–85. doi: 10.1038/nrd2487. [DOI] [PubMed] [Google Scholar]
- 3.Mayer AM, Glaser KB, Cuevas C, Jacobs RS, Kem W, Little RD, McIntosh JM, Newman DJ, Potts BC, Shuster DE. Trends Pharmacol Sci. 2010;31:255–265. doi: 10.1016/j.tips.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Donia MS, Schmidt EW. In: Comprehensive Natural Products II Chemistry and Biology. Moore BS, Crews P, editors. Vol. 2. Elsevier; Oxford: 2010. pp. 539–558. [Google Scholar]
- 5.Caba JM, Rodriguez IM, Manzanares I, Giralt E, Albericio F. J Org Chem. 2001;66:7568–7574. doi: 10.1021/jo015703t. [DOI] [PubMed] [Google Scholar]
- 6.Wipf P, Uto Y. J Org Chem. 2000;65:1037–1049. doi: 10.1021/jo9914566. [DOI] [PubMed] [Google Scholar]
- 7.Carroll AR, Coll JC, Bourne DJ, MacLeod JK, Zabriskie TM, Ireland CM, Bowden BF. Aust J Chem. 1996;49(6):659–667. [Google Scholar]
- 8.Donia MS, Wang B, Dunbar DC, Desai PV, Patny A, Avery M, Hamann MT. J Nat Prod. 2008;71(6):941–945. doi: 10.1021/np700718p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson AD, Aalbersberg W, Ireland CM. Anticancer Drugs. 2005;16:533–541. doi: 10.1097/00001813-200506000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Zabriskie TM, Mayne CL, Ireland CM. J Am Chem Soc. 1988;110:7919–7920. [Google Scholar]
- 11.Corley DG, Moore RE, Paul VJ. J Am Chem Soc. 1988;110:7920–7922. [Google Scholar]
- 12.Donia MS, Fricke WF, Ravel J, Schmidt EW. PLoS ONE. 2011 doi: 10.1371/journal.pone.0017897. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donia MS, Hathaway BJ, Sudek S, Haygood MG, Rosovitz MJ, Ravel J, Schmidt EW. Nat Chem Biol. 2006;2:729–735. doi: 10.1038/nchembio829. [DOI] [PubMed] [Google Scholar]
- 14.Donia MS, Ravel J, Schmidt EW. Nat Chem Biol. 2008;4:341–343. doi: 10.1038/nchembio.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt EW, Nelson JT, Rasko DA, Sudek S, Eisen JA, Haygood MG, Ravel J. Proc Natl Acad Sci U S A. 2005;102:7315–7320. doi: 10.1073/pnas.0501424102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Zhang F, Kelly WL. Mol Biosyst. 2010;7:82–90. doi: 10.1039/c0mb00129e. [DOI] [PubMed] [Google Scholar]
- 17.Cowan ME. Mar Ecol Prog Ser. 1981;6:335–337. [Google Scholar]
- 18.Schmidt EW. BMC Biol. 2010;8:83. doi: 10.1186/1741-7007-8-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Widdick DA, Dodd HM, Barraille P, White J, Stein TH, Chater KF, Gasson MJ, Bibb MJ. Proc Natl Acad Sci U S A. 2003;100:4316–4321. doi: 10.1073/pnas.0230516100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, van der Donk WA. Proc Natl Acad Sci U S A. 2010;107:10430–10435. doi: 10.1073/pnas.0913677107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowers AA, Acker MG, Koglin A, Walsh CT. J Am Chem Soc. 2010;132:7519–7527. doi: 10.1021/ja102339q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi Y, Yang X, Garg N, van der Donk WA. J Am Chem Soc. 2011;133:2338–2341. doi: 10.1021/ja109044r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisch KM, Gurgui C, Heycke N, van der Sar SA, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. Nat Chem Biol. 2009;5:494–501. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]
- 24.Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. J Nat Prod. 2007;70:67–74. doi: 10.1021/np060361d. [DOI] [PubMed] [Google Scholar]
- 25.Ma H, Kunes S, Schatz PJ, Botstein D. Gene. 1987;58:201–216. doi: 10.1016/0378-1119(87)90376-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.